
Multifactorial Diseases of the Heart, Kidneys, Lungs, and Liver and Incident Cancer: Epidemiology and Shared Mechanisms

 , , and
, , and
Abstract
Simple Summary
Abstract
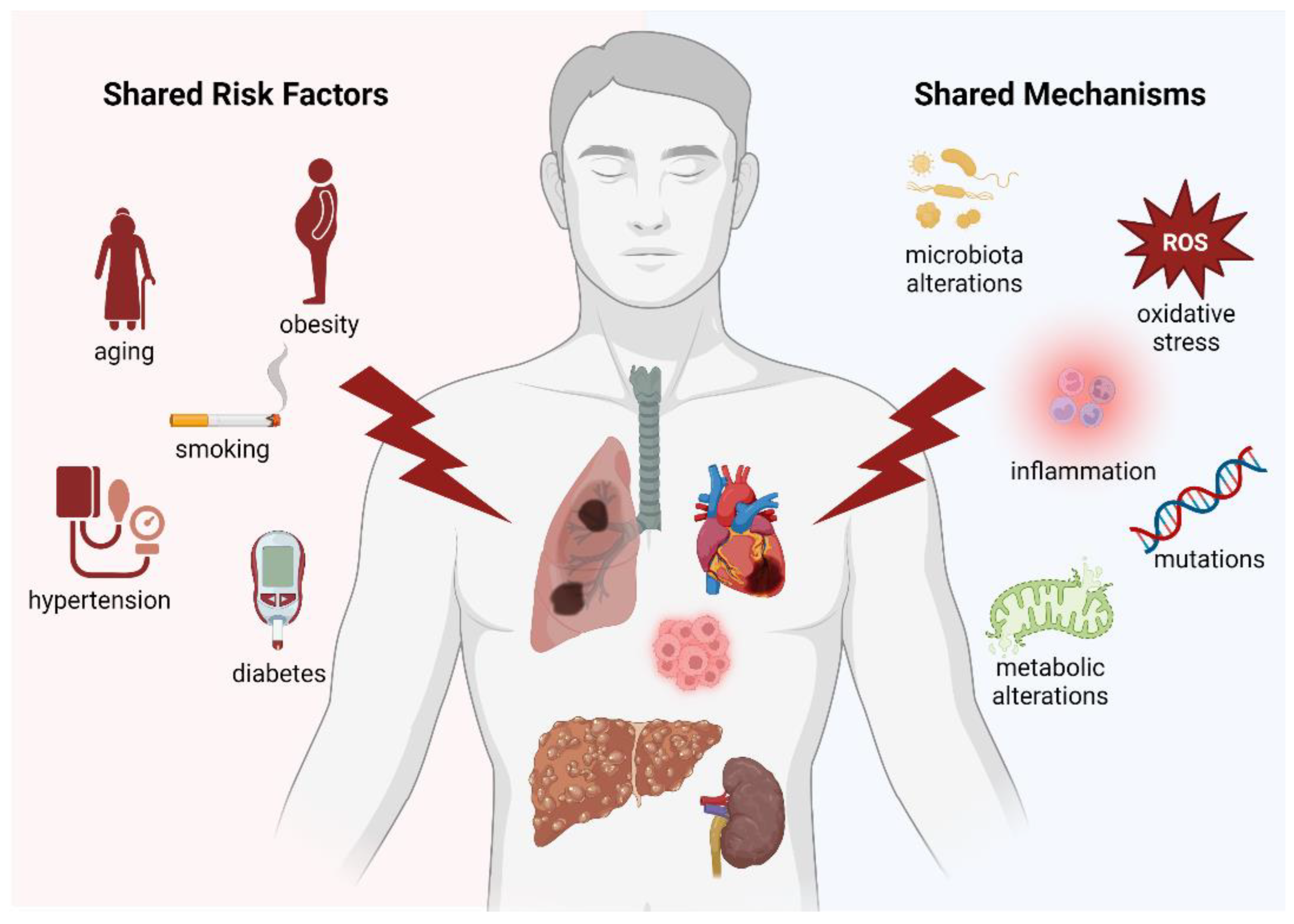
1. Introduction
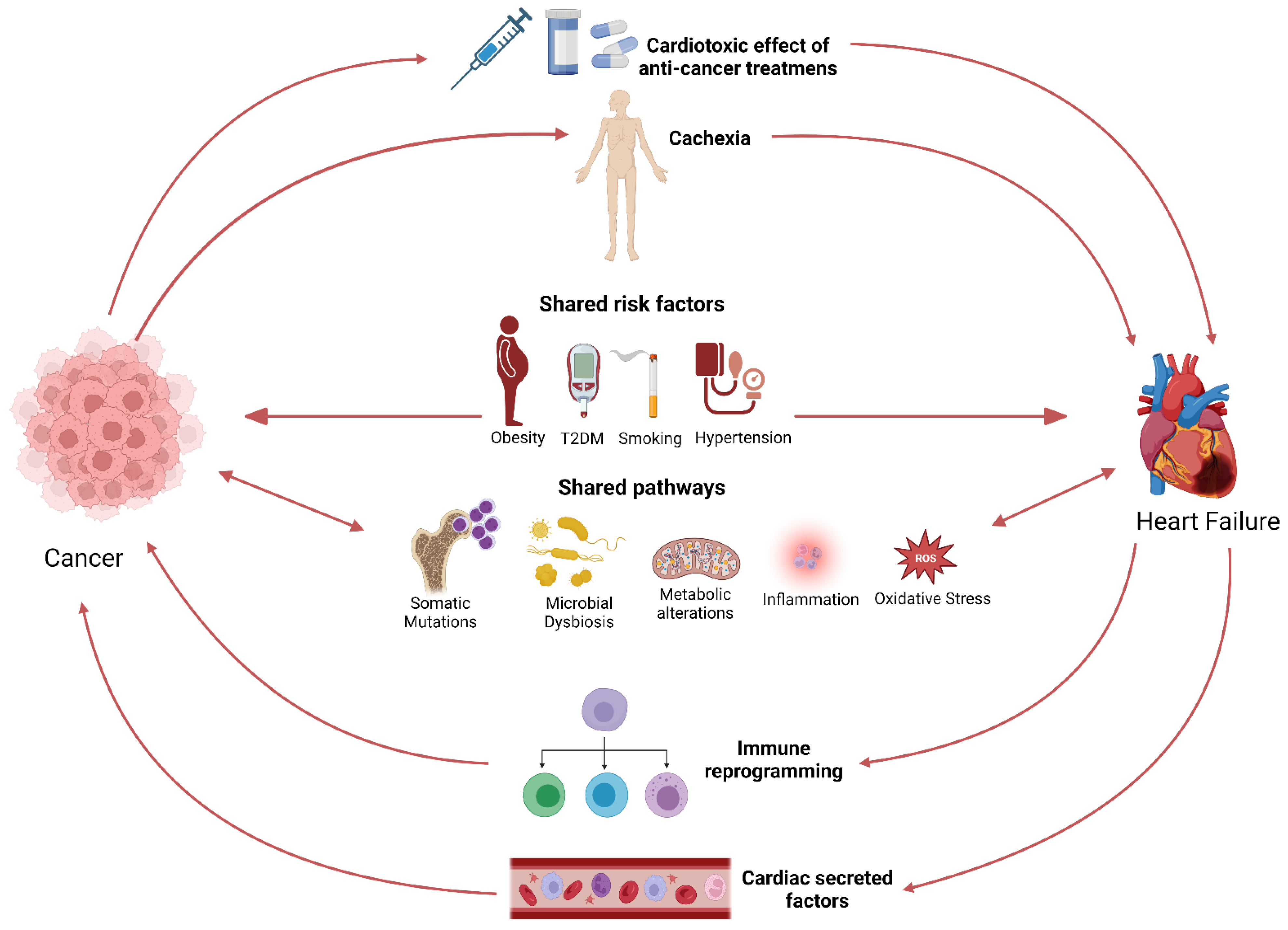
2. Heart Failure Triggers a Pro-Oncogenic Milieu
2.1. The Bidirectional Relationship between Cancer and Heart Failure
2.2. Preclinical Data: Heart Failure Accelerates Tumour Growth
2.3. Common Risk Factors and Signalling Pathways
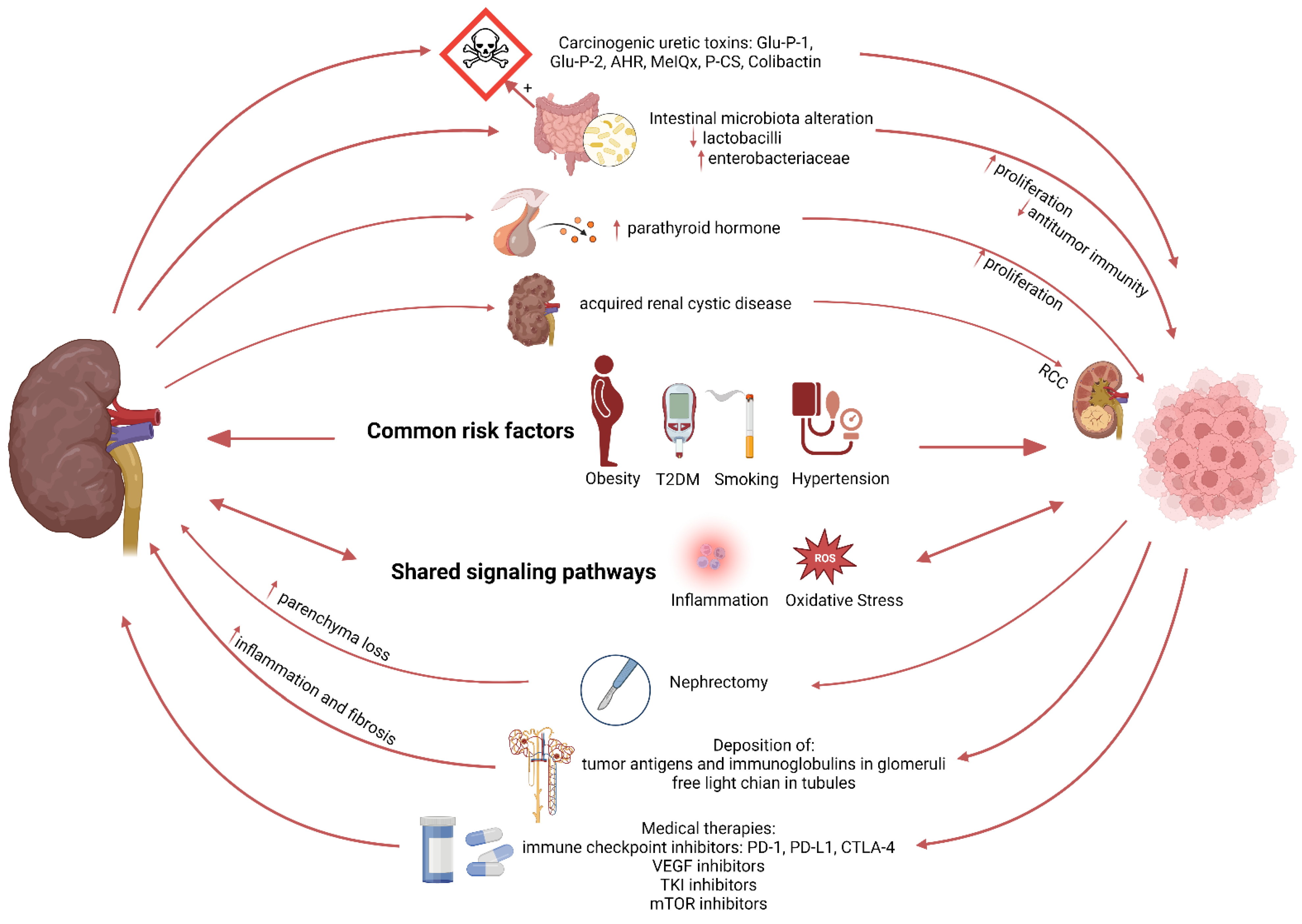
3. Kidney Disease: A Potential Risk Factor for Cancer
3.1. Cancer Risk in Chronic Kidney Disease Patients
3.2. Mechanisms Linking Kidney Disease to Cancer Development
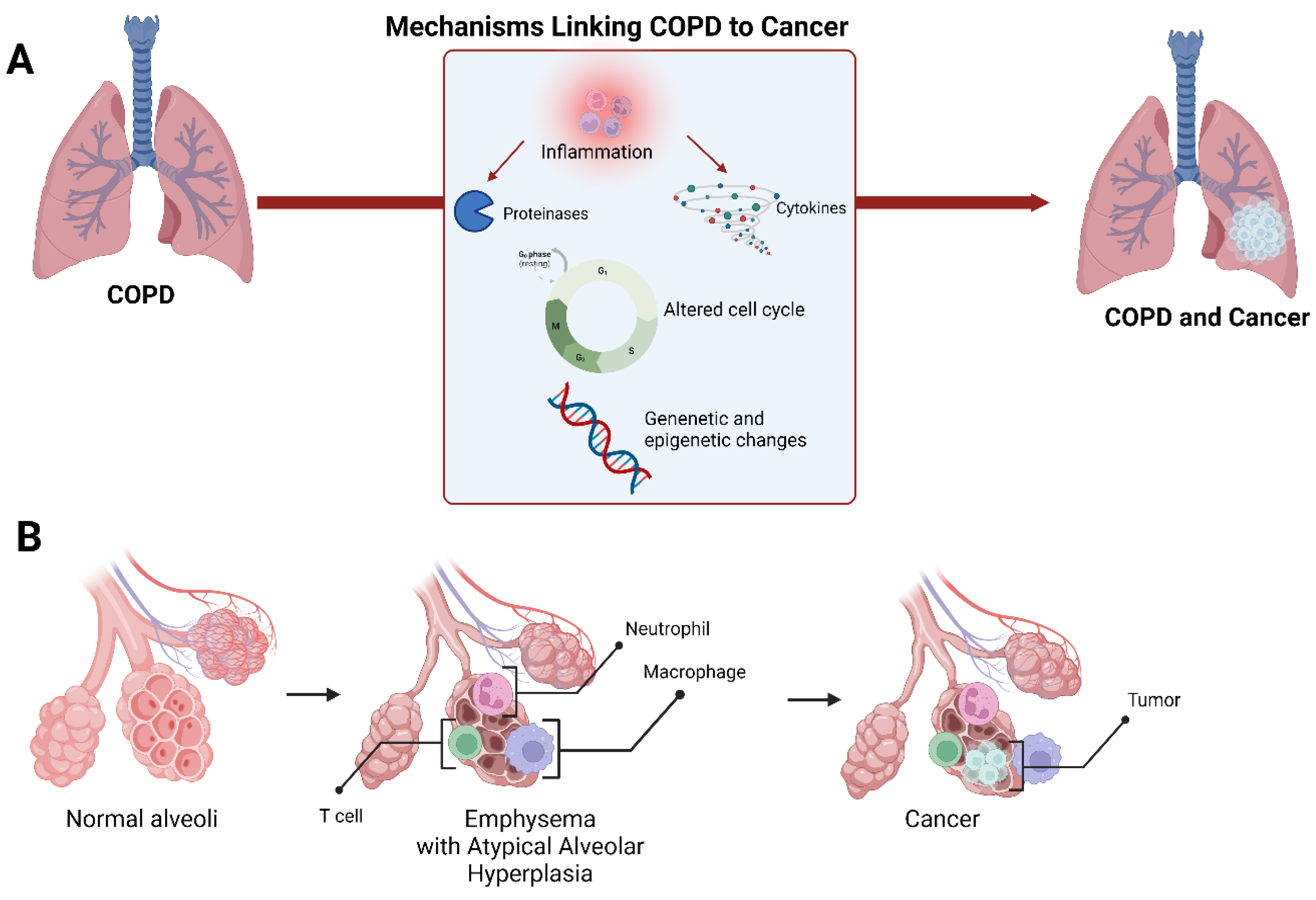
4. Lung Cancer Development in Chronic Obstructive Pulmonary Disease
4.1. Shared Susceptibility Loci and DNA Epigenetic Modification in COPD and Lung Cancer
4.2. Inflammation and Oxidative Stress: Two Shared Signalling Pathways
5. The Progression of Metabolic Associated Fatty Liver Disease to (Extra)-Hepatic Cancers
5.1. Metabolic (Dysfunction) Associated Fatty Liver Disease
5.2. MAFLD: A Multisystem Disease
5.3. Mechanisms Linking MAFLD to HCC
6. Clinical Considerations
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CEBP | CCAAT enhancer binding protein |
| CHRNA | Cholinergic receptor nicotinic alpha |
| CKD | Chronic kidney disease |
| COPD | Chronic obstructive pulmonary disease |
| COS | Cardio-oncology syndromes |
| CRC | Colorectal cancer |
| CVD | Cardiovascular disease |
| eGFR | Estimated glomerular filtration rate |
| EGFR | Epidermal growth factor receptor |
| ER | Endoplasmic reticulum |
| ESRD | End-stage renal disease |
| FAM13A | Family with sequence similarity 13, member A |
| FXR | Farnesoid X receptor |
| Glu-P-1 | 2-amino-6-methyldipyrido [1,2-a: 3′,2′-d]imidazole |
| Glu-P-2 | 2-aminodipyrido [1,2-a:3′,2′- d]imidazole |
| GWAS | Genome-wide association studies |
| GYPA | Glycophorin A |
| HCC | Hepatocellular carcinoma |
| HF | Heart failure |
| HHIP | Hedgehog interacting protein |
| HR | Hazard ratio |
| IRF | Interferon regulatory factor |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| MAFLD | Metabolic (dysfunction)-associated fatty liver disease |
| MGMT | O-6-methylguanine-DNA methyltransferase |
| MI | Myocardial infarction |
| mMDSCs | Monocytic myeloid-derived suppressor cells |
| MMP | Matrix metalloproteinases |
| MPO | Myeloperoxidase |
| NA | Not available |
| nAChRs | Nicotinic acetylcholine receptors |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NF-κB | NF-kappa-B transcription complex |
| RIT | Renal replacement therapy |
| ROS | Reactive oxygen species |
| SIR | Standardized incidence ratio |
| SIRT1 | Sirtuin-1 |
| SNPs | Single nucleotide polymorphisms |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAC | Transverse aortic constriction |
| T2DM | Type 2 diabetes mellitus |
| TNF-α | Tumour necrosis factor α |
| US | United states |
| VEGF | Vascular endothelial growth factor |
| VLDL | Very low-density lipoprotein |
References
- United Nations. World Population Ageing 2019; United Nations: New York, NY, USA, 2020. [Google Scholar]
- Khan, S.S.; Singer, B.D.; Vaughan, D.E. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell 2017, 16, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Bleumink, G.S.; Knetsch, A.M.; Sturkenboom, M.C.; Straus, S.M.; Hofman, A.; Deckers, J.W.; Witteman, J.C.; Stricker, B.H. Quantifying the heart failure epidemic: Prevalence, incidence rate, lifetime risk and prognosis of heart failure The Rotterdam Study. Eur. Heart J. 2004, 25, 1614–1619. [Google Scholar] [CrossRef]
- Williams, M.E. Diabetic kidney disease in elderly individuals. Med. Clin. N. Am. 2013, 97, 75–89. [Google Scholar] [CrossRef]
- Balogun, S.A.; Balogun, R.; Philbrick, J.; Abdel-Rahman, E. Quality of Life, Perceptions, and Health Satisfaction of Older Adults with End-Stage Renal Disease: A Systematic Review. J. Am. Geriatr. Soc. 2017, 65, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Chronic obstructive pulmonary disease among adults—United States, 2011. Morb. Mortal. Wkly. Rep. 2012, 61, 938–943. [Google Scholar]
- Gan, L.; Chitturi, S.; Farrell, G.C. Mechanisms and implications of age-related changes in the liver: Nonalcoholic Fatty liver disease in the elderly. Curr. Gerontol. Geriatr. Res. 2011, 2011, 831536. [Google Scholar] [CrossRef]
- Cancer Incidence by Age. 2020. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/incidence/age (accessed on 31 December 2020).
- White, M.C.; Holman, D.M.; Boehm, J.E.; Peipins, L.A.; Grossman, M.; Henley, S.J. Age and cancer risk: A potentially modifiable relationship. Am. J. Prev Med. 2014, 46, S7–S15. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Pennesi, G.; Donatelli, F.; Cammarota, R.; De Flora, S.; Noonan, D.M. Cardiotoxicity of anticancer drugs: The need for cardio-oncology and cardio-oncological prevention. J. Natl. Cancer Inst. 2010, 102, 14–25. [Google Scholar] [CrossRef]
- Aboumsallem, J.P.; Moslehi, J.; de Boer, R.A. Reverse Cardio-Oncology: Cancer Development in Patients With Cardiovascular Disease. J. Am. Heart Assoc. 2020, 9, e013754. [Google Scholar] [CrossRef]
- Rashdan, S.; Minna, J.D.; Gerber, D.E. Diagnosis and management of pulmonary toxicity associated with cancer immunotherapy. Lancet Respir. Med. 2018, 6, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.H.; Perazella, M.A. Acute Kidney Injury in Patients with Cancer. N. Engl. J. Med. 2017, 377, 500–501. [Google Scholar] [CrossRef] [PubMed]
- Meunier, L.; Larrey, D. Chemotherapy-associated steatohepatitis. Ann. Hepatol. 2020, 19, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Munoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef] [PubMed]
- Zaorsky, N.G.; Churilla, T.M.; Egleston, B.L.; Fisher, S.G.; Ridge, J.A.; Horwitz, E.M.; Meyer, J.E. Causes of death among cancer patients. Ann. Oncol. 2017, 28, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Koelwyn, G.J.; Aboumsallem, J.P.; Moore, K.J.; de Boer, R.A. Reverse cardio-oncology: Exploring the effects of cardiovascular disease on cancer pathogenesis. J. Mol. Cell Cardiol. 2021, 163, 1–8. [Google Scholar] [CrossRef] [PubMed]
- de Boer, R.A.; Hulot, J.S.; Tocchetti, C.G.; Aboumsallem, J.P.; Ameri, P.; Anker, S.D.; Bauersachs, J.; Bertero, E.; Coats, A.J.S.; Celutkiene, J.; et al. Common mechanistic pathways in cancer and heart failure. A scientific roadmap on behalf of the Translational Research Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2020, 22, 2272–2289. [Google Scholar] [CrossRef]
- de Boer, R.A.; Aboumsallem, J.P.; Bracun, V.; Leedy, D.; Cheng, R.; Patel, S.; Rayan, D.; Zaharova, S.; Rymer, J.; Kwan, J.M.; et al. A new classification of cardio-oncology syndromes. Cardiooncology 2021, 7, 24. [Google Scholar] [CrossRef]
- Banke, A.; Schou, M.; Videbaek, L.; Møller, J.E.; Torp-Pedersen, C.; Gustafsson, F.; Dahl, J.S.; Køber, L.; Hildebrandt, P.R.; Gislason, G.H. Incidence of cancer in patients with chronic heart failure: A long-term follow-up study. Eur. J. Heart Fail. 2016, 18, 260–266. [Google Scholar] [CrossRef]
- Bertero, E.; Robusto, F.; Rulli, E.; D′Ettorre, A.; Bisceglia, L.; Staszewsky, L.; Maack, C.; Lepore, V.; Latini, R.; Ameri, P. Cancer Incidence and Mortality According to Pre-Existing Heart Failure in a Community-Based Cohort. JACC CardioOncol. 2022, 4, 98–109. [Google Scholar] [CrossRef]
- Hasin, T.; Gerber, Y.; McNallan, S.M.; Weston, S.A.; Kushwaha, S.S.; Nelson, T.J.; Cerhan, J.R.; Roger, V.L. Patients with heart failure have an increased risk of incident cancer. J. Am. Coll. Cardiol. 2013, 62, 881–886. [Google Scholar] [CrossRef]
- Meijers, W.C.; Maglione, M.; Bakker, S.J.L.; Oberhuber, R.; Kieneker, L.M.; de Jong, S.; Haubner, B.J.; Nagengast, W.B.; Lyon, A.R.; van der Vegt, B.; et al. Heart Failure Stimulates Tumor Growth by Circulating Factors. Circulation 2018, 138, 678–691. [Google Scholar] [CrossRef] [PubMed]
- de Boer, R.A.; Meijers, W.C.; van der Meer, P.; van Veldhuisen, D.J. Cancer and heart disease: Associations and relations. Eur. J. Heart Fail. 2019, 21, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Koelwyn, G.J.; Newman, A.A.C.; Afonso, M.S.; van Solingen, C.; Corr, E.M.; Brown, E.J.; Albers, K.B.; Yamaguchi, N.; Narke, D.; Schlegel, M.; et al. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat. Med. 2020, 26, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Avraham, S.; Abu-Sharki, S.; Shofti, R.; Haas, T.; Korin, B.; Kalfon, R.; Friedman, T.; Shiran, A.; Saliba, W.; Shaked, Y.; et al. Early Cardiac Remodeling Promotes Tumor Growth and Metastasis. Circulation 2020, 142, 670–683. [Google Scholar] [CrossRef]
- Bertero, E.; Canepa, M.; Maack, C.; Ameri, P. Linking Heart Failure to Cancer: Background Evidence and Research Perspectives. Circulation 2018, 138, 735–742. [Google Scholar] [CrossRef]
- Yu, B.; Roberts, M.B.; Raffield, L.M.; Zekavat, S.M.; Nguyen, N.Q.H.; Biggs, M.L.; Brown, M.R.; Griffin, G.; Desai, P.; Correa, A.; et al. Supplemental Association of Clonal Hematopoiesis With Incident Heart Failure. J. Am. Coll. Cardiol. 2021, 78, 42–52. [Google Scholar] [CrossRef]
- Shi, C.; Aboumsallem, J.P.; Suthahar, N.; de Graaf, A.O.; Jansen, J.H.; van Zeventer, I.A.; Bracun, V.; de Wit, S.; Screever, E.M.; van den Berg, P.F.; et al. Clonal haematopoiesis of indeterminate potential: Associations with heart failure incidence, clinical parameters and biomarkers. Eur. J. Heart Fail. 2022. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; van der Wal, H.H.; Silljé, H.H.W.; Dokter, M.M.; van den Berg, F.; Huizinga, L.; Vriesema, M.; Post, J.; Anker, S.D.; Cleland, J.G.; et al. Tumour biomarkers: Association with heart failure outcomes. J. Intern. Med. 2020, 288, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Bracun, V.; Suthahar, N.; Shi, C.; de Wit, S.; Meijers, W.C.; Klip, I.T.; de Boer, R.A.; Aboumsallem, J.P. Established Tumour Biomarkers Predict Cardiovascular Events and Mortality in the General Population. Front. Cardiovasc. Med. 2021, 8, 753885. [Google Scholar] [CrossRef]
- de Wit, S.; Glen, C.; de Boer, R.A.; Lang, N.N. Mechanisms shared between cancer, heart failure, and targeted anti-cancer therapies. Cardiovasc. Res. 2022, cvac132. [Google Scholar] [CrossRef] [PubMed]
- Gagnière, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Jin, Y.; Chen, G.; Ma, X.; Zhang, L. Gut Microbiota Dysbiosis Drives the Development of Colorectal Cancer. Digestion 2021, 102, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Mima, K.; Nakagawa, S.; Sawayama, H.; Ishimoto, T.; Imai, K.; Iwatsuki, M.; Hashimoto, D.; Baba, Y.; Yamashita, Y.i.; Yoshida, N.; et al. The microbiome and hepatobiliary-pancreatic cancers. Cancer Lett. 2017, 402, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Luedde, M.; Winkler, T.; Heinsen, F.A.; Rühlemann, M.C.; Spehlmann, M.E.; Bajrovic, A.; Lieb, W.; Franke, A.; Ott, S.J.; Frey, N. Heart failure is associated with depletion of core intestinal microbiota. ESC Heart Fail. 2017, 4, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef]
- Pasini, E.; Aquilani, R.; Testa, C.; Baiardi, P.; Angioletti, S.; Boschi, F.; Verri, M.; Dioguardi, F. Pathogenic Gut Flora in Patients with Chronic Heart Failure. JACC Heart Fail. 2016, 4, 220–227. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef]
- Bae, S.; Ulrich, C.M.; Neuhouser, M.L.; Malysheva, O.; Bailey, L.B.; Xiao, L.; Brown, E.C.; Cushing-Haugen, K.L.; Zheng, Y.; Cheng, T.Y.D.; et al. Plasma choline metabolites and colorectal cancer risk in the women’s health initiative observational study. Cancer Res. 2014, 74, 7442–7452. [Google Scholar] [CrossRef] [PubMed]
- Mondul, A.M.; Moore, S.C.; Weinstein, S.J.; Karoly, E.D.; Sampson, J.N.; Albanes, D. Metabolomic analysis of prostate cancer risk in a prospective cohort: The alpha-tocopherol, beta-carotene cancer prevention (ATBC) study. Int. J. Cancer 2015, 137, 2124–2132. [Google Scholar] [CrossRef]
- Stengel, B. Chronic kidney disease and cancer: A troubling connection. J. Nephrol. 2010, 23, 253–262. [Google Scholar] [PubMed]
- Ronco, P.M. Paraneoplastic glomerulopathies: New insights into an old entity. Kidney Int. 1999, 56, 355–377. [Google Scholar] [CrossRef]
- Audard, V.; Larousserie, F.; Grimbert, P.; Abtahi, M.; Sotto, J.J.; Delmer, A.; Boue, F.; Nochy, D.; Brousse, N.; Delarue, R.; et al. Minimal change nephrotic syndrome and classical Hodgkin’s lymphoma: Report of 21 cases and review of the literature. Kidney Int. 2006, 69, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Levey, A.S.; Serio, A.M.; Snyder, M.; Vickers, A.J.; Raj, G.V.; Scardino, P.T.; Russo, P. Chronic kidney disease after nephrectomy in patients with renal cortical tumours: A retrospective cohort study. Lancet Oncol. 2006, 7, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Saly, D.L.; Eswarappa, M.S.; Street, S.E.; Deshpande, P. Renal Cell Cancer and Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2021, 28, 460–468.e461. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, Q.; Liu, B.; Ma, Q.; Zhang, T.; Huang, T.; Lv, Z.; Wang, R. Chronic Kidney Disease and Cancer: Inter-Relationships and Mechanisms. Front. Cell Dev. Biol. 2022, 10, 868715. [Google Scholar] [CrossRef]
- Wong, G.; Hayen, A.; Chapman, J.R.; Webster, A.C.; Wang, J.J.; Mitchell, P.; Craig, J.C. Association of CKD and cancer risk in older people. J. Am. Soc. Nephrol. 2009, 20, 1341–1350. [Google Scholar] [CrossRef]
- Lowrance, W.T.; Ordonez, J.; Udaltsova, N.; Russo, P.; Go, A.S. CKD and the risk of incident cancer. J. Am. Soc. Nephrol. 2014, 25, 2327–2334. [Google Scholar] [CrossRef]
- Jorgensen, L.; Heuch, I.; Jenssen, T.; Jacobsen, B.K. Association of albuminuria and cancer incidence. J. Am. Soc. Nephrol. 2008, 19, 992–998. [Google Scholar] [CrossRef]
- Wong, G.; Staplin, N.; Emberson, J.; Baigent, C.; Turner, R.; Chalmers, J.; Zoungas, S.; Pollock, C.; Cooper, B.; Harris, D.; et al. Chronic kidney disease and the risk of cancer: An individual patient data meta-analysis of 32,057 participants from six prospective studies. BMC Cancer 2016, 16, 488. [Google Scholar] [CrossRef]
- Vajdic, C.M.; McDonald, S.P.; McCredie, M.R.; van Leeuwen, M.T.; Stewart, J.H.; Law, M.; Chapman, J.R.; Webster, A.C.; Kaldor, J.M.; Grulich, A.E. Cancer incidence before and after kidney transplantation. JAMA 2006, 296, 2823–2831. [Google Scholar] [CrossRef]
- Maisonneuve, P.; Agodoa, L.; Gellert, R.; Stewart, J.H.; Buccianti, G.; Lowenfels, A.B.; Wolfe, R.A.; Jones, E.; Disney, A.P.; Briggs, D.; et al. Cancer in patients on dialysis for end-stage renal disease: An international collaborative study. Lancet 1999, 354, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.H.; Vajdic, C.M.; van Leeuwen, M.T.; Amin, J.; Webster, A.C.; Chapman, J.R.; McDonald, S.P.; Grulich, A.E.; McCredie, M.R. The pattern of excess cancer in dialysis and transplantation. Nephrol. Dial. Transpl. 2009, 24, 3225–3231. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.Y.; Chan, G.C.; Chan, S.K.; Ng, F.; Lam, M.F.; Wong, S.S.; Chak, W.L.; Chau, K.F.; Lui, S.L.; Lo, W.K.; et al. Cancer Incidence and Mortality in Chronic Dialysis Population: A Multicenter Cohort Study. Am. J. Nephrol. 2016, 43, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.F.; Li, Y.H.; Wang, C.H.; Chou, C.L.; Kuo, D.J.; Fang, T.C. Increased risk of cancer in chronic dialysis patients: A population-based cohort study in Taiwan. Nephrol. Dial. Transpl. 2012, 27, 1585–1590. [Google Scholar] [CrossRef]
- Stewart, J.H.; Buccianti, G.; Agodoa, L.; Gellert, R.; McCredie, M.R.; Lowenfels, A.B.; Disney, A.P.; Wolfe, R.A.; Boyle, P.; Maisonneuve, P. Cancers of the kidney and urinary tract in patients on dialysis for end-stage renal disease: Analysis of data from the United States, Europe, and Australia and New Zealand. J. Am. Soc. Nephrol. 2003, 14, 197–207. [Google Scholar] [CrossRef]
- Cheung, C.Y.; Lam, M.F.; Chu, K.H.; Chow, K.M.; Tsang, K.Y.; Yuen, S.K.; Wong, P.N.; Chan, S.K.; Leung, K.T.; Chan, C.K.; et al. Malignancies after kidney transplantation: Hong Kong renal registry. Am. J. Transpl. 2012, 12, 3039–3046. [Google Scholar] [CrossRef]
- Li, W.H.; Chen, Y.J.; Tseng, W.C.; Lin, M.W.; Chen, T.J.; Chu, S.Y.; Hwang, C.Y.; Chen, C.C.; Lee, D.D.; Chang, Y.T.; et al. Malignancies after renal transplantation in Taiwan: A nationwide population-based study. Nephrol. Dial. Transpl. 2012, 27, 833–839. [Google Scholar] [CrossRef]
- Komaki, Y.; Komaki, F.; Micic, D.; Ido, A.; Sakuraba, A. Risk of Colorectal Cancer in Chronic Kidney Disease: A Systematic Review and Meta-Analysis. J. Clin. Gastroenterol. 2018, 52, 796–804. [Google Scholar] [CrossRef]
- Locatelli, F.; Canaud, B.; Eckardt, K.U.; Stenvinkel, P.; Wanner, C.; Zoccali, C. Oxidative stress in end-stage renal disease: An emerging threat to patient outcome. Nephrol. Dial. Transpl. 2003, 18, 1272–1280. [Google Scholar] [CrossRef]
- Loft, S.; Poulsen, H.E. Cancer risk and oxidative DNA damage in man. J. Mol. Med. 1996, 74, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Masereeuw, R.; Mutsaers, H.A.; Toyohara, T.; Abe, T.; Jhawar, S.; Sweet, D.H.; Lowenstein, J. The kidney and uremic toxin removal: Glomerulus or tubule? Semin. Nephrol. 2014, 34, 191–208. [Google Scholar] [CrossRef]
- Syed-Ahmed, M.; Narayanan, M. Immune Dysfunction and Risk of Infection in Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2019, 26, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.K. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef] [PubMed]
- Punturieri, A.; Szabo, E.; Croxton, T.L.; Shapiro, S.D.; Dubinett, S.M. Lung cancer and chronic obstructive pulmonary disease: Needs and opportunities for integrated research. J. Natl. Cancer Inst. 2009, 101, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Wasswa-Kintu, S.; Gan, W.Q.; Man, S.F.; Pare, P.D.; Sin, D.D. Relationship between reduced forced expiratory volume in one second and the risk of lung cancer: A systematic review and meta-analysis. Thorax 2005, 60, 570–575. [Google Scholar] [CrossRef]
- Sekine, Y.; Katsura, H.; Koh, E.; Hiroshima, K.; Fujisawa, T. Early detection of COPD is important for lung cancer surveillance. Eur. Respir. J. 2012, 39, 1230–1240. [Google Scholar] [CrossRef]
- Bozinovski, S.; Vlahos, R.; Anthony, D.; McQualter, J.; Anderson, G.; Irving, L.; Steinfort, D. COPD and squamous cell lung cancer: Aberrant inflammation and immunity is the common link. Br. J. Pharm. 2016, 173, 635–648. [Google Scholar] [CrossRef]
- Young, R.P.; Hopkins, R.J.; Gamble, G.D.; Etzel, C.; El-Zein, R.; Crapo, J.D. Genetic evidence linking lung cancer and COPD: A new perspective. Appl. Clin. Genet. 2011, 4, 99–111. [Google Scholar] [CrossRef]
- Wilk, J.B.; Chen, T.H.; Gottlieb, D.J.; Walter, R.E.; Nagle, M.W.; Brandler, B.J.; Myers, R.H.; Borecki, I.B.; Silverman, E.K.; Weiss, S.T.; et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009, 5, e1000429. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Boutaoui, N.; Klanderman, B.J.; Sylvia, J.S.; Ziniti, J.P.; Hersh, C.P.; DeMeo, D.L.; Hunninghake, G.M.; Litonjua, A.A.; Sparrow, D.; et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat. Genet. 2010, 42, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Young, R.P.; Whittington, C.F.; Hopkins, R.J.; Hay, B.A.; Epton, M.J.; Black, P.N.; Gamble, G.D. Chromosome 4q31 locus in COPD is also associated with lung cancer. Eur. Respir. J. 2010, 36, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.G.; Kong, X.; Edwards, L.D.; Cho, M.H.; Anderson, W.H.; Coxson, H.O.; Lomas, D.A.; Silverman, E.K. Loci identified by genome-wide association studies influence different disease-related phenotypes in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Thorgeirsson, T.E.; Geller, F.; Sulem, P.; Rafnar, T.; Wiste, A.; Magnusson, K.P.; Manolescu, A.; Thorleifsson, G.; Stefansson, H.; Ingason, A.; et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 2008, 452, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Hung, R.J.; McKay, J.D.; Gaborieau, V.; Boffetta, P.; Hashibe, M.; Zaridze, D.; Mukeria, A.; Szeszenia-Dabrowska, N.; Lissowska, J.; Rudnai, P.; et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 2008, 452, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Lawless, M.W.; O′Byrne, K.J.; Gray, S.G. Oxidative stress induced lung cancer and COPD: Opportunities for epigenetic therapy. J. Cell. Mol. Med. 2009, 13, 2800–2821. [Google Scholar] [CrossRef]
- Belinsky, S.A.; Palmisano, W.A.; Gilliland, F.D.; Crooks, L.A.; Divine, K.K.; Winters, S.A.; Grimes, M.J.; Harms, H.J.; Tellez, C.S.; Smith, T.M.; et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 2002, 62, 2370–2377. [Google Scholar]
- Krushkal, J.; Silvers, T.; Reinhold, W.C.; Sonkin, D.; Vural, S.; Connelly, J.; Varma, S.; Meltzer, P.S.; Kunkel, M.; Rapisarda, A.; et al. Epigenome-wide DNA methylation analysis of small cell lung cancer cell lines suggests potential chemotherapy targets. Clin. Epigenetics 2020, 12, 93. [Google Scholar] [CrossRef]
- Hu, X.; Estecio, M.R.; Chen, R.; Reuben, A.; Wang, L.; Fujimoto, J.; Carrot-Zhang, J.; McGranahan, N.; Ying, L.; Fukuoka, J.; et al. Evolution of DNA methylome from precancerous lesions to invasive lung adenocarcinomas. Nat. Commun. 2021, 12, 687. [Google Scholar] [CrossRef]
- Liang, R.; Li, X.; Li, W.; Zhu, X.; Li, C. DNA methylation in lung cancer patients: Opening a "window of life" under precision medicine. Biomed Pharm. 2021, 144, 112202. [Google Scholar] [CrossRef] [PubMed]
- Guzman, L.; Depix, M.S.; Salinas, A.M.; Roldan, R.; Aguayo, F.; Silva, A.; Vinet, R. Analysis of aberrant methylation on promoter sequences of tumor suppressor genes and total DNA in sputum samples: A promising tool for early detection of COPD and lung cancer in smokers. Diagn. Pathol. 2012, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Baccarelli, A.; Carey, V.J.; Boutaoui, N.; Bacherman, H.; Klanderman, B.; Rennard, S.; Agusti, A.; Anderson, W.; Lomas, D.A.; et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am. J. Respir. Crit. Care Med. 2012, 185, 373–381. [Google Scholar] [CrossRef]
- Kachroo, P.; Morrow, J.D.; Kho, A.T.; Vyhlidal, C.A.; Silverman, E.K.; Weiss, S.T.; Tantisira, K.G.; DeMeo, D.L. Co-methylation analysis in lung tissue identifies pathways for fetal origins of COPD. Eur. Respir. J. 2020, 56, 1902347. [Google Scholar] [CrossRef] [PubMed]
- King, P.T. Inflammation in chronic obstructive pulmonary disease and its role in cardiovascular disease and lung cancer. Clin. Transl. Med. 2015, 4, 68. [Google Scholar] [CrossRef]
- Tan, Z.; Xue, H.; Sun, Y.; Zhang, C.; Song, Y.; Qi, Y. The Role of Tumor Inflammatory Microenvironment in Lung Cancer. Front. Pharm. 2021, 12, 688625. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Shang, G.S.; Liu, L.; Qin, Y.W. IL-6 and TNF-alpha promote metastasis of lung cancer by inducing epithelial-mesenchymal transition. Oncol. Lett. 2017, 13, 4657–4660. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef]
- Wells, J.M.; Parker, M.M.; Oster, R.A.; Bowler, R.P.; Dransfield, M.T.; Bhatt, S.P.; Cho, M.H.; Kim, V.; Curtis, J.L.; Martinez, F.J.; et al. Elevated circulating MMP-9 is linked to increased COPD exacerbation risk in SPIROMICS and COPDGene. JCI Insight 2018, 3, e123614. [Google Scholar] [CrossRef]
- Mahor, D.; Kumari, V.; Vashisht, K.; Galgalekar, R.; Samarth, R.M.; Mishra, P.K.; Banerjee, N.; Dixit, R.; Saluja, R.; De, S.; et al. Elevated serum matrix metalloprotease (MMP-2) as a candidate biomarker for stable COPD. BMC Pulm. Med. 2020, 20, 302. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Sheng, B.; Zeng, Q.; Yao, W.; Jiang, Q. Correlation between MMP2 expression in lung cancer tissues and clinical parameters: A retrospective clinical analysis. BMC Pulm. Med. 2020, 20, 283. [Google Scholar] [CrossRef] [PubMed]
- Rollin, J.; Regina, S.; Vourc′h, P.; Iochmann, S.; Blechet, C.; Reverdiau, P.; Gruel, Y. Influence of MMP-2 and MMP-9 promoter polymorphisms on gene expression and clinical outcome of non-small cell lung cancer. Lung Cancer 2007, 56, 273–280. [Google Scholar] [CrossRef]
- Decock, J.; Thirkettle, S.; Wagstaff, L.; Edwards, D.R. Matrix metalloproteinases: Protective roles in cancer. J. Cell. Mol. Med. 2011, 15, 1254–1265. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; MacNee, W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic. Biol. Med. 1996, 21, 669–681. [Google Scholar] [CrossRef]
- Durham, A.L.; Adcock, I.M. The relationship between COPD and lung cancer. Lung Cancer 2015, 90, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Karin, M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef]
- Zaynagetdinov, R.; Sherrill, T.P.; Gleaves, L.A.; Hunt, P.; Han, W.; McLoed, A.G.; Saxon, J.A.; Tanjore, H.; Gulleman, P.M.; Young, L.R.; et al. Chronic NF-kappaB activation links COPD and lung cancer through generation of an immunosuppressive microenvironment in the lungs. Oncotarget 2016, 7, 5470–5482. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S.; et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-kappaB activity. PLoS ONE 2012, 7, e46364. [Google Scholar] [CrossRef]
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 177, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.A.; Tsai, M.J.; Jian, S.F.; Sheu, C.C.; Kuo, P.L. Systematic analysis of transcriptomic profiles of COPD airway epithelium using next-generation sequencing and bioinformatics. Int J. Chron. Obs. Pulmon. Dis. 2018, 13, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Yang, J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein. Cell 2013, 4, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. Backgr. Aims 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Wehmeyer, M.H.; Zyriax, B.-C.; Jagemann, B.; Roth, E.; Windler, E.; Wiesch, J.S.z.; Lohse, A.W.; Kluwe, J. Nonalcoholic fatty liver disease is associated with excessive calorie intake rather than a distinctive dietary pattern. Medicine 2016, 95, e3887. [Google Scholar] [CrossRef]
- Herman, M.A.; Samuel, V.T. The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol. Metab. 2016, 27, 719–730. [Google Scholar] [CrossRef]
- Flannery, C.; Dufour, S.; Rabøl, R.; Shulman, G.I.; Petersen, K.F. Skeletal Muscle Insulin Resistance Promotes Increased Hepatic De Novo Lipogenesis, Hyperlipidemia, and Hepatic Steatosis in the Elderly. Diabetes 2012, 61, 2711–2717. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Adams, L.A.; Canbay, A.; Syn, W.-K. Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology 2014, 59, 1174–1197. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. Backgr. Aims 2015, 62, 547–564. [Google Scholar] [CrossRef]
- Liu, Z.; Lin, C.; Suo, C.; Zhao, R.; Jin, L.; Zhang, T.; Chen, X. Metabolic dysfunction–associated fatty liver disease and the risk of 24 specific cancers. Metabolism 2022, 127, 154955. [Google Scholar] [CrossRef]
- Duell, P.B.; Welty, F.K.; Miller, M.; Chait, A.; Hammond, G.; Ahmad, Z.; Cohen, D.E.; Horton, J.D.; Pressman, G.S.; Toth, P.P. Nonalcoholic Fatty Liver Disease and Cardiovascular Risk: A Scientific Statement From the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2022, 42, e168–e185. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Lombardi, R.; Cattazzo, F.; Zusi, C.; Cappelli, D.; Dalbeni, A. MAFLD and CKD: An Updated Narrative Review. Int. J. Mol. Sci. 2022, 23, 7007. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.E.; Ng, C.H.; Fu, C.E.; Quek, J.; Kong, G.; Goh, Y.J.; Zeng, R.W.; Tseng, M.; Aggarwal, M.; Nah, B.; et al. The Spectrum and Impact of Metabolic Dysfunction in MAFLD: A Longitudinal Cohort Analysis of 32,683 Overweight and Obese Individuals. Clin. Gastroenterol. Hepatol. 2022, in press. [CrossRef] [PubMed]
- Lee, H.; Lee, Y.h.; Kim, S.U.; Kim, H.C. Metabolic Dysfunction-Associated Fatty Liver Disease and Incident Cardiovascular Disease Risk: A Nationwide Cohort Study. Clin. Gastroenterol. Hepatol. 2021, 19, 2138–2147. [Google Scholar] [CrossRef]
- Deng, Y.; Zhao, Q.; Gong, R. Association Between Metabolic Associated Fatty Liver Disease and Chronic Kidney Disease: A Cross-Sectional Study from NHANES 2017–2018. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 1751. [Google Scholar] [CrossRef]
- Kim, G.-A.; Lee, H.C.; Choe, J.; Kim, M.-J.; Lee, M.J.; Chang, H.-S.; Bae, I.Y.; Kim, H.-K.; An, J.; Shim, J.H.; et al. Association between non-alcoholic fatty liver disease and cancer incidence rate. Backgr. Aims 2018, 68, 140–146. [Google Scholar] [CrossRef]
- Lee, H.; Lee, H.W.; Kim, S.U.; Chang Kim, H. Metabolic Dysfunction-Associated Fatty Liver Disease Increases Colon Cancer Risk: A Nationwide Cohort Study. Clin. Transl. Gastroenterol. 2022, 13, E00435. [Google Scholar] [CrossRef]
- van Kleef, L.A.; Choi, H.S.J.; Brouwer, W.P.; Hansen, B.E.; Patel, K.; de Man, R.A.; Janssen, H.L.A.; de Knegt, R.J.; Sonneveld, M.J. Metabolic dysfunction-associated fatty liver disease increases risk of adverse outcomes in patients with chronic hepatitis B. JHEP Rep. 2021, 3, 100350. [Google Scholar] [CrossRef]
- Yun, B.; Ahn, S.H.; Oh, J.; Yoon, J.H.; Kim, B.K. Effect of metabolic dysfunction-associated fatty liver disease on liver cancer risk in a population with chronic hepatitis B virus infection: A nationwide study. Hepatol. Res. 2022, 52, 975–984. [Google Scholar] [CrossRef]
- Xue, J.; Wang, Q.X.; Xiao, H.M.; Shi, M.J.; Xie, Y.B.; Li, S.; Lin, M.; Chi, X.L. Impact of Metabolic Dysfunction Associated Fatty Liver Disease on the Prognosis of Patients with Hepatitis B Virus-Related Hepatocellular Carcinoma Based on Propensity Score Matching Analysis. Cancer Manag. Res. 2022, 14, 2193–2202. [Google Scholar] [CrossRef]
- Xie, X.; Zheng, M.; Guo, W.; Zhou, Y.; Xiang, Z.; Li, Y. Correlation analysis of metabolic characteristics and the risk of metabolic-associated fatty liver disease—Related hepatocellular carcinoma. Sci. Rep. 2022, 12, 13969. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Li, L.; Dai, J.; Natarajan, Y.; Yu, X.; Asch, S.M.; El-Serag, H.B. Effect of Metabolic Traits on the Risk of Cirrhosis and Hepatocellular Cancer in Nonalcoholic Fatty Liver Disease. Hepatology 2020, 71, 808–819. [Google Scholar] [CrossRef] [PubMed]
- Yatsuji, S.; Hashimoto, E.; Tobari, M.; Taniai, M.; Tokushige, K.; Shiratori, K. Clinical features and outcomes of cirrhosis due to non-alcoholic steatohepatitis compared with cirrhosis caused by chronic hepatitis C. J. Gastroenterol. Hepatol. 2009, 24, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Poklepovic, A.; Moyneur, E.; Barghout, V. Population-based risk factors and resource utilization for HCC: US perspective. Curr. Med. Res. Opin. 2010, 26, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.M.; Sagar, V.M.; Shah, T.; Shetty, S. Carcinogenesis on the background of liver fibrosis: Implications for the management. World J. Gastroenterol. 2018, 24, 4436. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2020, 18, 223–238. [Google Scholar] [CrossRef]
- Margini, C.; Dufour, J.F. The story of HCC in NAFLD: From epidemiology, across pathogenesis, to prevention and treatment. Liver Int. Off. J. Int. Assoc. Study Liver 2016, 36, 317–324. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and Cancer: A consensus report. Diabetes Care 2010, 33, 1674. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in US Veterans is Associated with Non-Alcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124. [Google Scholar] [CrossRef] [PubMed]
- Gawrieh, S.; Dakhoul, L.; Miller, E.; Scanga, A.; DeLemos, A.; Kettler, C.; Burney, H.; Liu, H.; Abu-Sbeih, H.; Chalasani, N.; et al. Characteristics, aetiologies and trends of hepatocellular carcinoma in patients without cirrhosis: A United States multicentre study. Aliment. Pharmacol. Ther. 2019, 50, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Ertle, J.; Dechêne, A.; Sowa, J.P.; Penndorf, V.; Herzer, K.; Kaiser, G.; Schlaak, J.F.; Gerken, G.; Syn, W.K.; Canbay, A. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int. J. Cancer 2011, 128, 2436–2443. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxidative Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. Backgr. Aims 2002, 37, 56–62. [Google Scholar] [CrossRef]
- Uchida, D.; Takaki, A.; Oyama, A.; Adachi, T.; Wada, N.; Onishi, H.; Okada, H. Oxidative Stress Management in Chronic Liver Diseases and Hepatocellular Carcinoma. Nutrients 2020, 12, 1576. [Google Scholar] [CrossRef]
- Maurel, M.; Samali, A.; Chevet, E. Endoplasmic Reticulum Stress: At the Crossroads of Inflammation and Metabolism in Hepatocellular Carcinoma Development. Cancer Cell 2014, 26, 301–303. [Google Scholar] [CrossRef]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER Stress Cooperates with Hypernutrition to Trigger TNF-Dependent Spontaneous HCC Development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef]
- Stickel, F.; Hellerbrand, C. Non-alcoholic fatty liver disease as a risk factor for hepatocellular carcinoma: Mechanisms and implications. Gut 2010, 59, 1303–1307. [Google Scholar] [CrossRef]
- WANG, X.; LIN, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Yoo, W.; Stevenson, H.L.; Deshpande, D.; Shen, H.; Gagea, M.; Yoo, S.-Y.; Wang, J.; Eckols, T.K.; Bharadwaj, U.; et al. Multifunctional Effects of a Small-Molecule STAT3 Inhibitor on NASH and Hepatocellular Carcinoma in Mice. Clin. Cancer Res. 2017, 23, 5537–5546. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Lee, J.H.; Yu, G.-Y.; He, G.; Ali, S.R.; Holzer, R.G.; Österreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197. [Google Scholar] [CrossRef] [PubMed]
- Wieland, A.; Frank, D.N.; Harnke, B.; Bambha, K. Systematic review: Microbial dysbiosis and nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2015, 42, 1051–1063. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef]
- Su, H.; Ma, C.; Liu, J.; Li, N.; Gao, M.; Huang, A.; Wang, X.; Huang, W.; Huang, X. Downregulation of nuclear receptor FXR is associated with multiple malignant clinicopathological characteristics in human hepatocellular carcinoma. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Lythgoe, M.P.; Prasad, V. Repositioning canakinumab for non-small cell lung cancer-important lessons for drug repurposing in oncology. Br. J. Cancer 2022, 127, 785–787. [Google Scholar] [CrossRef]
- Ellis, P.M.; Coakley, N.; Feld, R.; Kuruvilla, S.; Ung, Y.C. Use of the epidermal growth factor receptor inhibitors gefitinib, erlotinib, afatinib, dacomitinib, and icotinib in the treatment of non-small-cell lung cancer: A systematic review. Curr. Oncol. 2015, 22, e183–e215. [Google Scholar] [CrossRef] [PubMed]
- Nadal, E.; Horinouchi, H.; Shih, J.Y.; Nakagawa, K.; Reck, M.; Garon, E.B.; Wei, Y.F.; Kollmeier, J.; Frimodt-Moller, B.; Barrett, E.; et al. RELAY, Ramucirumab Plus Erlotinib Versus Placebo Plus Erlotinib in Patients with Untreated, Epidermal Growth Factor Receptor Mutation-Positive, Metastatic Non-Small-Cell Lung Cancer: Safety Profile and Manageability. Drug Saf. 2022, 45, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhou, L.; Huang, Z.; Li, B.; Nice, E.C.; Xu, J.; Huang, C. Antioxidant Therapy in Cancer: Rationale and Progress. Antioxidants 2022, 11, 1128. [Google Scholar] [CrossRef] [PubMed]
- Balea, S.S.; Parvu, A.E.; Parvu, M.; Vlase, L.; Dehelean, C.A.; Pop, T.I. Antioxidant, Anti-Inflammatory and Antiproliferative Effects of the Vitis vinifera L. var. Feteasca Neagra and Pinot Noir Pomace Extracts. Front Pharm. 2020, 11, 990. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, M.; Giacomini, I.; Morandin, G.; Cocetta, V.; Ragazzi, E.; Orso, G.; Carnevali, I.; Berretta, M.; Mancini, M.; Pagano, F.; et al. Serenoa repens and Urtica dioica Fixed Combination: In-Vitro Validation of a Therapy for Benign Prostatic Hyperplasia (BPH). Int. J. Mol. Sci. 2020, 21, 9178. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.E.; Suthahar, N.; Paniagua, S.M.; Wang, D.; Lau, E.S.; Li, S.X.; Jovani, M.; Takvorian, K.S.; Kreger, B.E.; Benjamin, E.J.; et al. Association of Cardiometabolic Disease With Cancer in the Community. JACC CardioOncol. 2022, 4, 69–81. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Study | Number of Participants | Follow-Up (Years) | Age (Years) |
Estimated Risk
(95% CI) |
|---|---|---|---|---|---|
| Overall | Australia and New Zealand Dialysis and Transplant Registry (ANZDATA) [55] | 23,764 | mean 2.7 | 57.5 (43.5–67.6) | SIR 1.45 (1.36–1.54) |
| Pancreatic | A multicenter retrospective cohort study [56] | 6254 | mean 2.4 | 64.0 ± 13.0 | SIR 1.17 (0.31–2.99) |
| Hepatocellular | National Health Institutes Research Database [57] | 92,348 | mean 4.4 | 60.4 ± 14.8 | SIR 1.4 (1.2–1.5) |
| Colorectal | A multicenter retrospective cohort study [56] | 6254 | mean 2.4 | 64.0 ± 13.0 | SIR 1.53 (1.11–2.05) |
| Bladder | a retrospective cohort including United States (USRDS), Europe (EDTA), Australia, and New Zealand (ANZDATA) [58] | 831,804 | mean 2.46 | mean 55.5 | SIR 1.5 (1.4–1.6) |
| Kidney | ANZDATA [55] | 23,764 | mean 2.7 | 57.5 (43.5–67.6) | SIR 5.4 (4.3–6.7) |
| Lung | National Health Institutes Research Database [57] | 92,348 | mean 4.4 | 60.4 ± 14.8 | SIR 0.5 (0.5–0.6) |
| Gastric | A multicenter retrospective cohort study [56] | 6254 | mean 2.4 | 64.0 ± 13.0 | SIR 1.10 (0.47–2.17) |
| Thyroid | A multicenter retrospective cohort study [56] | 6254 | mean 2.4 | 64.0 ± 13.0 | SIR 3.42 (1.25–7.46) |
| Breast | Meta-analysis including 6 studies [52] | 32,057 | 4.4 (3.2–5.4) for dialysis patients | 60 ± 12 for dialysis patients | HR 1.03 (0.50–2.12) |
| Prostate | Meta-analysis including 6 studies [52] | 32,057 | 4.4 (3.2–5.4) for dialysis patients | 60 ± 12 for dialysis patients | HR 0.38 (0.19–0.77) |
| Leukaemia | National Health Institutes Research Database [57] | 92,348 | mean 4.4 | 60.4 ± 14.8 | SIR 0.4 (0.2–0.7) |
| Myeloma | A multicenter retrospective cohort study [56] | 6254 | mean 2.4 | 64.0 ± 13.0 | SIR 1.31 (0.15–4.72) |
| Cancer | Study | Number of Patients with RRT | Follow-Up (Years) | Age (Years) | Estimated Risk (95% CI) |
|---|---|---|---|---|---|
| Overall | Australia and New Zealand Dialysis and Transplant Registry (ANZDATA) [55] | 8173 | mean 6.0 | 43.4 (31.1–53.9) | SIR 3.03 (2.82–3.25) |
| Pancreatic | Hong Kong Renal Registry [59] | 4674 | 8.2 ± 6.2 | 43.7 ± 12.6 | SIR 1.57 (0.51–4.87) |
| Hepatocellular | National Health Insurance Database in Taiwan [60] | 4716 | 4.8 ± 3.1 | 44.1 ± 12.4 | SIR 5.07 (3.89–6.42) |
| Colorectal | Meta-analysis including 54 studies [61] | 1,208,767 | NA | NA | SIR 1.40 (1.15–1.71) |
| Gallbladder | National Health Insurance Database in Taiwan [60] | 4716 | 4.8 ± 3.1 | 44.1 ± 12.4 | SIR 3.02 (0.76–11.99) |
| Bladder | ANZDATA [55] | 8173 | mean 6.0 | 43.4 (31.1–53.9) | SIR 2.6 (1.5–4.2) |
| Kidney | ANZDATA [55] | 8173 | mean 6.0 | 43.4 (31.1–53.9) | SIR 5.0 (3.4–7.1) |
| Lung cancer | Hong Kong Renal Registry [59] | 4674 | 8.2 ± 6.2 | 43.7 ± 12.6 | SIR 1.68 (1.17–2.42) |
| Gastric | Hong Kong Renal Registry [59] | 4674 | 8.2 ± 6.2 | 43.7 ± 12.6 | SIR 2.85 (1.62–5.02) |
| Thyroid | ANZDATA [55] | 8173 | mean 6.0 | 43.4 (31.1–53.9) | SIR 3.5 (1.7–6.4) |
| Breast | Hong Kong Renal Registry [59] | 4674 | 8.2 ± 6.2 | 43.7 ± 12.6 | SIR 1.66 (1–2.75) |
| Ovarian | Hong Kong Renal Registry [59] | 4674 | 8.2 ± 6.2 | 43.7 ± 12.6 | SIR 3.29 (1.37–7.9) |
| Leukaemia | Hong Kong Renal Registry [59] | 4674 | 8.2 ± 6.2 | 43.7 ± 12.6 | SIR 2.15 (0.89–5.15) |
| Myeloma | ANZDATA [55] | 8173 | mean 6.0 | 43.4 (31.1–53.9) | SIR 1.8 (0.6–4.2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, C.; de Wit, S.; Učambarlić, E.; Markousis-Mavrogenis, G.; Screever, E.M.; Meijers, W.C.; de Boer, R.A.; Aboumsallem, J.P. Multifactorial Diseases of the Heart, Kidneys, Lungs, and Liver and Incident Cancer: Epidemiology and Shared Mechanisms. Cancers 2023, 15, 729. https://doi.org/10.3390/cancers15030729
Shi C, de Wit S, Učambarlić E, Markousis-Mavrogenis G, Screever EM, Meijers WC, de Boer RA, Aboumsallem JP. Multifactorial Diseases of the Heart, Kidneys, Lungs, and Liver and Incident Cancer: Epidemiology and Shared Mechanisms. Cancers. 2023; 15(3):729. https://doi.org/10.3390/cancers15030729
Chicago/Turabian StyleShi, Canxia, Sanne de Wit, Emina Učambarlić, George Markousis-Mavrogenis, Elles M. Screever, Wouter C. Meijers, Rudolf A. de Boer, and Joseph Pierre Aboumsallem. 2023. "Multifactorial Diseases of the Heart, Kidneys, Lungs, and Liver and Incident Cancer: Epidemiology and Shared Mechanisms" Cancers 15, no. 3: 729. https://doi.org/10.3390/cancers15030729
APA StyleShi, C., de Wit, S., Učambarlić, E., Markousis-Mavrogenis, G., Screever, E. M., Meijers, W. C., de Boer, R. A., & Aboumsallem, J. P. (2023). Multifactorial Diseases of the Heart, Kidneys, Lungs, and Liver and Incident Cancer: Epidemiology and Shared Mechanisms. Cancers, 15(3), 729. https://doi.org/10.3390/cancers15030729