
Osimertinib Resistance: Molecular Mechanisms and Emerging Treatment Options



Abstract
Simple Summary
Abstract
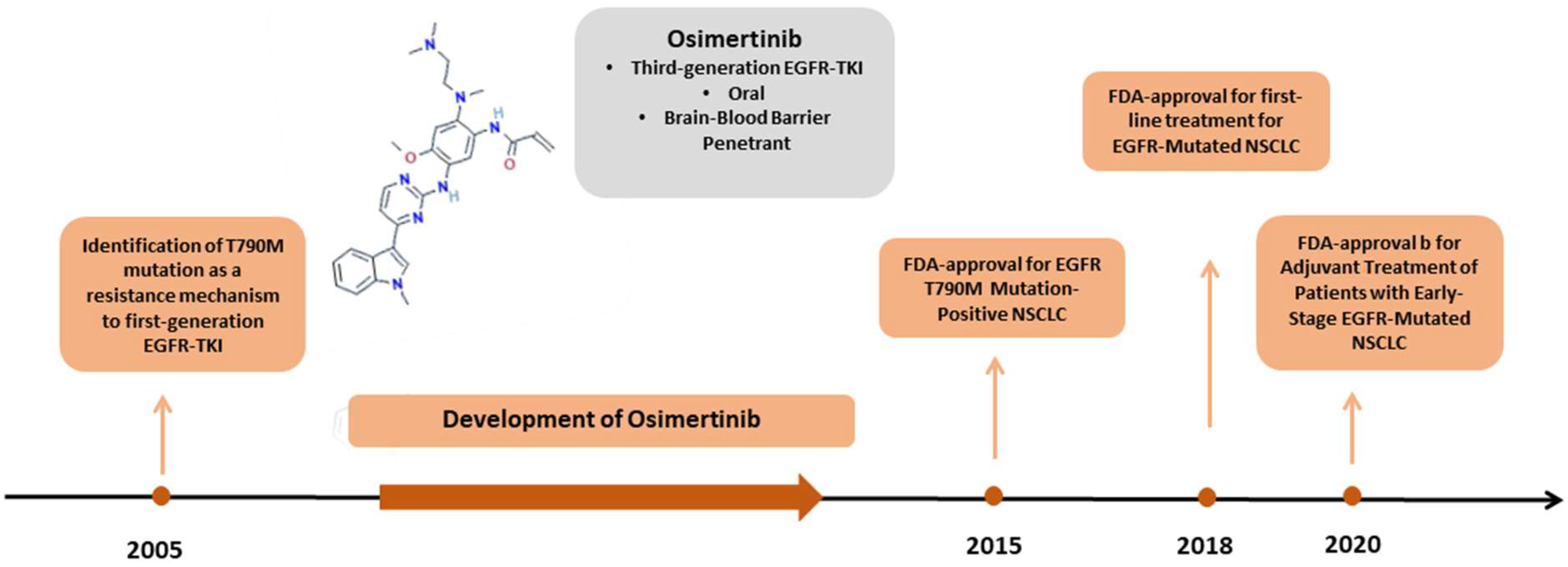
1. Introduction
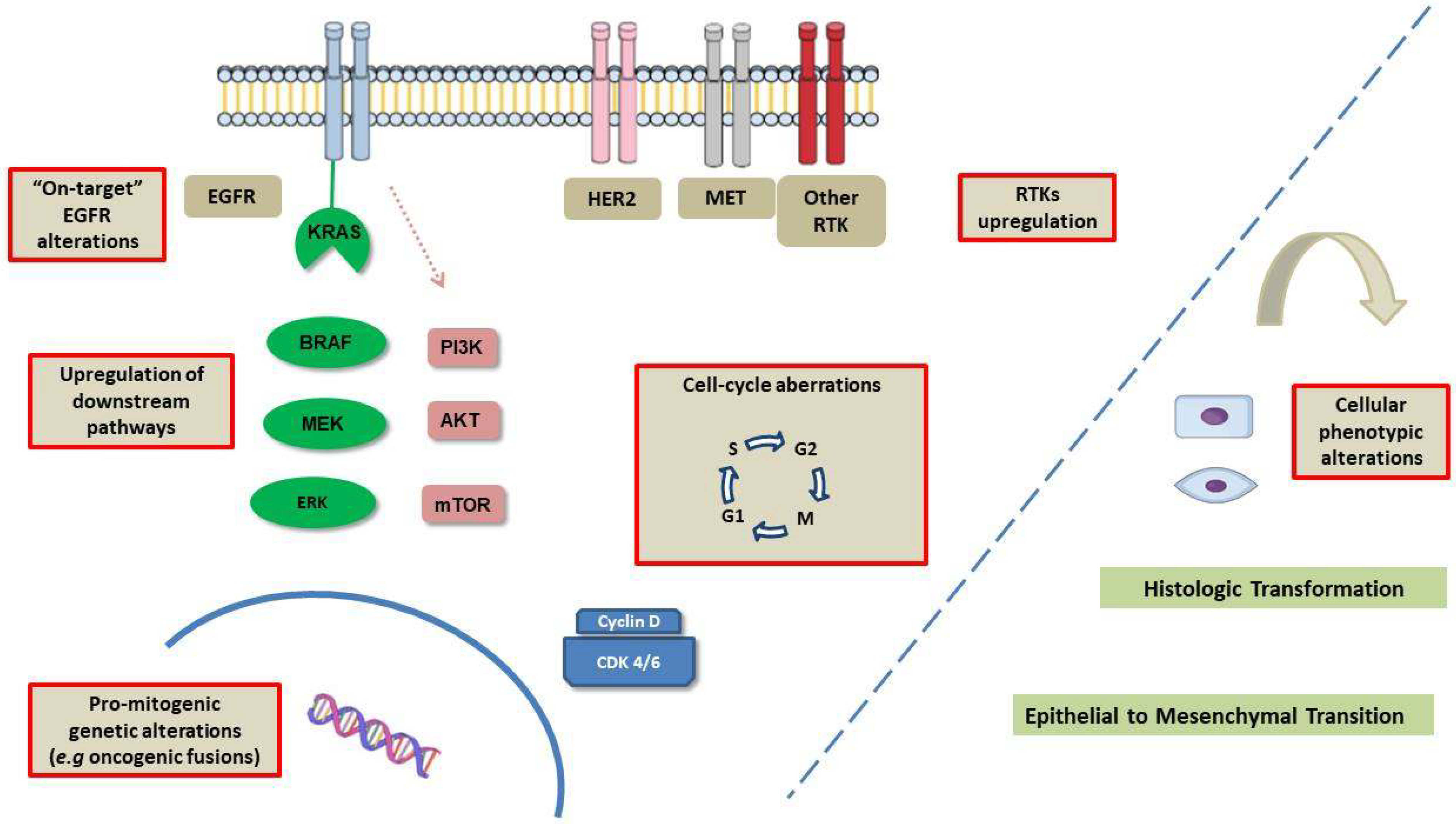
2. Mechanisms of Resistance
2.1. EGFR-Dependent (on-Target) Resistance Mechanisms
2.1.1. T790M Loss
2.1.2. C797S Mutation
2.1.3. Rare EGFR Mutations
2.2. EGFR-Independent (off-Target Resistance Mechanisms)
2.2.1. MET Amplification
2.2.2. HER2 Amplification and HER2 Point Mutations
2.2.3. HER3 Upregulation
2.2.4. RAS-RAF Pathway
2.2.5. PI3K Pathway
2.2.6. Oncogenic Fusions
2.2.7. Cell Cycle Aberrations
2.2.8. Histologic Transformation
2.2.9. Epithelial to Mesenchymal Transition (EMT)
3. Management of Osimertinib-Resistant NSCLC
3.1. Targeted Therapy for EGFR-Dependent Alterations
3.2. Targeted Therapy for EGFR-Independent Alterations
3.2.1. MET Amplification
3.2.2. HER2 Amplification
3.2.3. Osimertinib in Combination with Inhibitors of Oncogenic Genetic Alterations
3.3. Beyond Targeted Therapy
3.3.1. Chemotherapy
3.3.2. Immunotherapy
3.3.3. Combination of Chemotherapy and Antiangiogenic Therapy
3.3.4. Combination of Chemotherapy/Immunotherapy/Antiangiogenic Therapy
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Janne, P.A.; Engelman, J.A.; Johnson, B.E. Epidermal growth factor receptor mutations in non-small-cell lung cancer: Implications for treatment and tumor biology. J. Clin. Oncol. 2005, 23, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Red Brewer, M.; Yun, C.H.; Lai, D.; Lemmon, M.A.; Eck, M.J.; Pao, W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E3595–E3604. [Google Scholar] [CrossRef] [PubMed]
- Koulouris, A.; Tsagkaris, C.; Corriero, A.C.; Metro, G.; Mountzios, G. Resistance to TKIs in EGFR-Mutated Non-Small Cell Lung Cancer: From Mechanisms to New Therapeutic Strategies. Cancers 2022, 14, 3337. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Wu, Y.L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.W.; Kato, T.; et al. Osimertinib in Resected EGFR-Mutated Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.; Wu, Y.-L.; Han, J.-Y.; Ahn, M.-J.; Ramalingam, S.; John, T.; Okamoto, I.; Yang, J.-H.; Bulusu, K.; Laus, G. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29, viii741. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Enrico, D.H.; Lacroix, L.; Rouleau, E.; Scoazec, J.Y.; Loriot, Y.; Tselikas, L.; Jovelet, C.; Planchard, D.; Gazzah, A.; Mezquita, L.; et al. 1526P—Multiple synchronous mechanisms may contribute to osimertinib resistance in non-small cell lung cancer (NSCLC) patients: Insights of the MATCH-R study. Ann. Oncol. 2019, 30, v627. [Google Scholar] [CrossRef]
- Mehlman, C.; Cadranel, J.; Rousseau-Bussac, G.; Lacave, R.; Pujals, A.; Girard, N.; Callens, C.; Gounant, V.; Theou-Anton, N.; Friard, S.; et al. Resistance mechanisms to osimertinib in EGFR-mutated advanced non-small-cell lung cancer: A multicentric retrospective French study. Lung Cancer 2019, 137, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kim, D.; Yoon, S.; Lee, D.H.; Kim, S.W. Exploring the resistance mechanisms of second-line osimertinib and their prognostic implications using next-generation sequencing in patients with non-small-cell lung cancer. Eur. J. Cancer 2021, 148, 202–210. [Google Scholar] [CrossRef]
- Akli, A.; Girard, N.; Fallet, V.; Rousseau-Bussac, G.; Gounant, V.; Friard, S.; Tredaniel, J.; Dujon, C.; Wislez, M.; Duchemann, B.; et al. Histomolecular Resistance Mechanisms to First-Line Osimertinib in EGFR-Mutated Advanced Non-Small Cell Lung Cancer: A Multicentric Retrospective French Study. Target. Oncol. 2022, 17, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Nie, K.; Jiang, H.; Zhang, C.; Geng, C.; Xu, X.; Zhang, L.; Zhang, H.; Zhang, Z.; Lan, K.; Ji, Y. Mutational Profiling of Non-Small-Cell Lung Cancer Resistant to Osimertinib Using Next-Generation Sequencing in Chinese Patients. BioMed Res. Int. 2018, 2018, 9010353. [Google Scholar] [CrossRef]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef]
- Lin, C.C.; Shih, J.Y.; Yu, C.J.; Ho, C.C.; Liao, W.Y.; Lee, J.H.; Tsai, T.H.; Su, K.Y.; Hsieh, M.S.; Chang, Y.L.; et al. Outcomes in patients with non-small-cell lung cancer and acquired Thr790Met mutation treated with osimertinib: A genomic study. Lancet. Respir. Med. 2018, 6, 107–116. [Google Scholar] [CrossRef]
- Passaro, A.; Janne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [PubMed]
- Arulananda, S.; Do, H.; Musafer, A.; Mitchell, P.; Dobrovic, A.; John, T. Combination Osimertinib and Gefitinib in C797S and T790M EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1728–1732. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, J.J.; Huang, J.; Ye, J.Y.; Zhang, X.C.; Tu, H.Y.; Han-Zhang, H.; Wu, Y.L. Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S Responds to Combination Therapy of First- and Third-Generation EGFR TKIs and Shifts Allelic Configuration at Resistance. J. Thorac. Oncol. 2017, 12, 1723–1727. [Google Scholar] [CrossRef]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef]
- Nishino, M.; Suda, K.; Kobayashi, Y.; Ohara, S.; Fujino, T.; Koga, T.; Chiba, M.; Shimoji, M.; Tomizawa, K.; Takemoto, T.; et al. Effects of secondary EGFR mutations on resistance against upfront osimertinib in cells with EGFR-activating mutations in vitro. Lung Cancer 2018, 126, 149–155. [Google Scholar] [CrossRef]
- Rangachari, D.; To, C.; Shpilsky, J.E.; VanderLaan, P.A.; Kobayashi, S.S.; Mushajiang, M.; Lau, C.J.; Paweletz, C.P.; Oxnard, G.R.; Janne, P.A.; et al. EGFR-Mutated Lung Cancers Resistant to Osimertinib through EGFR C797S Respond to First-Generation Reversible EGFR Inhibitors but Eventually Acquire EGFR T790M/C797S in Preclinical Models and Clinical Samples. J. Thorac. Oncol. 2019, 14, 1995–2002. [Google Scholar] [CrossRef]
- Ma, L.; Chen, R.; Wang, F.; Ma, L.L.; Yuan, M.M.; Chen, R.R.; Liu, J. EGFR L718Q mutation occurs without T790M mutation in a lung adenocarcinoma patient with acquired resistance to osimertinib. Ann. Transl. Med. 2019, 7, 207. [Google Scholar] [CrossRef]
- Liu, J.; Jin, B.; Su, H.; Qu, X.; Liu, Y. Afatinib helped overcome subsequent resistance to osimertinib in a patient with NSCLC having leptomeningeal metastasis baring acquired EGFR L718Q mutation: A case report. BMC Cancer 2019, 19, 702. [Google Scholar] [CrossRef]
- Fassunke, J.; Muller, F.; Keul, M.; Michels, S.; Dammert, M.A.; Schmitt, A.; Plenker, D.; Lategahn, J.; Heydt, C.; Bragelmann, J.; et al. Overcoming EGFR(G724S)-mediated osimertinib resistance through unique binding characteristics of second-generation EGFR inhibitors. Nat. Commun. 2018, 9, 4655. [Google Scholar] [CrossRef]
- Minari, R.; Leonetti, A.; Gnetti, L.; Zielli, T.; Ventura, L.; Bottarelli, L.; Lagrasta, C.; La Monica, S.; Petronini, P.G.; Alfieri, R.; et al. Afatinib therapy in case of EGFR G724S emergence as resistance mechanism to osimertinib. Anticancer Drugs 2021, 32, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.P.; Zhang, Y.K.; Westover, D.; Yan, Y.; Qiao, H.; Huang, V.; Du, Z.; Smith, J.A.; Ross, J.S.; Miller, V.A.; et al. On-target Resistance to the Mutant-Selective EGFR Inhibitor Osimertinib Can Develop in an Allele-Specific Manner Dependent on the Original EGFR-Activating Mutation. Clin. Cancer Res. 2019, 25, 3341–3351. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.F.; Zhu, M.L.; Liu, M.M.; Xu, Y.T.; Yuan, L.L.; Bian, J.; Xia, Y.Z.; Kong, L.Y. EGFR mutation mediates resistance to EGFR tyrosine kinase inhibitors in NSCLC: From molecular mechanisms to clinical research. Pharmacol. Res. 2021, 167, 105583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, B.; Zhou, D.; Li, M.; Hu, C. Newly emergent acquired EGFR exon 18 G724S mutation after resistance of a T790M specific EGFR inhibitor osimertinib in non-small-cell lung cancer: A case report. OncoTargets Ther. 2019, 12, 51–56. [Google Scholar] [CrossRef]
- Chen, K.; Zhou, F.; Shen, W.; Jiang, T.; Wu, X.; Tong, X.; Shao, Y.W.; Qin, S.; Zhou, C. Novel Mutations on EGFR Leu792 Potentially Correlate to Acquired Resistance to Osimertinib in Advanced NSCLC. J. Thorac. Oncol. 2017, 12, e65–e68. [Google Scholar] [CrossRef]
- Ou, S.I.; Cui, J.; Schrock, A.B.; Goldberg, M.E.; Zhu, V.W.; Albacker, L.; Stephens, P.J.; Miller, V.A.; Ali, S.M. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer 2017, 108, 228–231. [Google Scholar] [CrossRef]
- Castellano, G.M.; Aisner, J.; Burley, S.K.; Vallat, B.; Yu, H.A.; Pine, S.R.; Ganesan, S. A Novel Acquired Exon 20 EGFR M766Q Mutation in Lung Adenocarcinoma Mediates Osimertinib Resistance but is Sensitive to Neratinib and Poziotinib. J. Thorac. Oncol. 2019, 14, 1982–1988. [Google Scholar] [CrossRef]
- Rosario, M.; Birchmeier, W. How to make tubes: Signaling by the Met receptor tyrosine kinase. Trends Cell Biol. 2003, 13, 328–335. [Google Scholar] [CrossRef]
- Coleman, N.; Hong, L.; Zhang, J.; Heymach, J.; Hong, D.; Le, X. Beyond epidermal growth factor receptor: MET amplification as a general resistance driver to targeted therapy in oncogene-driven non-small-cell lung cancer. ESMO Open 2021, 6, 100319. [Google Scholar] [CrossRef]
- Piper-Vallillo, A.J.; Sequist, L.V.; Piotrowska, Z. Emerging Treatment Paradigms for EGFR-Mutant Lung Cancers Progressing on Osimertinib: A Review. J. Clin. Oncol. 2020, JCO1903123. [Google Scholar] [CrossRef]
- Fuchs, V.; Roisman, L.; Kian, W.; Daniel, L.; Dudnik, J.; Nechushtan, H.; Goldstein, I.; Dvir, A.; Soussan-Gutman, L.; Grinberg, R.; et al. The impact of osimertinib’ line on clonal evolution in EGFRm NSCLC through NGS-based liquid biopsy and overcoming strategies for resistance. Lung Cancer 2021, 153, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Nie, N.; Li, J.; Zhang, J.; Dai, J.; Liu, Z.; Ding, Z.; Wang, Y.; Zhu, M.; Hu, C.; Han, R.; et al. First-Line Osimertinib in Patients with EGFR-Mutated Non-Small Cell Lung Cancer: Effectiveness, Resistance Mechanisms, and Prognosis of Different Subsequent Treatments. Clin. Med. Insights Oncol. 2022, 16, 11795549221134735. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Marti, A.; Felip, E.; Matito, J.; Mereu, E.; Navarro, A.; Cedres, S.; Pardo, N.; Martinez de Castro, A.; Remon, J.; Miquel, J.M.; et al. Dual MET and ERBB inhibition overcomes intratumor plasticity in osimertinib-resistant-advanced non-small-cell lung cancer (NSCLC). Ann. Oncol. 2017, 28, 2451–2457. [Google Scholar] [CrossRef] [PubMed]
- Suzawa, K.; Offin, M.; Schoenfeld, A.J.; Plodkowski, A.J.; Odintsov, I.; Lu, D.; Lockwood, W.W.; Arcila, M.E.; Rudin, C.M.; Drilon, A.; et al. Acquired MET Exon 14 Alteration Drives Secondary Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor in EGFR-Mutated Lung Cancer. JCO Precis. Oncol. 2019, 3, PO.19.00011. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, E.; De Carlo, E.; Del Conte, A.; Stanzione, B.; Revelant, A.; Fassetta, K.; Spina, M.; Bearz, A. Acquired Resistance to Osimertinib in EGFR-Mutated Non-Small Cell Lung Cancer: How Do We Overcome It? Int. J. Mol. Sci. 2022, 23, 6936. [Google Scholar] [CrossRef]
- Friedlaender, A.; Subbiah, V.; Russo, A.; Banna, G.L.; Malapelle, U.; Rolfo, C.; Addeo, A. EGFR and HER2 exon 20 insertions in solid tumours: From biology to treatment. Nat. Rev. Clin. Oncol. 2022, 19, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Liao, B.C.; Liao, W.Y.; Markovets, A.; Stetson, D.; Thress, K.; Yang, J.C. Exon 16-Skipping HER2 as a Novel Mechanism of Osimertinib Resistance in EGFR L858R/T790M-Positive Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2020, 15, 50–61. [Google Scholar] [CrossRef]
- Romaniello, D.; Marrocco, I.; Belugali Nataraj, N.; Ferrer, I.; Drago-Garcia, D.; Vaknin, I.; Oren, R.; Lindzen, M.; Ghosh, S.; Kreitman, M.; et al. Targeting HER3, a Catalytically Defective Receptor Tyrosine Kinase, Prevents Resistance of Lung Cancer to a Third-Generation EGFR Kinase Inhibitor. Cancers 2020, 12, 2394. [Google Scholar] [CrossRef]
- Yonesaka, K.; Takegawa, N.; Watanabe, S.; Haratani, K.; Kawakami, H.; Sakai, K.; Chiba, Y.; Maeda, N.; Kagari, T.; Hirotani, K.; et al. An HER3-targeting antibody-drug conjugate incorporating a DNA topoisomerase I inhibitor U3-1402 conquers EGFR tyrosine kinase inhibitor-resistant NSCLC. Oncogene 2019, 38, 1398–1409. [Google Scholar] [CrossRef]
- Haikala, H.M.; Lopez, T.; Kohler, J.; Eser, P.O.; Xu, M.; Zeng, Q.; Teceno, T.J.; Ngo, K.; Zhao, Y.; Ivanova, E.V.; et al. EGFR Inhibition Enhances the Cellular Uptake and Antitumor-Activity of the HER3 Antibody-Drug Conjugate HER3-DXd. Cancer Res. 2022, 82, 130–141. [Google Scholar] [CrossRef]
- Vicencio, J.M.; Evans, R.; Green, R.; An, Z.; Deng, J.; Treacy, C.; Mustapha, R.; Monypenny, J.; Costoya, C.; Lawler, K.; et al. Osimertinib and anti-HER3 combination therapy engages immune dependent tumor toxicity via STING activation in trans. Cell Death Dis. 2022, 13, 274. [Google Scholar] [CrossRef] [PubMed]
- Tsamis, I.; Gomatou, G.; Chachali, S.P.; Trontzas, I.P.; Patriarcheas, V.; Panagiotou, E.; Kotteas, E. BRAF/MEK inhibition in NSCLC: Mechanisms of resistance and how to overcome it. Clin. Transl. Oncol. 2023, 25, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, D.M.; Stehr, H.; Scherer, F.; Karlovich, C.A.; et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun. 2016, 7, 11815. [Google Scholar] [CrossRef]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- Minari, R.; Bordi, P.; La Monica, S.; Squadrilli, A.; Leonetti, A.; Bottarelli, L.; Azzoni, C.; Lagrasta, C.A.M.; Gnetti, L.; Campanini, N.; et al. Concurrent Acquired BRAF V600E Mutation and MET Amplification as Resistance Mechanism of First-Line Osimertinib Treatment in a Patient with EGFR-Mutated NSCLC. J. Thorac. Oncol. 2018, 13, e89–e91. [Google Scholar] [CrossRef]
- Ho, C.C.; Liao, W.Y.; Lin, C.A.; Shih, J.Y.; Yu, C.J.; Yang, J.C. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Osimertinib. J. Thorac. Oncol. 2017, 12, 567–572. [Google Scholar] [CrossRef]
- Xie, Z.; Gu, Y.; Xie, X.; Lin, X.; Ouyang, M.; Qin, Y.; Zhang, J.; Lizaso, A.; Chen, S.; Zhou, C. Lung Adenocarcinoma Harboring Concomitant EGFR Mutations and BRAF V600E Responds to a Combination of Osimertinib and Vemurafenib to Overcome Osimertinib Resistance. Clin. Lung Cancer 2021, 22, e390–e394. [Google Scholar] [CrossRef]
- Meng, P.; Koopman, B.; Kok, K.; Ter Elst, A.; Schuuring, E.; van Kempen, L.C.; Timens, W.; Hiltermann, T.J.N.; Groen, H.J.M.; van den Berg, A.; et al. Combined osimertinib, dabrafenib and trametinib treatment for advanced non-small-cell lung cancer patients with an osimertinib-induced BRAF V600E mutation. Lung Cancer 2020, 146, 358–361. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.S.; Lee, B.; Kim, H.K.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Lee, S.H. Genomic landscape of acquired resistance to third-generation EGFR tyrosine kinase inhibitors in EGFR T790M-mutant non-small cell lung cancer. Cancer 2020, 126, 2704–2712. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef]
- Park, S.; Shim, J.H.; Lee, B.; Cho, I.; Park, W.Y.; Kim, Y.; Lee, S.H.; Choi, Y.; Han, J.; Ahn, J.S.; et al. Paired genomic analysis of squamous cell carcinoma transformed from EGFR-mutated lung adenocarcinoma. Lung Cancer 2019, 134, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.; Woo, K.M.; Sima, C.S.; Plodkowski, A.; Hellmann, M.D.; Chaft, J.E.; Kris, M.G.; Arcila, M.E.; Ladanyi, M.; Drilon, A. Impact of Concurrent PIK3CA Mutations on Response to EGFR Tyrosine Kinase Inhibition in EGFR-Mutant Lung Cancers and on Prognosis in Oncogene-Driven Lung Adenocarcinomas. J. Thorac. Oncol. 2015, 10, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.J.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef]
- Zeng, L.; Yang, N.; Zhang, Y. GOPC-ROS1 Rearrangement as an Acquired Resistance Mechanism to Osimertinib and Responding to Crizotinib Combined Treatments in Lung Adenocarcinoma. J. Thorac. Oncol. 2018, 13, e114–e116. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, Z.; Isozaki, H.; Lennerz, J.K.; Gainor, J.F.; Lennes, I.T.; Zhu, V.W.; Marcoux, N.; Banwait, M.K.; Digumarthy, S.R.; Su, W.; et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov. 2018, 8, 1529–1539. [Google Scholar] [CrossRef]
- Rotow, J.; Patel, J.; Hanley, M.; Yu, H.; Goldman, J.; Nechustan, H.; Scheffler, M.; Awad, M.; Clifford, S.; Santucci, A.; et al. FP14.07 Combination Osimertinib plus Selpercatinib for EGFR-mutant Non-Small Cell Lung Cancer (NSCLC) with Acquired RET fusions. J. Thorac. Oncol. 2021, 16, S230. [Google Scholar] [CrossRef]
- Freydman, J.; Henshaw, L.; Patel, J.V.; Smith, C.E.; Everett, P.C. Combination EGFR and RET Inhibition in Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC. Ann. Pharmacother. 2022, 56, 503–504. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.B.; Zhu, V.W.; Hsieh, W.S.; Madison, R.; Creelan, B.; Silberberg, J.; Costin, D.; Bharne, A.; Bonta, I.; Bosemani, T.; et al. Receptor Tyrosine Kinase Fusions and BRAF Kinase Fusions are Rare but Actionable Resistance Mechanisms to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2018, 13, 1312–1323. [Google Scholar] [CrossRef]
- Offin, M.; Somwar, R.; Rekhtman, N.; Benayed, R.; Chang, J.C.; Plodkowski, A.; Lui, A.J.W.; Eng, J.; Rosenblum, M.; Li, B.T.; et al. Acquired ALK and RET Gene Fusions as Mechanisms of Resistance to Osimertinib in EGFR-Mutant Lung Cancers. JCO Precis Oncol 2018, 2, PO.18.00126. [Google Scholar] [CrossRef]
- Panagiotou, E.; Gomatou, G.; Trontzas, I.P.; Syrigos, N.; Kotteas, E. Cyclin-dependent kinase (CDK) inhibitors in solid tumors: A review of clinical trials. Clin. Transl. Oncol. 2022, 24, 161–192. [Google Scholar] [CrossRef]
- La Monica, S.; Fumarola, C.; Cretella, D.; Bonelli, M.; Minari, R.; Cavazzoni, A.; Digiacomo, G.; Galetti, M.; Volta, F.; Mancini, M.; et al. Efficacy of the CDK4/6 Dual Inhibitor Abemaciclib in EGFR-Mutated NSCLC Cell Lines with Different Resistance Mechanisms to Osimertinib. Cancers 2020, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. CDK4/6 inhibitor palbociclib overcomes acquired resistance to third-generation EGFR inhibitor osimertinib in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 2389–2397. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Chan, J.M.; Kubota, D.; Sato, H.; Rizvi, H.; Daneshbod, Y.; Chang, J.C.; Paik, P.K.; Offin, M.; Arcila, M.E.; et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations As Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2020, 26, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Marcoux, N.; Gettinger, S.N.; O’Kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; Bonomi, P.D.; et al. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J. Clin. Oncol. 2019, 37, 278–285. [Google Scholar] [CrossRef]
- Hakozaki, T.; Kitazono, M.; Takamori, M.; Kiriu, T. Combined Small and Squamous Transformation in EGFR-mutated Lung Adenocarcinoma. Intern. Med. 2020, 59, 1291–1294. [Google Scholar] [CrossRef]
- Lee, J.K.; Lee, J.; Kim, S.; Kim, S.; Youk, J.; Park, S.; An, Y.; Keam, B.; Kim, D.W.; Heo, D.S.; et al. Clonal History and Genetic Predictors of Transformation Into Small-Cell Carcinomas From Lung Adenocarcinomas. J. Clin. Oncol. 2017, 35, 3065–3074. [Google Scholar] [CrossRef]
- Offin, M.; Chan, J.M.; Tenet, M.; Rizvi, H.A.; Shen, R.; Riely, G.J.; Rekhtman, N.; Daneshbod, Y.; Quintanal-Villalonga, A.; Penson, A.; et al. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at risk for Histologic Transformation and Inferior Clinical Outcomes. J. Thorac. Oncol. 2019, 14, 1784–1793. [Google Scholar] [CrossRef]
- Roca, E.; Gurizzan, C.; Amoroso, V.; Vermi, W.; Ferrari, V.; Berruti, A. Outcome of patients with lung adenocarcinoma with transformation to small-cell lung cancer following tyrosine kinase inhibitors treatment: A systematic review and pooled analysis. Cancer Treat. Rev. 2017, 59, 117–122. [Google Scholar] [CrossRef]
- Jiang, X.M.; Xu, Y.L.; Yuan, L.W.; Zhang, L.L.; Huang, M.Y.; Ye, Z.H.; Su, M.X.; Chen, X.P.; Zhu, H.; Ye, R.D.; et al. TGFbeta2-mediated epithelial-mesenchymal transition and NF-kappaB pathway activation contribute to osimertinib resistance. Acta Pharmacol. Sin. 2021, 42, 451–459. [Google Scholar] [CrossRef]
- Weng, C.H.; Chen, L.Y.; Lin, Y.C.; Shih, J.Y.; Lin, Y.C.; Tseng, R.Y.; Chiu, A.C.; Yeh, Y.H.; Liu, C.; Lin, Y.T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef]
- Castosa, R.; Martinez-Iglesias, O.; Roca-Lema, D.; Casas-Pais, A.; Díaz-Díaz, A.; Iglesias, P.; Santamarina, I.; Graña, B.; Calvo, L.; Valladares-Ayerbes, M.; et al. Hakai overexpression effectively induces tumour progression and metastasis in vivo. Sci. Rep. 2018, 8, 3466. [Google Scholar] [CrossRef]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. Targeting the EMT transcription factor Snail overcomes resistance to osimertinib in EGFR-mutant non-small cell lung cancer. Thorac. Cancer 2021, 12, 1708–1715. [Google Scholar] [CrossRef]
- Yochum, Z.A.; Cades, J.; Wang, H.; Chatterjee, S.; Simons, B.W.; O’Brien, J.P.; Khetarpal, S.K.; Lemtiri-Chlieh, G.; Myers, K.V.; Huang, E.H.; et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 2019, 38, 656–670. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- Pascual, J.; Attard, G.; Bidard, F.C.; Curigliano, G.; De Mattos-Arruda, L.; Diehn, M.; Italiano, A.; Lindberg, J.; Merker, J.D.; Montagut, C.; et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2022, 33, 750–768. [Google Scholar] [CrossRef]
- Chic, N.; Mayo-de-Las-Casas, C.; Reguart, N. Successful Treatment with Gefitinib in Advanced Non-Small Cell Lung Cancer after Acquired Resistance to Osimertinib. J. Thorac. Oncol. 2017, 12, e78–e80. [Google Scholar] [CrossRef]
- Fang, W.; Huang, Y.; Gan, J.; Zheng, Q.; Zhang, L. Emergence of EGFR G724S After Progression on Osimertinib Responded to Afatinib Monotherapy. J. Thorac. Oncol. 2020, 15, e36–e37. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhao, Y.; Shen, S.; Gu, L.; Niu, X.; Xu, Y.; Zhang, T.; Xiang, J.; Mao, X.; Lu, S. Durable Clinical Response of Lung Adenocarcinoma Harboring EGFR 19Del/T790M/in trans-C797S to Combination Therapy of First- and Third-Generation EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2019, 14, e157–e159. [Google Scholar] [CrossRef]
- Liu, Y.; Lai, M.; Li, S.; Wang, Y.; Feng, F.; Zhang, T.; Tong, L.; Zhang, M.; Chen, H.; Chen, Y.; et al. LS-106, a novel EGFR inhibitor targeting C797S, exhibits antitumor activities both in vitro and in vivo. Cancer Sci. 2022, 113, 709–720. [Google Scholar] [CrossRef]
- Eno, M.S.; Brubaker, J.D.; Campbell, J.E.; De Savi, C.; Guzi, T.J.; Williams, B.D.; Wilson, D.; Wilson, K.; Brooijmans, N.; Kim, J.; et al. Discovery of BLU-945, a Reversible, Potent, and Wild-Type-Sparing Next-Generation EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung Cancer. J. Med. Chem. 2022, 65, 9662–9677. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef]
- To, C.; Beyett, T.S.; Jang, J.; Feng, W.W.; Bahcall, M.; Haikala, H.M.; Shin, B.H.; Heppner, D.E.; Rana, J.K.; Leeper, B.A.; et al. An allosteric inhibitor against the therapy-resistant mutant forms of EGFR in non-small cell lung cancer. Nat. Cancer 2022, 3, 402–417. [Google Scholar] [CrossRef]
- Haura, E.B.; Cho, B.C.; Lee, J.S.; Han, J.-Y.; Lee, K.H.; Sanborn, R.E.; Govindan, R.; Cho, E.K.; Kim, S.-W.; Reckamp, K.L.; et al. JNJ-61186372 (JNJ-372), an EGFR-cMet bispecific antibody, in EGFR-driven advanced non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2019, 37, 9009. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, N.; Zhang, Y.; Li, L.; Han, R.; Zhu, M.; Feng, M.; Chen, H.; Lizaso, A.; Qin, T.; et al. Effective Treatment of Lung Adenocarcinoma Harboring EGFR-Activating Mutation, T790M, and cis-C797S Triple Mutations by Brigatinib and Cetuximab Combination Therapy. J. Thorac. Oncol. 2020, 15, 1369–1375. [Google Scholar] [CrossRef]
- Deng, L.; Kiedrowski, L.A.; Ravera, E.; Cheng, H.; Halmos, B. Response to Dual Crizotinib and Osimertinib Treatment in a Lung Cancer Patient with MET Amplification Detected by Liquid Biopsy Who Acquired Secondary Resistance to EGFR Tyrosine Kinase Inhibition. J. Thorac. Oncol. 2018, 13, e169–e172. [Google Scholar] [CrossRef]
- York, E.R.; Varella-Garcia, M.; Bang, T.J.; Aisner, D.L.; Camidge, D.R. Tolerable and Effective Combination of Full-Dose Crizotinib and Osimertinib Targeting MET Amplification Sequentially Emerging after T790M Positivity in EGFR-Mutant Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, e85–e88. [Google Scholar] [CrossRef]
- Wang, Y.; Tian, P.; Xia, L.; Li, L.; Han, R.; Zhu, M.; Lizaso, A.; Qin, T.; Li, M.; Yu, B.; et al. The clinical efficacy of combinatorial therapy of EGFR-TKI and crizotinib in overcoming MET amplification-mediated resistance from prior EGFR-TKI therapy. Lung Cancer 2020, 146, 165–173. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Yang, J.C.; Yu, H.; Kim, S.W.; Saka, H.; Horn, L.; Goto, K.; Ohe, Y.; Mann, H.; Thress, K.S.; et al. TATTON: A multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 2020, 31, 507–516. [Google Scholar] [CrossRef]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Li, B.T.; Michelini, F.; Misale, S.; Cocco, E.; Baldino, L.; Cai, Y.; Shifman, S.; Tu, H.Y.; Myers, M.L.; Xu, C.; et al. HER2-Mediated Internalization of Cytotoxic Agents in ERBB2 Amplified or Mutant Lung Cancers. Cancer Discov. 2020, 10, 674–687. [Google Scholar] [CrossRef]
- Nakagawa, K.; Nagasaka, M.; Felip, E.; Pacheco, J.; Baik, C.; Goto, Y.; Saltos, A.; Li, B.; Udagawa, H.; Gadgeel, S.; et al. OA04.05 Trastuzumab Deruxtecan in HER2-Overexpressing Metastatic Non-Small Cell Lung Cancer: Interim Results of DESTINY-Lung01. J. Thorac. Oncol. 2021, 16, S109–S110. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, H.; Wang, Y.; Yang, L.; Zhou, C.; Yin, W.; Wang, G.; Mao, X.; Xiang, J.; Li, B.; et al. Clinical Characteristics and Molecular Patterns of RET-Rearranged Lung Cancer in Chinese Patients. Oncol. Res. 2019, 27, 575–582. [Google Scholar] [CrossRef]
- Nelson, A.W.; Schrock, A.B.; Pavlick, D.C.; Ali, S.M.; Atkinson, E.C.; Chachoua, A. Novel SPECC1L-MET Fusion Detected in Circulating Tumor DNA in a Patient with Lung Adenocarcinoma following Treatment with Erlotinib and Osimertinib. J. Thorac. Oncol. 2019, 14, e27–e29. [Google Scholar] [CrossRef]
- Batra, U.; Sharma, M.; Amrith, B.P.; Mehta, A.; Jain, P. EML4-ALK Fusion as a Resistance Mechanism to Osimertinib and Its Successful Management with Osimertinib and Alectinib: Case Report and Review of the Literature. Clin. Lung Cancer 2020, 21, e597–e600. [Google Scholar] [CrossRef]
- Mu, Y.; Hao, X.; Yang, K.; Ma, D.; Wang, S.; Xu, Z.; Li, J.; Xing, P. Clinical Modality of Resistance and Subsequent Management of Patients with Advanced Non-small Cell Lung Cancer Failing Treatment with Osimertinib. Target. Oncol. 2019, 14, 335–342. [Google Scholar] [CrossRef]
- Fu, K.; Xie, F.; Wang, F.; Fu, L. Therapeutic strategies for EGFR-mutated non-small cell lung cancer patients with osimertinib resistance. J. Hematol. Oncol. 2022, 15, 173. [Google Scholar] [CrossRef]
- Lee, C.K.; Man, J.; Lord, S.; Cooper, W.; Links, M.; Gebski, V.; Herbst, R.S.; Gralla, R.J.; Mok, T.; Yang, J.C. Clinical and Molecular Characteristics Associated with Survival Among Patients Treated with Checkpoint Inhibitors for Advanced Non-Small Cell Lung Carcinoma: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 210–216. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Kim, C.G.; Kim, K.H.; Pyo, K.H.; Xin, C.F.; Hong, M.H.; Ahn, B.C.; Kim, Y.; Choi, S.J.; Yoon, H.I.; Lee, J.G.; et al. Hyperprogressive disease during PD-1/PD-L1 blockade in patients with non-small-cell lung cancer. Ann. Oncol. 2019, 30, 1104–1113. [Google Scholar] [CrossRef]
- Zhou, Q.; Xu, C.R.; Cheng, Y.; Liu, Y.P.; Chen, G.Y.; Cui, J.W.; Yang, N.; Song, Y.; Li, X.L.; Lu, S.; et al. Bevacizumab plus erlotinib in Chinese patients with untreated, EGFR-mutated, advanced NSCLC (ARTEMIS-CTONG1509): A multicenter phase 3 study. Cancer Cell 2021, 39, 1279–1291.e3. [Google Scholar] [CrossRef]
- Kenmotsu, H.; Wakuda, K.; Mori, K.; Kato, T.; Sugawara, S.; Kirita, K.; Yoneshima, Y.; Azuma, K.; Nishino, K.; Teraoka, S.; et al. Randomized Phase 2 Study of Osimertinib Plus Bevacizumab Versus Osimertinib for Untreated Patients with Nonsquamous NSCLC Harboring EGFR Mutations: WJOG9717L Study. J. Thorac. Oncol. 2022, 17, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Mok, T.S.K.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sibilot, D.; et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): Key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet. Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]
- Nogami, N.; Barlesi, F.; Socinski, M.A.; Reck, M.; Thomas, C.A.; Cappuzzo, F.; Mok, T.S.K.; Finley, G.; Aerts, J.G.; Orlandi, F.; et al. IMpower150 Final Exploratory Analyses for Atezolizumab Plus Bevacizumab and Chemotherapy in Key NSCLC Patient Subgroups with EGFR Mutations or Metastases in the Liver or Brain. J. Thorac. Oncol. 2022, 17, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.C.; Tsang, K.C.; Choi, H.C.; Lee, V.H.; Lam, K.O.; Chiang, C.L.; So, T.H.; Chan, W.W.; Nyaw, S.F.; Lim, F.; et al. Combination atezolizumab, bevacizumab, pemetrexed and carboplatin for metastatic EGFR mutated NSCLC after TKI failure. Lung Cancer 2021, 159, 18–26. [Google Scholar] [CrossRef]
- Lu, S.; Wu, L.; Jian, H.; Chen, Y.; Wang, Q.; Fang, J.; Wang, Z.; Hu, Y.; Sun, M.; Han, L.; et al. Sintilimab plus bevacizumab biosimilar IBI305 and chemotherapy for patients with EGFR-mutated non-squamous non-small-cell lung cancer who progressed on EGFR tyrosine-kinase inhibitor therapy (ORIENT-31): First interim results from a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2022, 23, 1167–1179. [Google Scholar] [CrossRef]
- Passaro, A.; Leighl, N.; Blackhall, F.; Popat, S.; Kerr, K.; Ahn, M.J.; Arcila, M.E.; Arrieta, O.; Planchard, D.; de Marinis, F.; et al. ESMO expert consensus statements on the management of EGFR mutant non-small-cell lung cancer. Ann. Oncol. 2022, 33, 466–487. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Author (Year) | Number of Patients | Line of Therapy | EGFR-Dependent Mechanisms (On-Target) | EGFR-Independent Mechanisms (Off-Target) | [Ref] |
|---|---|---|---|---|---|
| Papadimitrakopoulou et al. (2018) | 73 | 2nd line | T790M loss (49%) C797 mutations (15%; 10 patients with C797S, 1 patient with C797G) | MET amplification (19%) HER2 amplification (5%) PIK3CA amplification (4%) BRAFmut (V600E) (4%) KRAS mutation (1%) PIK3CA mut (E545K) (1%) FGFR/RET/NTRK fusions (4%) | [11] |
| Oxnard et al. (2018) | 41 | 2nd line | T790Mloss (63%) C797S (22%) | SCLC transformation (15%) MET amplification (10%) BRAF mutation (5%) PIK3CA mutation (5%) KRAS mutation (2%) CCDC6-RET fusion (2%) FGFR fusion (2%) BRAF fusion (2%) | [12] |
| Ramalingam et al. (2018) | 91 | 1st line | C797S (7%) | MET amplification (15%) HER2 ampl, PIK3CAmut, RAS mut (2–7%) | [13] |
| Enrico et al. (2019) | 31 | Any | C797S (29%) L817Q (6%) EGFR amplification (3%) | Oncogenic fusions (RET, MET, BRAF, ALK, FGFR3, and NTRK1) 16% BRAF mutation (V600E) 6% (co-existing with C797S) MET amplification (3%) HER2 amplification (3%) KRAS mutation (3%) PIK3CA mutation (3%) | [14] |
| Mehlman et al. (2019) | 73 | Any | T790M loss (68%) C797S (12%) | MET amplification (11%) Histologic transformation (9% of patients who underwent a tissue biopsy) HER2 amplification (3%) BRAF mutation (V600E) (1%) | [15] |
| Lee et al. (2021) | 34 | 2nd line | T790Mloss (65%) C797S (12%) | SCLC transformation (9%) Squamous cell carcinoma transformation (5%) MET amplification (15%) | [16] |
| Akli et al. (2022) | 27 | 1st line | C797S (11%) | MET amplification (15%) HER2 amplification (4%) SCLC transformation RET fusion (4%) | [17] |
| Nie et al. (2022) | 21 | 1st line | C797S (24%) L718Q (5%) EGFR amplification (1%) | MET amplification (29%) HER2 amplification (10%) PTEN loss (5%) PIK3CA mutation (5%) | [18] |
| EGFR Mutations | 3rd-Generation EGFR-TKI Alone | 1st/2nd Generation with or without 3rd Generation EGFR-TKI | [Ref] |
|---|---|---|---|
| del_19 or L858R/T790M/C797S (in cis) | Resistance | Resistance | [25] |
| del_19 or L858R/T790M/C797S (in trans) | Resistance | Sensitivity (1st-gen plus 3rd-gen EGFR-TKIs) | [22,23,24] |
| del_19 or L858R/C797S | Resistance | Sensitivity * (1st-gen plus 3rd-gen EGFR-TKIs) | [26] |
| L858R/T790M/L718 | Resistance | Resistance | [28] |
| L858R/L718 | Resistance | Sensitivity (2nd-gen EGFR-TKI) | [29] |
| del_19/G724S | Resistance | Sensitivity (2nd-gen EGFR-TKI) | [30,31] |
| L858R/G724S | Sensitivity ** | Sensitivity | [32] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomatou, G.; Syrigos, N.; Kotteas, E. Osimertinib Resistance: Molecular Mechanisms and Emerging Treatment Options. Cancers 2023, 15, 841. https://doi.org/10.3390/cancers15030841
Gomatou G, Syrigos N, Kotteas E. Osimertinib Resistance: Molecular Mechanisms and Emerging Treatment Options. Cancers. 2023; 15(3):841. https://doi.org/10.3390/cancers15030841
Chicago/Turabian StyleGomatou, Georgia, Nikolaos Syrigos, and Elias Kotteas. 2023. "Osimertinib Resistance: Molecular Mechanisms and Emerging Treatment Options" Cancers 15, no. 3: 841. https://doi.org/10.3390/cancers15030841
APA StyleGomatou, G., Syrigos, N., & Kotteas, E. (2023). Osimertinib Resistance: Molecular Mechanisms and Emerging Treatment Options. Cancers, 15(3), 841. https://doi.org/10.3390/cancers15030841