
Molecular Mechanisms in Murine Syngeneic Leukemia Stem Cells

 and
and 
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Acute Myeloid Leukemia (AML)
1.2. Leukemic Stem Cells
1.3. Murine Models for AML
1.4. A Novel Murine Model of AML LSC
2. Materials and Methods
2.1. Animals
2.2. Blood Sampling and FACS Processing
2.3. RNA Sequencing and Analysis
3. Results
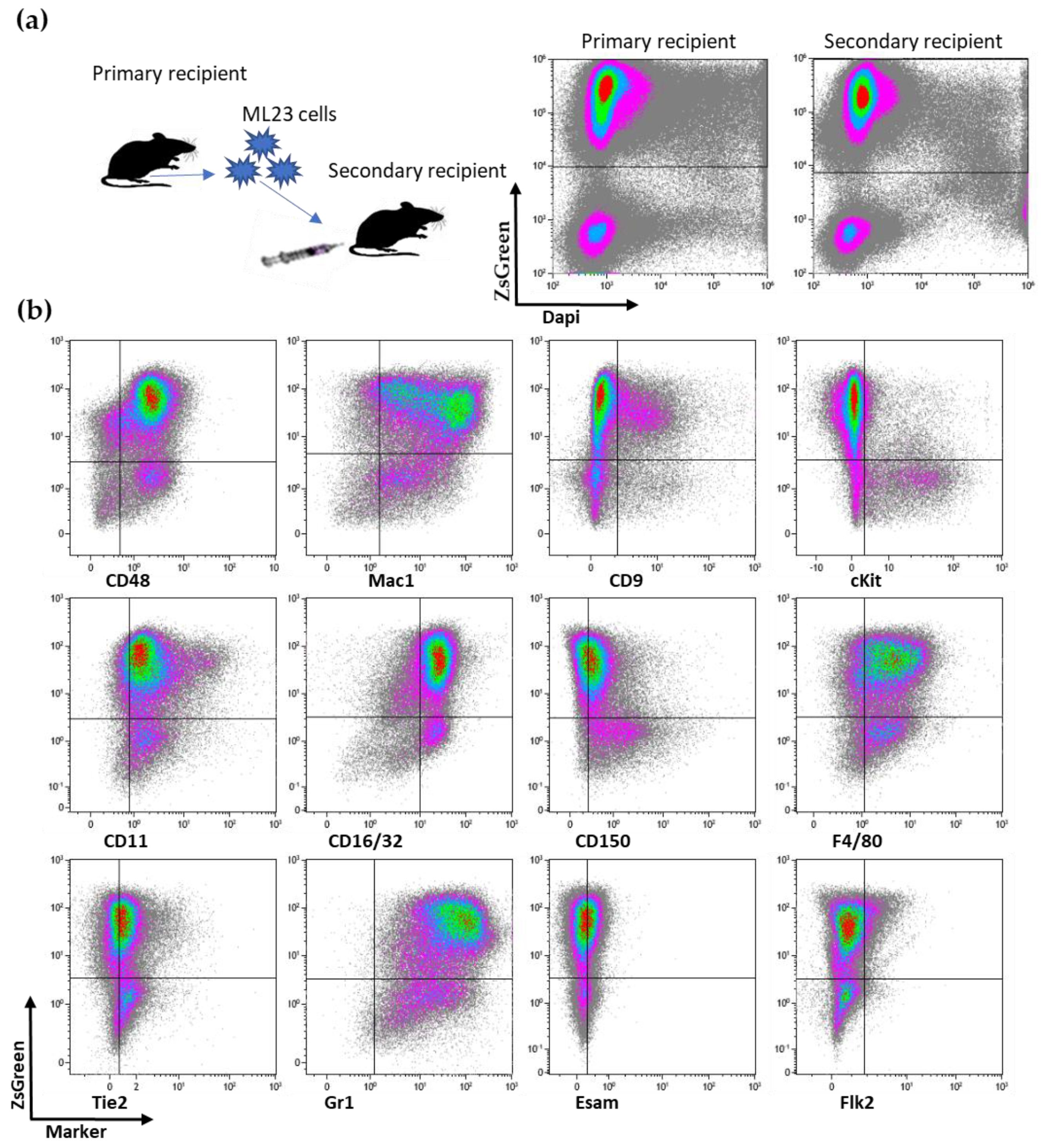
3.1. ML23 Leukemia Line Can Passage Serially and Present Heterogeneity
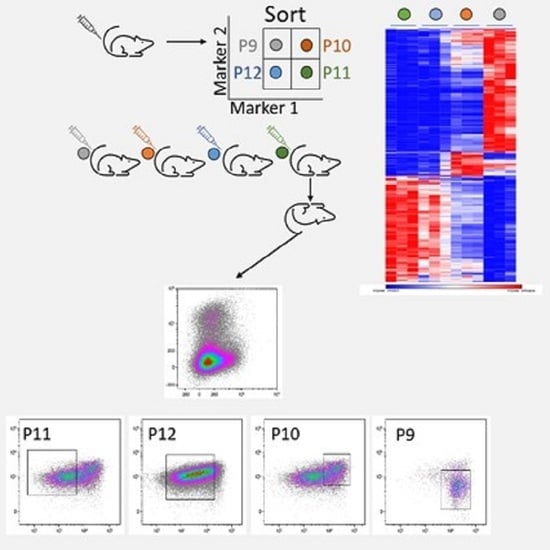
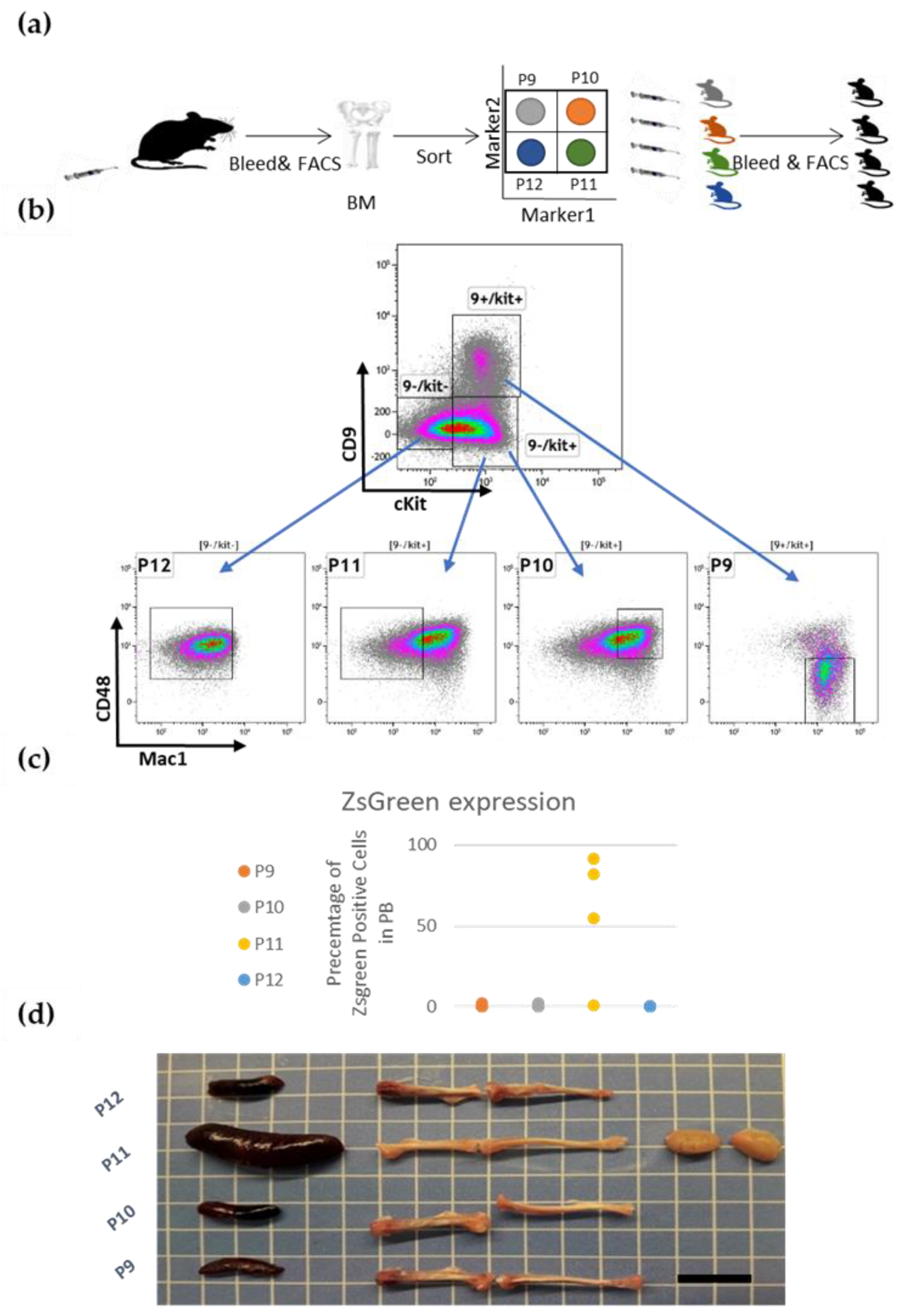
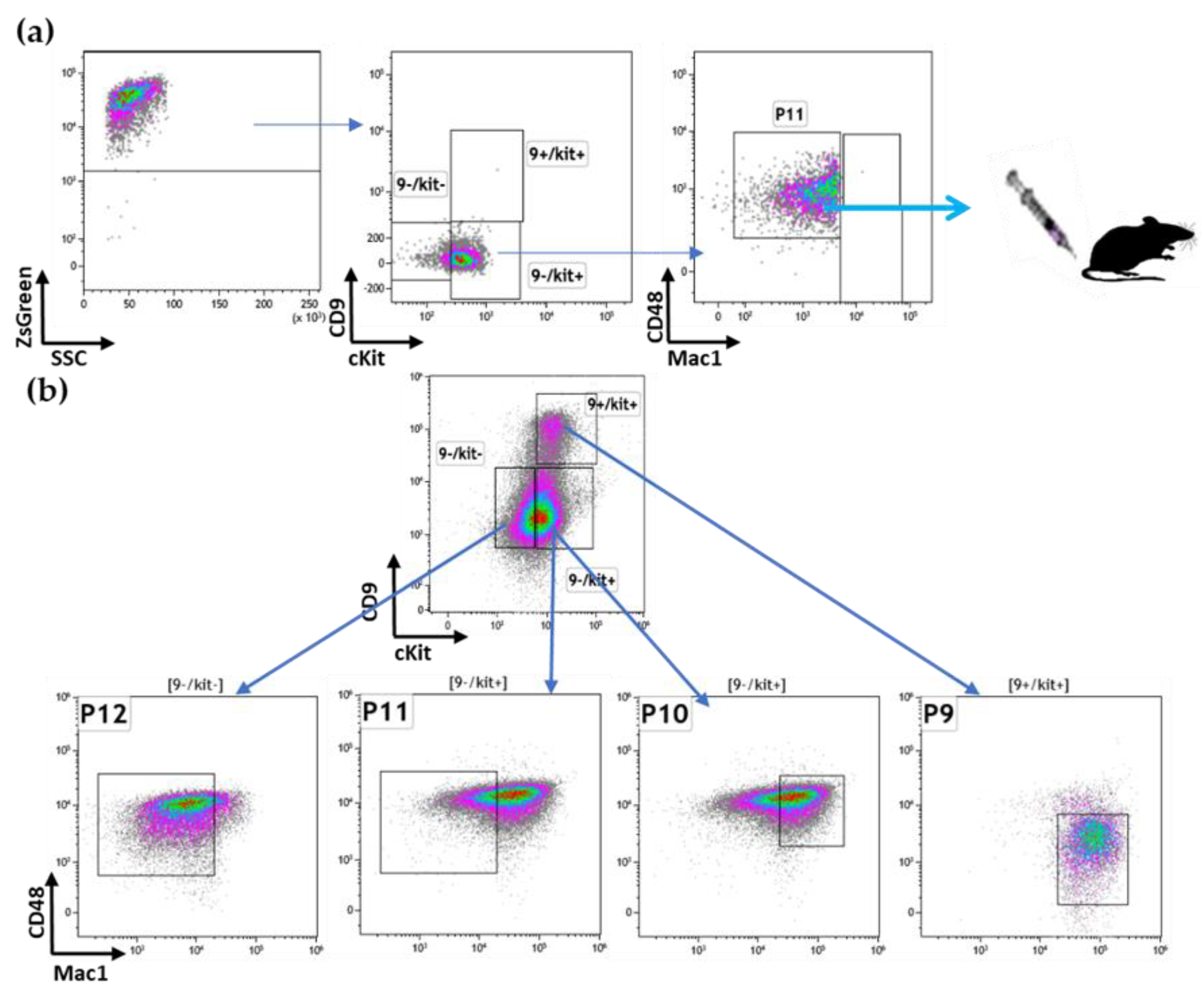
3.2. Prospective Isolation of LSC in the ML23 Leukemia Line
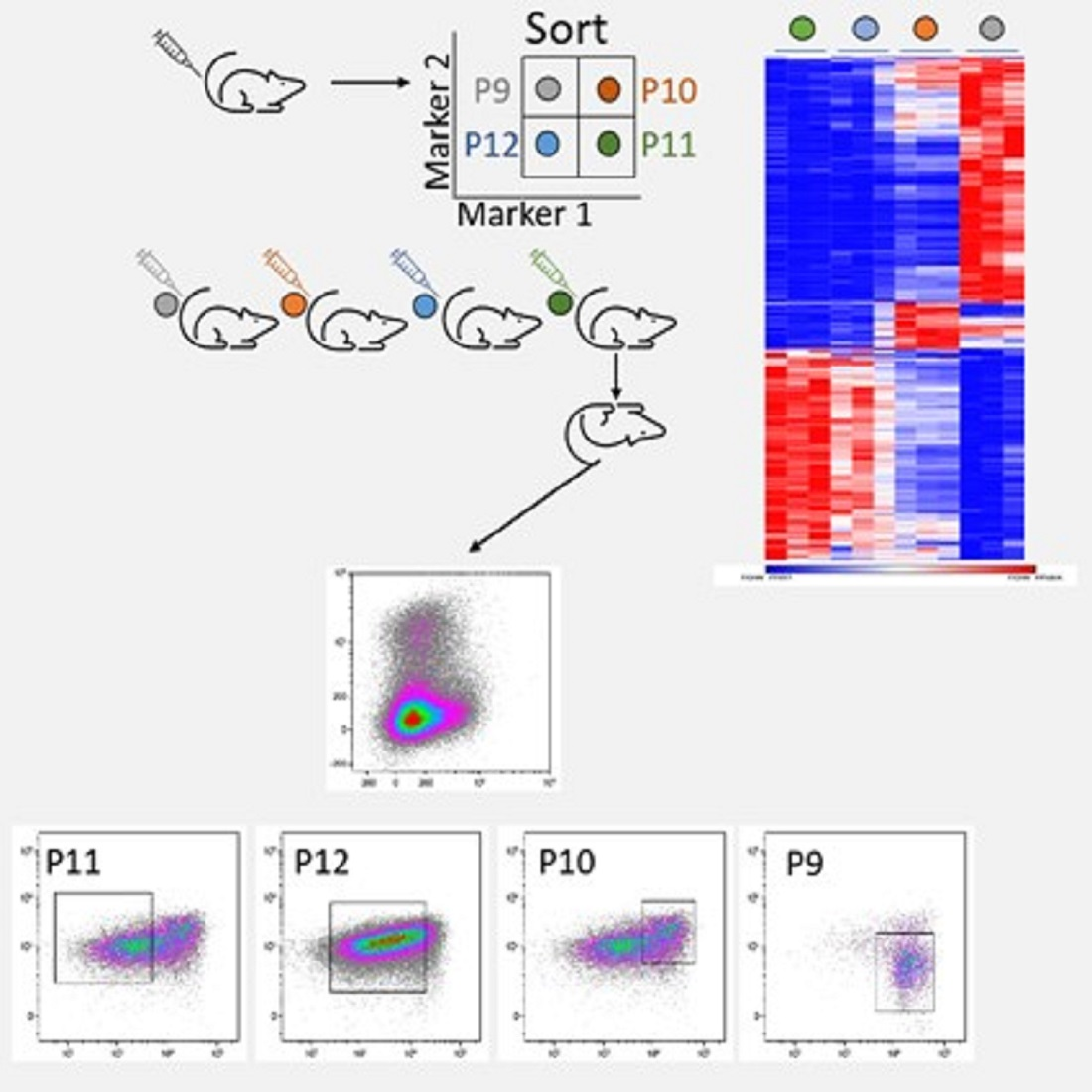
3.3. The P11 LSC Sub-Population Reconstitutes the Heterogeneity of ML23
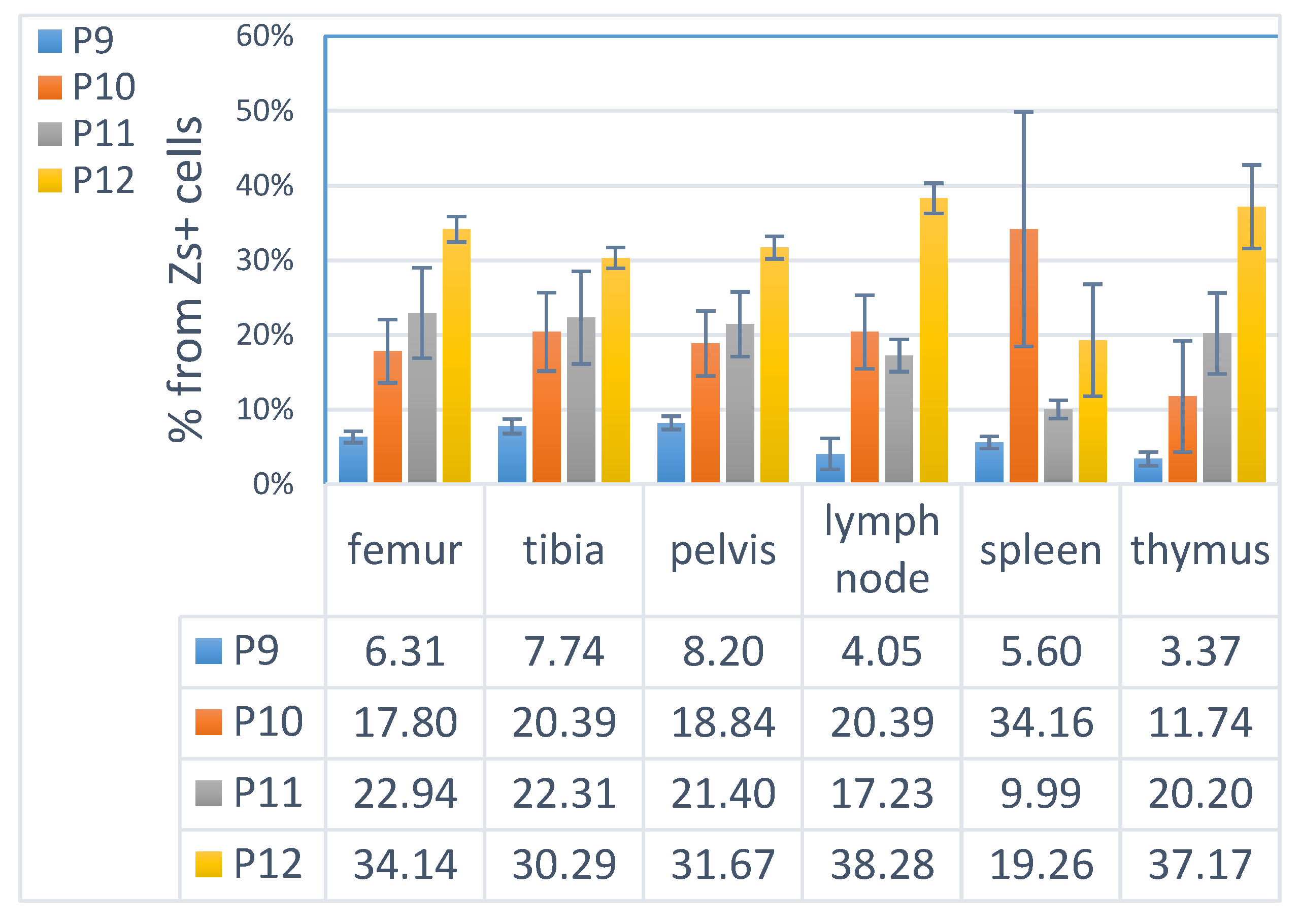
3.4. ML23 Sub-Populations Distribute in Various Organs
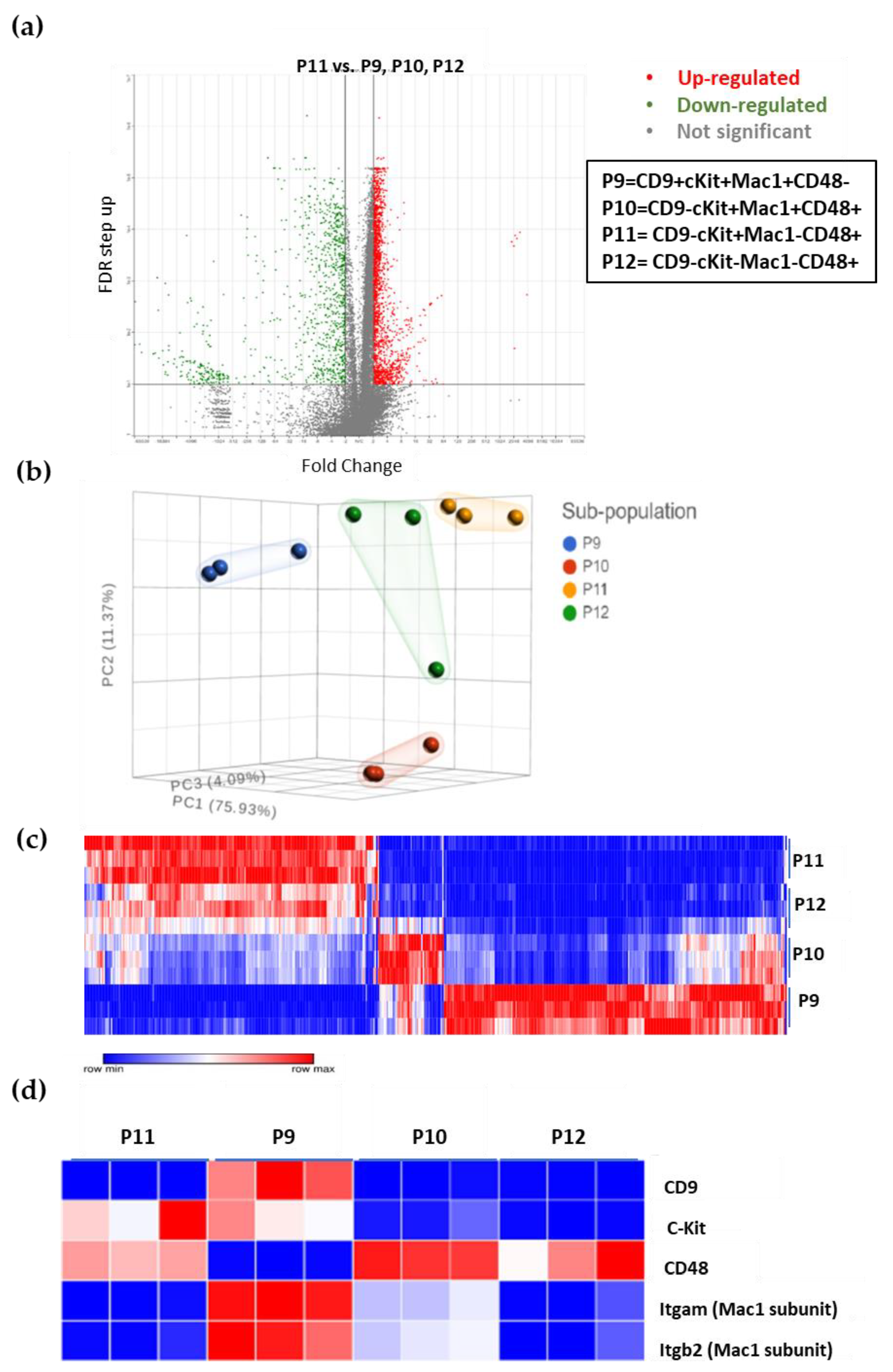
3.5. The Unique Gene Expression Signature of LSC
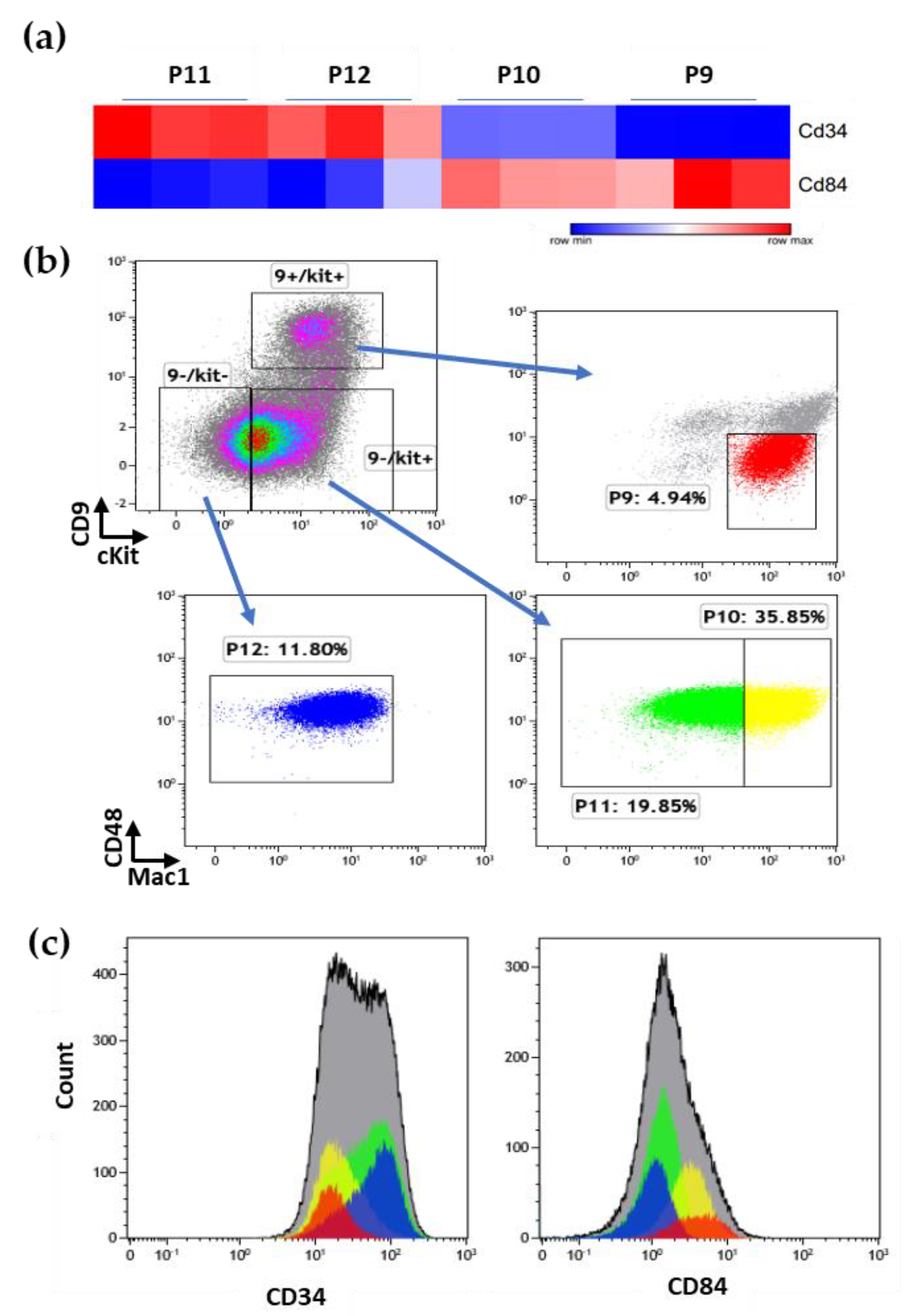
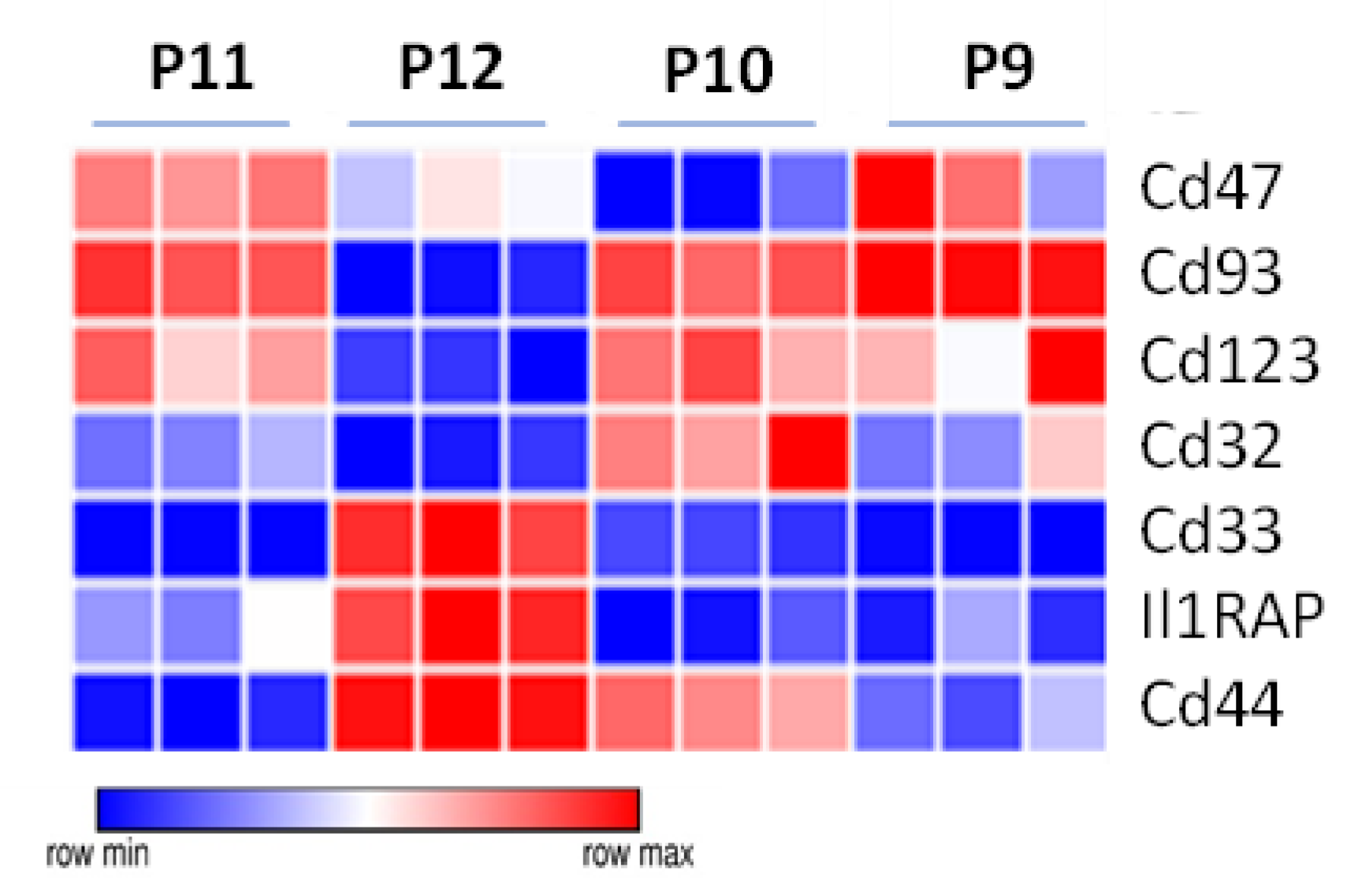
3.6. Additional Surface Markers Validate Differential Expression on LSCs vs. Other ML23 Sub-Populations
3.7. Specific Drug-Targeted Genes Expressed in ML23 Cells
4. Discussion
4.1. Overview
4.2. Heterogeneity of ML23 Cells
4.3. Prospective Isolation of LSCs and the Reconstitution of the Disease
4.4. Distribution of Leukemia and LSC
4.5. Molecular Signature of LSCs
4.6. Possible Treatments to Eradicate Leukemia and LSCs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cook, G.J.; Pardee, T.S. Animal models of leukemia: Any closer to the real thing? Cancer Metastasis Rev. 2013, 32, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 2, 41. [Google Scholar] [CrossRef] [PubMed]
- Hanekamp, D.; Denys, B.; Kaspers, G.J.L.; Te Marvelde, J.G.; Schuurhuis, G.J.; De Haas, V.; De Moerloose, B.; de Bont, E.S.; Zwaan, C.M.; de Jong, A.; et al. Leukaemic stem cell load at diagnosis predicts the development of relapse in young acute myeloid leukaemia patients. Br. J. Haematol. 2018, 183, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Schiller, G. How thinly can one slice the AML diagnostic pie? Blood 2022, 140, 1330–1331. [Google Scholar] [CrossRef]
- Estey, E.; Döhner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Kelly, L.M.; Gilliland, D.G. Genetics of Myeloid Leukemias. Annu. Rev. Genom. Hum. Genet. 2003, 3, 179–198. [Google Scholar] [CrossRef]
- Fröhling, S.; Scholl, C.; Gilliland, D.G.; Levine, R.L. Genetics of myeloid malignancies: Pathogenetic and clinical implications. J. Clin. Oncol. 2005, 23, 6285–6295. [Google Scholar] [CrossRef]
- Keinan, N.; Scharff, Y.; Goldstein, O.; Chamo, M.; Ilic, S.; Gazit, R. Syngeneic leukemia models using lentiviral transgenics. Cell Death Dis. 2021, 12, 193. [Google Scholar] [CrossRef]
- Almosailleakh, M.; Schwaller, J. Murine Models of Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 453. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 7, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Mechanisms of Disease: Cancer Stem Cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef]
- Zeng, A.G.X.; Bansal, S.; Jin, L.; Mitchell, A.; Chen, W.C.; Abbas, H.A.; Chan-Seng-Yue, M.; Voisin, V.; van Galen, P.; Tierens, A.; et al. A cellular hierarchy framework for understanding heterogeneity and predicting drug response in acute myeloid leukemia. Nat. Med. 2022, 28, 1212–1223. [Google Scholar] [CrossRef]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef]
- Titov, A.; Kaminskiy, Y.; Ganeeva, I.; Zmievskaya, E.; Valiullina, A.; Rakhmatullina, A.; Petukhov, A.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Knowns and Unknowns about CAR-T Cell Dysfunction. Cancers 2022, 14, 1078. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A.; Chávez-Valencia, V.; Gómez-Guijosa, M.Á.; Cortes-Penagos, C. Acute Myeloid Leukemia-Genetic Alterations and Their Clinical Prognosis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 328–339. [Google Scholar] [PubMed]
- Terwijn, M.; Zeijlemaker, W.; Kelder, A.; Rutten, A.P.; Snel, A.N.; Scholten, W.J.; Pabst, T.; Verhoef, G.; Löwenberg, B.; Zweegman, S.; et al. Leukemic stem cell frequency: A strong biomarker for clinical outcome in acute myeloid leukemia. PLoS ONE 2014, 9, e107587. [Google Scholar] [CrossRef]
- Hahn, W.C.; Weinberg, R.A. Modelling the molecular circuitry of cancer. Nat. Rev. Cancer 2002, 2, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Friend, C. Cell-free transmission in adult Swiss mice of a disease having the character of a leukemia. J. Exp. Med. 1957, 105, 307–318. [Google Scholar] [CrossRef]
- Kunimoto, H.; Meydan, C.; Nazir, A.; Whitfield, J.; Shank, K.; Rapaport, F.; Maher, R.; Pronier, E.; Meyer, S.C.; Garrett-Bakelman, F.E.; et al. Cooperative Epigenetic Remodeling by TET2 Loss and NRAS Mutation Drives Myeloid Transformation and MEK Inhibitor Sensitivity. Cancer Cell 2018, 33, 44–59. [Google Scholar] [CrossRef]
- Hogenesch, H.; Nikitin, A.Y. Challenges in pre-clinical testing of anti-cancer drugs in cell culture and in animal models. J. Control. Release 2012, 164, 183–186. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Kim, D.Y.; Lee, S.; Kim, D.Y.; Lee, J.Y. Effective Murine Model Induction for Niche Study in Immune Cells Against Leukemia. Adv. Exp. Med. Biol. 2020, 1232, 415–420. [Google Scholar]
- Adams, J.M.; Kelly, P.N.; Dakic, A.; Carotta, S.; Nutt, S.L.; Strasser, A. Role of “cancer stem cells” and cell survival in tumor development and maintenance. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 451–459. [Google Scholar] [CrossRef]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T. Leukocyte Adhesion Molecules. Encycl. Immunobiol. 2016, 3, 505–511. [Google Scholar]
- Yoon, S.O.; Zhang, X.; Freedman, A.S.; Zahrieh, D.; Lossos, I.S.; Li, L.; Choi, Y.S. Down-regulation of CD9 expression and its correlation to tumor progression in B lymphomas. Am. J. Pathol. 2010, 177, 377–386. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, G.; Zhang, J.; Chen, X.; Xu, H.; Heng, G.; Chen, J.; Zhao, Y.; Li, J.; Ni, Y.; et al. CD9, a potential leukemia stem cell marker, regulates drug resistance and leukemia development in acute myeloid leukemia. Stem Cell Res. Ther. 2021, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Soda, Y.; Jinno, A.; Tanaka, Y.; Akagi, T.; Shimotohno, K.; Hoshino, H. Rapid tumor formation and development of neutrophilia and splenomegaly in nude mice transplanted with human cells expressing human T cell leukemia virus type I or Tax1. Leukemia 2000, 14, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Gollard, R.P.; Robbins, B.A.; Piro, L.; Saven, A. Acute myelogenous leukemia presenting with bulky lymphadenopathy. Case report and literature review. Acta Haematol. 1996, 95, 129–134. [Google Scholar] [CrossRef]
- Houshmand, M.; Blanco, T.M.; Circosta, P.; Yazdi, N.; Kazemi, A.; Saglio, G.; Zarif, M.N. Bone marrow microenvironment: The guardian of leukemia stem cells. World, J. Stem Cells 2019, 11, 476–490. [Google Scholar] [CrossRef]
- Somervaille, T.C.P.; Cleary, M.L. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006, 10, 257–268. [Google Scholar] [CrossRef]
- Zaiss, M.; Hirtreiter, C.; Rehli, M.; Rehm, A.; Kunz-Schughart, L.A.; Andreesen, R.; Hennemann, B. CD84 expression on human hematopoietic progenitor cells. Exp. Hematol. 2003, 31, 798–805. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Jordan, C.T. Therapeutic targeting of acute myeloid leukemia stem cells. Blood 2017, 129, 1627–1635. [Google Scholar] [CrossRef]
- Jin, L.; Lee, E.M.; Ramshaw, H.S.; Busfield, S.J.; Peoppl, A.G.; Wilkinson, L.; Guthridge, M.A.; Thomas, D.; Barry, E.F.; Boyd, A.; et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell 2009, 5, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006, 12, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Taussig, D.C.; Pearce, D.J.; Simpson, C.; Rohatiner, A.Z.; Lister, T.A.; Kelly, G.; Luongo, J.L.; Danet-Desnoyers, G.A.; Bonnet, D. Hematopoietic stem cells express multiple myeloid markers: Implications for the origin and targeted therapy of acute myeloid leukemia. Blood 2005, 106, 4086–4092. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef]
- Theocharides, A.P.; Jin, L.; Cheng, P.Y.; Prasolava, T.K.; Malko, A.V.; Ho, J.M.; Poeppl, A.G.; van Rooijen, N.; Minden, M.D.; Danska, J.S.; et al. Disruption of SIRPα signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J. Exp. Med. 2012, 209, 1883–1899. [Google Scholar] [CrossRef]
- Hosen, N.; Park, C.Y.; Tatsumi, N.; Oji, Y.; Sugiyama, H.; Gramatzki, M.; Krensky, A.M.; Weissman, I.L. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2007, 104, 11008–11013. [Google Scholar] [CrossRef]
- Iwasaki, M.; Liedtke, M.; Gentles, A.J.; Cleary, M.L. CD93 Marks a Non-Quiescent Human Leukemia Stem Cell Population and Is Required for Development of MLL-Rearranged Acute Myeloid Leukemia. Cell Stem Cell 2015, 17, 412–421. [Google Scholar] [CrossRef]
- Saito, Y.; Kitamura, H.; Hijikata, A.; Tomizawa-Murasawa, M.; Tanaka, S.; Takagi, S.; Uchida, N.; Suzuki, N.; Sone, A.; Najima, Y.; et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci. Transl. Med. 2010, 2, 17. [Google Scholar] [CrossRef]
- Kikushige, Y.; Shima, T.; Takayanagi, S.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 2010, 7, 708–717. [Google Scholar] [CrossRef]
- Chung, S.S.; Eng, W.S.; Hu, W.; Khalaj, M.; Garrett-Bakelman, F.E.; Tavakkoli, M.; Levine, R.L.; Carroll, M.; Klimek, V.M.; Melnick, A.M.; et al. CD99 is a therapeutic target on disease stem cells in myeloid malignancies. Sci. Transl. Med. 2017, 9, eaaj2025. [Google Scholar] [CrossRef] [PubMed]
- Askmyr, M.; Ågerstam, H.; Hansen, N.; Gordon, S.; Arvanitakis, A.; Rissler, M.; Juliusson, G.; Richter, J.; Järås, M.; Fioretos, T. Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood 2013, 121, 3709–3713. [Google Scholar] [CrossRef] [PubMed]
- Saadatpour, A.; Guo, G.; Orkin, S.H.; Yuan, G.C. Characterizing heterogeneity in leukemic cells using single-cell gene expression analysis. Genome Biol. 2014, 15, 525. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Luc, S.; Marco, E.; Lin, T.W.; Peng, C.; Kerenyi, M.A.; Beyaz, S.; Kim, W.; Xu, J.; Das, P.P.; et al. Mapping cellular hierarchy by single-cell analysis of the cell surface repertoire. Cell Stem Cell 2013, 13, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.; LaMere, M.; Stevens, B.M.; Ashton, J.M.; Myers, J.R.; O’Dwyer, K.M.; Liesveld, J.L.; Mendler, J.H.; Guzman, M.; Morrissette, J.D.; et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016, 128, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.W.; Scadden, D.T.; Gilliland, D.G. The leukemic stem cell niche: Current concepts and therapeutic opportunities. Blood 2009, 114, 1150–1157. [Google Scholar] [CrossRef]
- Vetrie, D.; Helgason, G.V.; Copland, M. The leukaemia stem cell: Similarities, differences and clinical prospects in CML and AML. Nat. Rev. Cancer 2020, 20, 158–173. [Google Scholar] [CrossRef]
- Abarrategi, A.; Foster, K.; Hamilton, A.; Mian, S.A.; Passaro, D.; Gribben, J.; Mufti, G.; Bonnet, D. Versatile humanized niche model enables study of normal and malignant human hematopoiesis. J. Clin. Investig. 2017, 127, 543–548. [Google Scholar] [CrossRef]
- Duarte, D.; Hawkins, E.D.; Lo Celso, C. The interplay of leukemia cells and the bone marrow microenvironment. Blood 2018, 131, 1507–1511. [Google Scholar] [CrossRef]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef]
- Bernasconi, P.; Borsani, O. Targeting Leukemia Stem Cell-Niche Dynamics: A New Challenge in AML Treatment. J. Oncol. 2019, 2019, 8323592. [Google Scholar] [CrossRef] [PubMed]
- Lutz, C.; Hoang, V.T.; Buss, E.; Ho, A.D. Identifying leukemia stem cells--is it feasible and does it matter? Cancer Lett. 2013, 338, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Thastrup, M.; Duguid, A.; Mirian, C.; Schmiegelow, K.; Halsey, C. Central nervous system involvement in childhood acute lymphoblastic leukemia: Challenges and solutions. Leukemia 2022, 36, 2751–2768. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Sadovnik, I.; Eisenwort, G.; Herrmann, H.; Bauer, K.; Mueller, N.; Sperr, W.R.; Wicklein, D.; Schumacher, U. Redistribution, homing and organ-invasion of neoplastic stem cells in myeloid neoplasms. Semin. Cancer Biol. 2020, 60, 191–201. [Google Scholar] [CrossRef]
- Perrera, C.; Colombo, R.; Valsasina, B.; Carpinelli, P.; Troiani, S.; Modugno, M.; Gianellini, L.; Cappella, P.; Isacchi, A.; Moll, J.; et al. Identification of Myb-binding protein 1A (MYBBP1A) as a novel substrate for aurora B kinase. J. Biol. Chem. 2010, 285, 11775–11785. [Google Scholar] [CrossRef]
- Thomas, D.; Majeti, R. Biology and relevance of human acute myeloid leukemia stem cells. Blood 2017, 129, 1577–1585. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamo, M.; Koren, O.; Goldstein, O.; Bujanover, N.; Keinan, N.; Scharff, Y.; Gazit, R. Molecular Mechanisms in Murine Syngeneic Leukemia Stem Cells. Cancers 2023, 15, 720. https://doi.org/10.3390/cancers15030720
Chamo M, Koren O, Goldstein O, Bujanover N, Keinan N, Scharff Y, Gazit R. Molecular Mechanisms in Murine Syngeneic Leukemia Stem Cells. Cancers. 2023; 15(3):720. https://doi.org/10.3390/cancers15030720
Chicago/Turabian StyleChamo, Michael, Omri Koren, Oron Goldstein, Nir Bujanover, Nurit Keinan, Ye’ela Scharff, and Roi Gazit. 2023. "Molecular Mechanisms in Murine Syngeneic Leukemia Stem Cells" Cancers 15, no. 3: 720. https://doi.org/10.3390/cancers15030720
APA StyleChamo, M., Koren, O., Goldstein, O., Bujanover, N., Keinan, N., Scharff, Y., & Gazit, R. (2023). Molecular Mechanisms in Murine Syngeneic Leukemia Stem Cells. Cancers, 15(3), 720. https://doi.org/10.3390/cancers15030720