
Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability


Abstract
Simple Summary
Abstract
1. Introduction
2. Mechanisms of ADC Toxicity
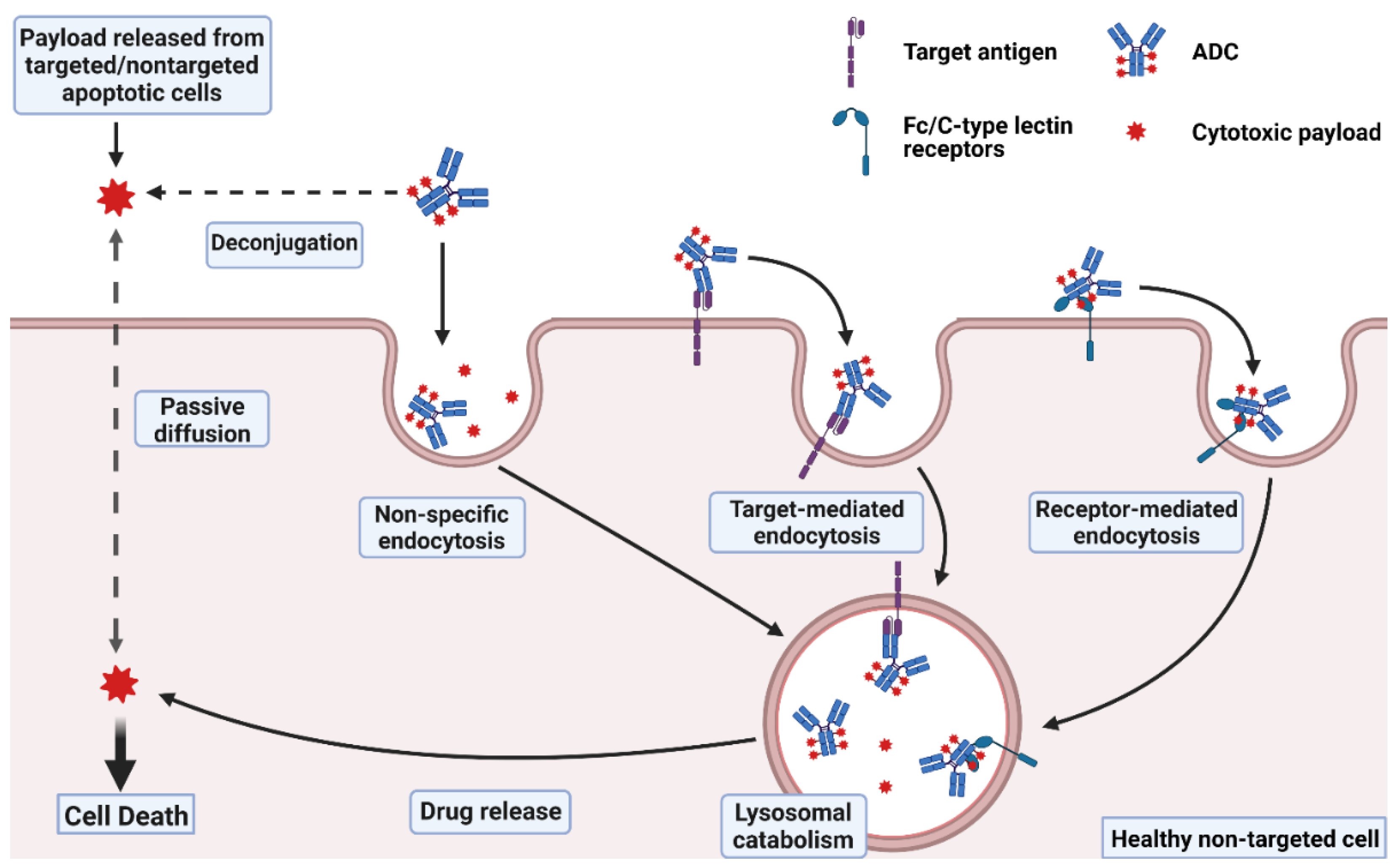
2.1. Target-Independent Toxicity: Off-Target, Off-Site Toxicity
2.1.1. Off-Target Delivery of ADC Payloads
2.1.2. Off-Target Receptor-Mediated Uptake of ADCs
2.2. Off-Site, On-Target Toxicity
3. Clinical Toxicity Profiles of Antibody-Drug Conjugates
3.1. Approved ADCs
3.1.1. ADCs with Calicheamicin Payload
Gemtuzumab Ozogamicin (Mylotarg)
Inotuzumab Ozogamicin (Besponsa)
3.1.2. ADCs with Auristatin Payloads
Brentuximab Vedotin (Adcetris)
Polatuzumab Vedotin (Polivy)
Enfortumab Vedotin (Padcev)
Tisotumab Vedotin (Tivdak)
Belantamab Mafodotin (Blenrep)
3.1.3. ADCs with Maytansinoid Payloads
Trastuzumab Emtansine (Kadcyla)
Mirvetuximab Soravtansine (Elahere)
3.1.4. ADCs with Camptothecin Payloads
Trastuzumab Deruxtecan (Enhertu)
Sacituzumab Govitecan (Trodelvy)
3.1.5. ADCs with Pyrrolobenzodiazepine Payloads
Loncastuximab Tesirine (Zynlonta)
3.2. Late-Stage ADCs
3.2.1. Trastuzumab Duocarmazine
3.2.2. Disitamab Vedotin
3.3. Frequently Reported ADC-Associated Dose-Limiting Toxicities
3.3.1. Neutropenia
3.3.2. Thrombocytopenia
3.3.3. Peripheral Neuropathy
3.3.4. Ocular Toxicity
4. Strategies to Reduce Toxicities
4.1. Modifying Conjugation Technology or Drug/Linker Chemistry
4.2. Antibody Modifications
4.3. Modifying Dosage Regimens
4.4. Inverse Targeting Strategy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Mecklenburg, L. A Brief Introduction to Antibody–Drug Conjugates for Toxicologic Pathologists. Toxicol. Pathol. 2018, 46, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Pysz, I.; Jackson, P.J.M.; Barlow, D.J.; Rahman, K.M.; Thurston, D.E. UPLC-based assay to assess the hydrophobicity of Antibody-Drug Conjugate (ADC) payloads. J. Chromatogr. B 2020, 1146, 122075. [Google Scholar] [CrossRef]
- Lobo, E.D.; Hansen, R.J.; Balthasar, J.P. Antibody pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 2004, 93, 2645–2668. [Google Scholar] [CrossRef]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef]
- Williams, R.J.; Tse, T.; Dipiazza, K.; Zarin, D.A. Terminated Trials in the ClinicalTrials.gov Results Database: Evaluation of Availability of Primary Outcome Data and Reasons for Termination. PLoS ONE 2015, 10, e0127242. [Google Scholar] [CrossRef]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016, 8, 659–671. [Google Scholar] [CrossRef]
- Nejadmoghaddam, M.-R.; Minai-Tehrani, A.; Ghahremanzadeh, R.; Mahmoudi, M.; Dinarvand, R.; Zarnani, A.-H. Antibody-Drug Conjugates: Possibilities and Challenges. Avicenna J. Med. Biotechnol. 2019, 11, 3–23. [Google Scholar]
- Coats, S.; Williams, M.; Kebble, B.; Dixit, R.; Tseng, L.; Yao, N.-S.; Tice, D.A.; Soria, J.-C. Antibody–Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin. Cancer Res. 2019, 25, 5441–5448. [Google Scholar] [CrossRef]
- Wolska-Washer, A.; Robak, T. Safety and Tolerability of Antibody-Drug Conjugates in Cancer. Drug Saf. 2019, 42, 295–314. [Google Scholar] [CrossRef]
- Masters, J.C.; Nickens, D.J.; Xuan, D.; Shazer, R.L.; Amantea, M. Clinical toxicity of antibody drug conjugates: A meta-analysis of payloads. Investig. New Drugs 2018, 36, 121–135. [Google Scholar] [CrossRef]
- Dokmanovic, M.; El Zarrad, K.; Hirsch, D.S.; Wu, W.J. Antibody-drug conjugates as therapeutic agents in oncology: Overview and perspectives. In Frontiers in Anti-Cancer Drug Discovery; Atta-Ur-Rahman, M., Choudhary, I., Eds.; Bentham Science Publishers: Oak Park, IL, USA, 2013; pp. 139–189. [Google Scholar]
- Lansita, J.A.; Burke, J.M.; Apgar, J.F.; Mounho-Zamora, B. An introduction to the regulatory and nonclinical aspects of the nonclinical development of antibody drug conjugates. Pharm. Res. 2015, 32, 3584–3592. [Google Scholar] [CrossRef] [PubMed]
- Saber, H.; Leighton, J.K. An FDA oncology analysis of antibody-drug conjugates. Regul. Toxicol. Pharmacol. 2015, 71, 444–452. [Google Scholar] [CrossRef]
- Casi, G.; Neri, D. Antibody–Drug Conjugates and Small Molecule–Drug Conjugates: Opportunities and Challenges for the Development of Selective Anticancer Cytotoxic Agents. J. Med. Chem. 2015, 58, 8751–8761. [Google Scholar] [CrossRef]
- Baker, M.P.; Reynolds, H.M.; Lumicisi, B.; Bryson, C.J. Immunogenicity of protein therapeutics: The key causes, consequences and challenges. Self Nonself 2010, 1, 314–322. [Google Scholar] [CrossRef]
- Guffroy, M.; Falahatpisheh, H.; Finkelstein, M. Improving the Safety Profile of ADCs. In Innovations for Next-Generation Antibody-Drug Conjugates; Damelin, M., Ed.; Springer International Publishing: Cham, Germany, 2018; pp. 45–71. [Google Scholar]
- Palanca-Wessels, M.C.A.; Czuczman, M.; Salles, G.; Assouline, S.; Sehn, L.H.; Flinn, I.; Patel, M.R.; Sangha, R.; Hagenbeek, A.; Advani, R.; et al. Safety and activity of the anti-CD79B antibody–drug conjugate polatuzumab vedotin in relapsed or refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukaemia: A phase 1 study. Lancet Oncol. 2015, 16, 704–715. [Google Scholar] [CrossRef]
- Rosenberg, J.; Sridhar, S.S.; Zhang, J.; Smith, D.; Ruether, D.; Flaig, T.W.; Baranda, J.; Lang, J.; Plimack, E.R.; Sangha, R.; et al. EV-101: A Phase I Study of Single-Agent Enfortumab Vedotin in Patients with Nectin-4–Positive Solid Tumors, Including Metastatic Urothelial Carcinoma. J. Clin. Oncol. 2020, 38, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Concin, N.; Hong, D.S.; Thistlethwaite, F.C.; Machiels, J.-P.; Arkenau, H.-T.; Plummer, R.; Jones, R.H.; Nielsen, D.; Windfeld, K.; et al. Tisotumab vedotin in patients with advanced or metastatic solid tumours (InnovaTV 201): A first-in-human, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Saber, H.; Simpson, N.; Ricks, T.K.; Leighton, J.K. An FDA oncology analysis of toxicities associated with PBD-containing antibody-drug conjugates. Regul. Toxicol. Pharm. 2019, 107, 104429. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, K.; Wang, K.; Zhu, H. Treatment-related adverse events of antibody–drug conjugates in clinical trials: A systematic review and meta-analysis. Cancer 2023, 129, 283–295. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers Having a Crucial Role in Antibody–Drug Conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Tumey, L.N.; Rago, B.; Han, X. In vivo biotransformations of antibody-drug conjugates. Bioanalysis 2015, 7, 1649–1664. [Google Scholar] [CrossRef]
- Drake, P.M.; Rabuka, D. Recent Developments in ADC Technology: Preclinical Studies Signal Future Clinical Trends. BioDrugs 2017, 31, 521–531. [Google Scholar] [CrossRef]
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.-T.; et al. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker Design. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Pillow, T.H.; Sadowsky, J.D.; Zhang, D.; Yu, S.-F.; Del Rosario, G.; Xu, K.; He, J.; Bhakta, S.; Ohri, R.; Kozak, K.R.; et al. Decoupling stability and release in disulfide bonds with antibody-small molecule conjugates. Chem. Sci. 2017, 8, 366–370. [Google Scholar] [CrossRef] [PubMed]
- McCombs, J.R.; Owen, S.C. Antibody Drug Conjugates: Design and Selection of Linker, Payload and Conjugation Chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Mahalingaiah, P.K.; Ciurlionis, R.; Durbin, K.R.; Yeager, R.L.; Philip, B.K.; Bawa, B.; Mantena, S.R.; Enright, B.P.; Liguori, M.J.; van Vleet, T.R.; et al. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol. Ther. 2019, 200, 110–125. [Google Scholar] [CrossRef]
- Singh, A.P.; Sharma, S.; Shah, D.K. Quantitative characterization of in vitro bystander effect of antibody-drug conjugates. J. Pharmacokinet. Pharmacodyn. 2016, 43, 567–582. [Google Scholar] [CrossRef]
- Polson, A.G.; Calemine-Fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; de Sauvage, F.J.; Eaton, D.; Elkins, K.; Elliott, J.M.; et al. Antibody-drug conjugates for the treatment of non-Hodgkin’s lymphoma: Target and linker-drug selection. Cancer Res. 2009, 69, 2358–2364. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of Drug–Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody–Maytansinoid Conjugates. Bioconjugate Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Boswell, C.A.; Tesar, D.B.; Mukhyala, K.; Theil, F.-P.; Fielder, P.J.; Khawli, L.A. Effects of Charge on Antibody Tissue Distribution and Pharmacokinetics. Bioconjugate Chem. 2010, 21, 2153–2163. [Google Scholar] [CrossRef] [PubMed]
- Stüber, J.C.; Rechberger, K.F.; Miladinović, S.M.; Pöschinger, T.; Zimmermann, T.; Villenave, R.; Eigenmann, M.J.; Kraft, T.E.; Shah, D.K.; Kettenberger, H.; et al. Impact of charge patches on tumor disposition and biodistribution of therapeutic antibodies. AAPS Open 2022, 8, 3. [Google Scholar] [CrossRef]
- Liu, S.; Verma, A.; Kettenberger, H.; Richter, W.F.; Shah, D.K. Effect of variable domain charge on in vitro and in vivo disposition of monoclonal antibodies. mAbs 2021, 13, 1993769. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Atkinson, J.; Gulesserian, S.; Zeng, Z.; Nater, J.; Ou, J.; Yang, P.; Morrison, K.; Coleman, J.; Malik, F.; et al. Modulation of Macropinocytosis-Mediated Internalization Decreases Ocular Toxicity of Antibody–Drug Conjugates. Cancer Res. 2018, 78, 2115–2126. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.; Einarsdottir, H.K.; Vidarsson, G. IgG-effector functions: “The Good, The Bad and The Ugly”. Immunol. Lett. 2014, 160, 139–144. [Google Scholar] [CrossRef]
- Uppal, H.; Doudement, E.; Mahapatra, K.; Darbonne, W.C.; Bumbaca, D.; Shen, B.-Q.; Du, X.; Saad, O.; Bowles, K.; Olsen, S.; et al. Potential Mechanisms for Thrombocytopenia Development with Trastuzumab Emtansine (T-DM1). Clin. Cancer Res. 2015, 21, 123–133. [Google Scholar] [CrossRef]
- Zhao, H.; Gulesserian, S.; Ganesan, S.K.; Ou, J.; Morrison, K.; Zeng, Z.; Robles, V.; Snyder, J.; Do, L.; Aviña, H.; et al. Inhibition of Megakaryocyte Differentiation by Antibody–Drug Conjugates (ADCs) is Mediated by Macropinocytosis: Implications for ADC-induced Thrombocytopenia. Mol. Cancer Ther. 2017, 16, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, M.; Tada, M.; Yokoo, H.; Demizu, Y.; Ishii-Watabe, A. Fcγ Receptor-Dependent Internalization and Off-Target Cytotoxicity of Antibody-Drug Conjugate Aggregates. Pharm. Res. 2022, 39, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Aoyama, M.; Ishii-Watabe, A. Fcγ Receptor Activation by Human Monoclonal Antibody Aggregates. J. Pharm. Sci. 2020, 109, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Bryson, C.J.; Cloake, E.A.; Welch, K.; Filipe, V.; Romeijn, S.; Hawe, A.; Jiskoot, W.; Baker, M.P.; Fogg, M.H. Small Amounts of Sub-Visible Aggregates Enhance the Immunogenic Potential of Monoclonal Antibody Therapeutics. Pharm. Res. 2015, 32, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Ohri, R.; Bhakta, S.; Fourie-O’Donohue, A.; dela Cruz-Chuh, J.; Tsai, S.P.; Cook, R.; Wei, B.; Ng, C.; Wong, A.W.; Bos, A.B.; et al. High-Throughput Cysteine Scanning To Identify Stable Antibody Conjugation Sites for Maleimide- and Disulfide-Based Linkers. Bioconjugate Chem. 2018, 29, 473–485. [Google Scholar] [CrossRef]
- Buecheler, J.W.; Winzer, M.; Tonillo, J.; Weber, C.; Gieseler, H. Impact of Payload Hydrophobicity on the Stability of Antibody–Drug Conjugates. Mol. Pharm. 2018, 15, 2656–2664. [Google Scholar] [CrossRef]
- Mills, B.J.; Kruger, T.; Bruncko, M.; Zhang, X.; Jameel, F. Effect of Linker-Drug Properties and Conjugation Site on the Physical Stability of ADCs. J. Pharm. Sci. 2020, 109, 1662–1672. [Google Scholar] [CrossRef]
- Bruggeman, C.W.; Houtzager, J.; Dierdorp, B.; Kers, J.; Pals, S.T.; Lutter, R.; van Gulik, T.; den Haan, J.M.M.; van den Berg, T.K.; van Bruggen, R.; et al. Tissue-specific expression of IgG receptors by human macrophages ex vivo. PLoS ONE 2019, 14, e0223264. [Google Scholar] [CrossRef]
- Hackshaw, M.D.; Danysh, H.E.; Singh, J.; Ritchey, M.E.; Ladner, A.; Taitt, C.; Camidge, D.R.; Iwata, H.; Powell, C.A. Incidence of pneumonitis/interstitial lung disease induced by HER2-targeting therapy for HER2-positive metastatic breast cancer. Breast Cancer Res. Treat. 2020, 183, 23–39. [Google Scholar] [CrossRef]
- Kumagai, K.; Aida, T.; Tsuchiya, Y.; Kishino, Y.; Kai, K.; Mori, K. Interstitial pneumonitis related to trastuzumab deruxtecan, a human epidermal growth factor receptor 2-targeting Ab-drug conjugate, in monkeys. Cancer Sci. 2020, 111, 4636–4645. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Norvell, T.M.; Tosi, M.F.; Emancipator, S.N.; Konstan, M.W.; Schreiber, J.R. Tissue-specific Fc gamma and complement receptor expression by alveolar macrophages determines relative importance of IgG and complement in promoting phagocytosis of Pseudomonas aeruginosa. Pediatr. Res. 1994, 35, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Conte, P.; Ascierto, P.A.; Patelli, G.; Danesi, R.; Vanzulli, A.; Sandomenico, F.; Tarsia, P.; Cattelan, A.; Comes, A.; De Laurentiis, M.; et al. Drug-induced interstitial lung disease during cancer therapies: Expert opinion on diagnosis and treatment. ESMO Open 2022, 7, 100404. [Google Scholar] [CrossRef] [PubMed]
- Gorovits, B.; Krinos-Fiorotti, C. Proposed mechanism of off-target toxicity for antibody–drug conjugates driven by mannose receptor uptake. Cancer Immunol. Immunother. 2013, 62, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Goetze, A.M.; Liu, Y.D.; Zhang, Z.; Shah, B.; Lee, E.; Bondarenko, P.V.; Flynn, G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 2011, 21, 949–959. [Google Scholar] [CrossRef]
- Kogelberg, H.; Tolner, B.; Sharma, S.K.; Lowdell, M.W.; Qureshi, U.; Robson, M.; Hillyer, T.; Pedley, R.B.; Vervecken, W.; Contreras, R.; et al. Clearance mechanism of a mannosylated antibody-enzyme fusion protein used in experimental cancer therapy. Glycobiology 2007, 17, 36–45. [Google Scholar] [CrossRef]
- Guffroy, M.; Falahatpisheh, H.; Biddle, K.; Kreeger, J.; Obert, L.; Walters, K.; Goldstein, R.; Boucher, G.; Coskran, T.; Reagan, W.; et al. Liver Microvascular Injury and Thrombocytopenia of Antibody–Calicheamicin Conjugates in Cynomolgus Monkeys—Mechanism and Monitoring. Clin. Cancer Res. 2017, 23, 1760–1770. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- S.A.A.P.U. Padcev Inc. (Enfortumab Vedotinejfv) for Injection [Prescribing Information]. Available online: https://astellas.us/docs/PADCEV_label.pdf (accessed on 2 April 2022).
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody–Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef]
- FDA: US Prescribing Information: Brentuximab Vedotin. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125388s100lbl.pdf (accessed on 2 April 2022).
- FDA: US Prescribing Information: Polatuzumab Vedotin. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761121s000lbl.pdf (accessed on 2 April 2022).
- TIVDAK (Tisotumab Vedoin-tftv): US Prescribing Information. Available online: https://seagendocs.com/Tivdak_Full_Ltr_Master.pdf (accessed on 2 April 2022).
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- FDA: US Prescribing Information: Trastuzumab Emtansine. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125427lbl.pdf (accessed on 2 April 2022).
- Banerji, U.; van Herpen, C.M.L.; Saura, C.; Thistlethwaite, F.; Lord, S.; Moreno, V.; Macpherson, I.R.; Boni, V.; Rolfo, C.; De Vries, E.G.E.; et al. Trastuzumab duocarmazine in locally advanced and metastatic solid tumours and HER2-expressing breast cancer: A phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2019, 20, 1124–1135. [Google Scholar] [CrossRef]
- Siegel, P.; Rose, A.; Maric, M.G.; Siegel, P.M. Glycoprotein non-metastatic b (GPNMB): A metastatic mediator and emerging therapeutic target in cancer. OncoTargets Ther. 2013, 2013, 839. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hamid, O.; Pavlick, A.C.; Kluger, H.; Kim, K.B.; Boasberg, P.D.; Simantov, R.; Crowley, E.; Green, J.A.; Hawthorne, T.; et al. Phase I/II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Patients with Advanced Melanoma. J. Clin. Oncol. 2014, 32, 3659–3666. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A.; Weaver, R.; Melisko, M.E.; Saleh, M.N.; Arena, F.P.; Forero, A.; Cigler, T.; Stopeck, A.; Citrin, D.; Oliff, I.; et al. EMERGE: A Randomized Phase II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Advanced Glycoprotein NMB–Expressing Breast Cancer. J. Clin. Oncol. 2015, 33, 1609–1619. [Google Scholar] [CrossRef]
- L’Italien, L.; Orozco, O.; Abrams, T.; Cantagallo, L.; Connor, A.; Desai, J.; Ebersbach, H.; Gelderblom, H.; Hoffmaster, K.; Lees, E.; et al. Mechanistic Insights of an Immunological Adverse Event Induced by an Anti-KIT Antibody Drug Conjugate and Mitigation Strategies. Clin. Cancer Res. 2018, 24, 3465–3474. [Google Scholar] [CrossRef]
- Liu, J.F.; Moore, K.N.; Birrer, M.J.; Berlin, S.; Matulonis, U.A.; Infante, J.R.; Wolpin, B.; Poon, K.A.; Firestein, R.; Xu, J.; et al. Phase I study of safety and pharmacokinetics of the anti-MUC16 antibody–drug conjugate DMUC5754A in patients with platinum-resistant ovarian cancer or unresectable pancreatic cancer. Ann. Oncol. 2016, 27, 2124–2130. [Google Scholar] [CrossRef]
- Argu’Eso, P.; Spurr-Michaud, S.; Russo, C.L.; Tisdale, A.; Gipson, I.K. MUC16 Mucin Is Expressed by the Human Ocular Surface Epithelia and Carries the H185 Carbohydrate Epitope. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2487–2495. [Google Scholar] [CrossRef] [PubMed]
- Stepan, L.P.; Trueblood, E.S.; Hale, K.; Babcook, J.; Borges, L.; Sutherland, C.L. Expression of Trop2 Cell Surface Glycoprotein in Normal and Tumor Tissues. J. Histochem. Cytochem. 2011, 59, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Diamond, J.R.; Moroose, R.L.; Isakoff, S.J.; Starodub, A.N.; Shah, N.C.; O’Shaughnessy, J.; Kalinsky, K.; Guarino, M.; et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients with Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2017, 35, 2141–2148. [Google Scholar] [CrossRef]
- Fenn, K.M.; Kalinsky, K. Sacituzumab govitecan: Antibody-drug conjugate in triple-negative breast cancer and other solid tumors. Drugs Today 2019, 55, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Hamann, P.R.; Hinman, L.M.; Beyer, C.F.; Lindh, D.; Upeslacis, J.; Flowers, D.A.; Bernstein, I. An anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Choice of linker. Bioconjugate Chem. 2002, 13, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.-R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab Ozogamicin, A Potent and Selective Anti-CD33 Antibody−Calicheamicin Conjugate for Treatment of Acute Myeloid Leukemia. Bioconjugate Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L.; Appelbaum, F.R.; Spielberger, R.T.; Forman, S.J.; Flowers, D.; Smith, F.O.; Shannon-Dorcy, K.; Berger, M.S.; Bernstein, I.D. Selective Ablation of Acute Myeloid Leukemia Using Antibody-Targeted Chemotherapy: A Phase I Study of an Anti-CD33 Calicheamicin Immunoconjugate. Blood 1999, 93, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukaemia in first relapse. Expert Opin. Biol. Ther. 2001, 1, 893–901. [Google Scholar] [CrossRef]
- Rowe, J.M.; Lowenberg, B. Gemtuzumab ozogamicin in acute myeloid leukemia: A remarkable saga about an active drug. Blood 2013, 121, 4838–4841. [Google Scholar] [CrossRef]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef]
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab Ozogamicin Versus Best Supportive Care in Older Patients with Newly Diagnosed Acute Myeloid Leukemia Unsuitable for Intensive Chemotherapy: Results of the Randomized Phase III Eortc-Gimema Aml-19 Trial. J. Clin. Oncol. 2016, 34, 972–979. [Google Scholar] [CrossRef]
- Lambert, J.; Pautas, C.; Terré, C.; Raffoux, E.; Turlure, P.; Caillot, D.; Legrand, O.; Thomas, X.; Gardin, C.; Gogat-Marchant, K.; et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: Final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 2019, 104, 113–119. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; Ko, C.-W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncolgist 2018, 23, 1103–1108. [Google Scholar] [CrossRef]
- FDA: US Prescribing Information: Gemtuzumab Ozogamicin. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761060lbl.pdf (accessed on 2 April 2022).
- Larson, R.A.; Sievers, E.L.; Stadtmauer, E.A.; Löwenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M.; et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Fenton, C.; Perry, C.M. Gemtuzumab Ozogamicin. Drugs 2005, 65, 2405–2427. [Google Scholar] [CrossRef]
- Maniecki, M.B.; Hasle, H.; Bendix, K.; Møller, H.J. Is hepatotoxicity in patients treated with gemtuzumabozogamicin due to specific targeting of hepatocytes? Leuk. Res. 2011, 35, e84–e86. [Google Scholar] [CrossRef]
- Rajvanshi, P.; Shulman, H.M.; Sievers, E.L.; McDonald, G.B. Hepatic sinusoidal obstruction after gemtuzumab ozogamicin (Mylotarg) therapy. Blood 2002, 99, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
- Godwin, C.D.; McDonald, G.B.; Walter, R.B. Sinusoidal obstruction syndrome following CD33-targeted therapy in acute myeloid leukemia. Blood 2017, 129, 2330–2332. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Hecht, K.; Reif, S.; Pelka-Fleischer, R.; Pfister, K.; Schmetzer, H. Expression and prognostic value of hemopoietic cytokine receptors in acute myeloid leukemia (AML): Implications for future therapeutical strategies. Eur. J. Haematol. 2004, 72, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Van Rhenen, A.; van Dongen, G.A.M.S.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Walsum, M.S.-V.; Zweegman, S.; Ossenkoppele, G.J.; Jan Schuurhuis, G. The novel AML stem cell–associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007, 110, 2659–2666. [Google Scholar] [CrossRef] [PubMed]
- Shor, B.; Gerber, H.-P.; Sapra, P. Preclinical and clinical development of inotuzumab-ozogamicin in hematological malignancies. Mol. Immunol. 2015, 67, 107–116. [Google Scholar] [CrossRef]
- Deangelo, D.J.; Stock, W.; Stein, A.S.; Shustov, A.; Liedtke, M.; Schiffer, C.A.; Vandendries, E.; Liau, K.; Ananthakrishnan, R.; Boni, J.; et al. Inotuzumab ozogamicin in adults with relapsed or refractory CD22-positive acute lymphoblastic leukemia: A phase 1/2 study. Blood Adv. 2017, 1, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Deangelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; de Angelo, D.J.; Advani, A.S.; Stelljes, M.; Kebriaei, P.; Cassaday, R.D.; Merchant, A.A.; Fujishima, N.; Uchida, T.; Calbacho, M.; et al. Hepatic adverse event profile of inotuzumab ozogamicin in adult patients with relapsed or refractory acute lymphoblastic leukaemia: Results from the open-label, randomised, phase 3 INO-VATE study. Lancet Haematol. 2017, 4, e387–e398. [Google Scholar] [CrossRef]
- FDA: US Prescribing Information: Inotuzumab Ozogamicin. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf (accessed on 2 April 2022).
- Ansell, S.M. Brentuximab vedotin. Blood 2014, 124, 3197–3200. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab Vedotin (SGN-35) for Relapsed CD30-Positive Lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a Pivotal Phase II Study of Brentuximab Vedotin for Patients with Relapsed or Refractory Hodgkin’s Lymphoma. J. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.H.; Chen, A.I.; Stiff, P.; Gianni, A.M.; Carella, A.; et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Polatuzumab Vedotin: First Global Approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef]
- Morschhauser, F.; Flinn, I.W.; Advani, R.; Sehn, L.H.; Diefenbach, C.; Kolibaba, K.; Press, O.W.; Salles, G.; Tilly, H.; Chen, A.I.; et al. Polatuzumab vedotin or pinatuzumab vedotin plus rituximab in patients with relapsed or refractory non-Hodgkin lymphoma: Final results from a phase 2 randomised study (ROMULUS). Lancet Haematol. 2019, 6, e254–e265. [Google Scholar] [CrossRef]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Terui, Y.; Rai, S.; Izutsu, K.; Yamaguchi, M.; Takizawa, J.; Kuroda, J.; Ishikawa, T.; Kato, K.; Suehiro, Y.; Fukuhara, N.; et al. A phase 2 study of polatuzumab vedotin + bendamustine + rituximab in relapsed/refractory diffuse large B-cell lymphoma. Cancer Sci. 2021, 112, 2845–2854. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; O’Donnell, P.H.; Balar, A.V.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.M.; et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma After Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J. Clin. Oncol. 2019, 37, 2592–2600. [Google Scholar] [CrossRef]
- Powles, T.; Rosenberg, J.E.; Sonpavde, G.P.; Loriot, Y.; Durán, I.; Lee, J.-L.; Matsubara, N.; Vulsteke, C.; Castellano, D.; Wu, C.; et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. N. Engl. J. Med. 2021, 384, 1125–1135. [Google Scholar] [CrossRef]
- Lacouture, M.E.; Patel, A.B.; Rosenberg, J.E.; O’Donnell, P.H. Management of Dermatologic Events Associated with the Nectin-4-directed Antibody-Drug Conjugate Enfortumab Vedotin. Oncolgist 2022, 27, e223–e232. [Google Scholar] [CrossRef]
- Ott, P.A.; Pavlick, A.C.; Johnson, D.B.; Hart, L.L.; Infante, J.R.; Luke, J.J.; Lutzky, J.; Rothschild, N.E.; Spitler, L.E.; Cowey, C.L.; et al. A phase 2 study of glembatumumab vedotin, an antibody-drug conjugate targeting glycoprotein NMB, in patients with advanced melanoma. Cancer 2019, 125, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Weinstock, C.; Zhang, L.; Charlab, R.; Dorff, S.E.; Gong, Y.; Hsu, V.; Li, F.; Ricks, T.K.; Song, P.; et al. FDA Approval Summary: Enfortumab Vedotin for Locally Advanced or Metastatic Urothelial Carcinoma. Clin. Cancer Res. 2021, 27, 922–927. [Google Scholar] [CrossRef]
- Markham, A. Tisotumab Vedotin: First Approval. Drugs 2021, 81, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Lorusso, D.; Gennigens, C.; González-Martín, A.; Randall, L.; Cibula, D.; Lund, B.; Woelber, L.; Pignata, S.; Forget, F.; et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 609–619. [Google Scholar] [CrossRef]
- Cho, Y.; Cao, X.; Shen, D.; Tuo, J.; Parver, L.M.; Rickles, F.R.; Chan, C.-C. Evidence for enhanced tissue factor expression in age-related macular degeneration. Lab. Investig. 2011, 91, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Ando, R.; Kase, S.; Ohashi, T.; Dong, Z.; Fukuhara, J.; Kanda, A.; Murata, M.; Noda, K.; Kitaichi, N.; Ishida, S. Tissue factor expression in human pterygium. Mol. Vis. 2011, 17, 63–69. [Google Scholar]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Anderson, L.D.; Sutherland, H.J.; Yong, K.; et al. Targeting B-cell maturation antigen with GSK2857916 antibody–drug conjugate in relapsed or refractory multiple myeloma (BMA117159): A dose escalation and expansion phase 1 trial. Lancet Oncol. 2018, 19, 1641–1653. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- GlaxoSmithKline. BLENREP (Belantamab Mafodotin-Blmf): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761158s000lbl.pdf (accessed on 2 April 2022).
- GlaxoSmithKline. GSK Provides an Update on Blenrep (Belantamab Mafodotin-Blmf) US Marketing Authorisation. 2022. Available online: https://www.gsk.com/en-gb/media/press-releases/gsk-provides-update-on-blenrep-us-marketing-authorisation/. (accessed on 17 January 2023).
- Ballantyne, A.; Dhillon, S. Trastuzumab Emtansine: First Global Approval. Drugs 2013, 73, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Beeram, M.; Krop, I.E.; Burris, H.A.; Girish, S.R.; Yu, W.; Lu, M.W.; Holden, S.N.; Modi, S. A phase 1 study of weekly dosing of trastuzumab emtansine (T-DM1) in patients with advanced human epidermal growth factor 2–positive breast cancer. Cancer 2012, 118, 5733–5740. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; Beeram, M.; Modi, S.; Jones, S.F.; Holden, S.N.; Yu, W.; Girish, S.; Tibbitts, J.; Yi, J.-H.; Sliwkowski, M.X.; et al. Phase I Study of Trastuzumab-DM1, an HER2 Antibody-Drug Conjugate, Given Every 3 Weeks to Patients With HER2-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef]
- Shi, J.-H.; Guo, W.-Z.; Jin, Y.; Zhang, H.-P.; Pang, C.; Li, J.; Line, P.-D.; Zhang, S.-J. Recognition of HER2 expression in hepatocellular carcinoma and its significance in postoperative tumor recurrence. Cancer Med. 2019, 8, 1269–1278. [Google Scholar] [CrossRef]
- Yan, H.; Endo, Y.; Shen, Y.; Rotstein, D.; Dokmanovic, M.; Mohan, N.; Mukhopadhyay, P.; Gao, B.; Pacher, P.; Wu, W.J. Ado-Trastuzumab Emtansine Targets Hepatocytes Via Human Epidermal Growth Factor Receptor 2 to Induce Hepatotoxicity. Mol. Cancer Ther. 2016, 15, 480–490. [Google Scholar] [CrossRef]
- Endo, Y.; Takeda, K.; Mohan, N.; Shen, Y.; Jiang, J.; Rotstein, D.; Wu, W.J. Payload of T-DM1 binds to cell surface cytoskeleton-associated protein 5 to mediate cytotoxicity of hepatocytes. Oncotarget 2018, 9, 37200–37215. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Mohan, N.; Dokmanovic, M.; Wu, W.J. Mechanisms contributing to ado-trastuzumab emtansine-induced toxicities: A gateway to better understanding of ADC-associated toxicities. Antib. Ther. 2021, 4, 55–59. [Google Scholar] [CrossRef]
- Moilanen, T.; Jokimäki, A.; Tenhunen, O.; Koivunen, J.P. Trastuzumab-induced cardiotoxicity and its risk factors in real-world setting of breast cancer patients. J. Cancer Res. Clin. Oncol. 2018, 144, 1613–1621. [Google Scholar] [CrossRef]
- National Toxicology Program. NTP Monograph: Developmental Effects and Pregnancy Outcomes Associated with Cancer Chemotherapy Use During Pregnancy. NTP Monogr. 2013, 2, i-214. [Google Scholar]
- Xia, L.-Y.; Hu, Q.-L.; Zhou, Q. Use of trastuzumab in treating breast cancer during pregnancy: A systematic review and meta-analysis. BMC Women’s Health 2021, 21, 169. [Google Scholar] [CrossRef] [PubMed]
- Ab, O.; Whiteman, K.R.; Bartle, L.M.; Sun, X.; Singh, R.; Tavares, D.; LaBelle, A.; Payne, G.; Lutz, R.J.; Pinkas, J.; et al. IMGN853, a Folate Receptor-alpha (FRalpha)-Targeting Antibody-Drug Conjugate, Exhibits Potent Targeted Antitumor Activity against FRalpha-Expressing Tumors. Mol. Cancer Ther. 2015, 14, 1605–1613. [Google Scholar] [CrossRef]
- Moore, K.N.; Borghaei, H.; O’Malley, D.M.; Jeong, W.; Seward, S.M.; Bauer, T.M.; Perez, R.P.; Matulonis, U.A.; Running, K.L.; Zhang, X.; et al. Phase 1 dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody-drug conjugate, in patients with solid tumors. Cancer 2017, 123, 3080–3087. [Google Scholar] [CrossRef]
- Moore, K.N.; Oza, A.M.; Colombo, N.; Oaknin, A.; Scambia, G.; Lorusso, D.; Konecny, G.E.; Banerjee, S.; Murphy, C.G.; Tanyi, J.L.; et al. Phase III, randomized trial of mirvetuximab soravtansine versus chemotherapy in patients with platinum-resistant ovarian cancer: Primary analysis of Forward I. Ann. Oncol. 2021, 32, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Osgood, C.L.; Singh, H.; Chiu, H.-J.; Ricks, T.K.; Chiu Yuen Chow, E.; Qiu, J.; Song, P.; Yu, J.; Namuswe, F.; et al. FDA Approval Summary: Fam-Trastuzumab Deruxtecan-Nxki for the Treatment of Unresectable or Metastatic HER2-Positive Breast Cancer. Clin. Cancer Res. 2021, 27, 4478–4485. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Iwata, H.; Tsurutani, J.; Takahashi, S.; Park, H.; Redfern, C.H.; Shitara, K.; Shimizu, C.; Taniguchi, H.; Iwasa, T.; et al. Single agent activity of DS-8201a, a HER2-targeting antibody-drug conjugate, in heavily pretreated HER2 expressing solid tumors. J. Clin. Oncol. 2017, 35, 108. [Google Scholar] [CrossRef]
- FDA: US Prescribing Information: Trastuzumab Deruxtecan. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761139s000lbl.pdf (accessed on 2 April 2022).
- Goldenberg, D.M.; Sharkey, R.M. Sacituzumab Govitecan, A Novel, Third-Generation, Antibody-drug Conjugate (ADC) for Cancer Therapy. Expert Opin. Biol. Ther. 2020, 20, 871–885. [Google Scholar] [CrossRef]
- Starodub, A.N.; Ocean, A.J.; Shah, M.A.; Guarino, M.J.; Picozzi, V.J.; Vahdat, L.T.; Thomas, S.S.; Govindan, S.V.; Maliakal, P.P.; Wegener, W.A.; et al. First-in-Human Trial of a Novel Anti-Trop-2 Antibody-SN-38 Conjugate, Sacituzumab Govitecan, for the Treatment of Diverse Metastatic Solid Tumors. Clin. Cancer Res. 2015, 21, 3870–3878. [Google Scholar] [CrossRef] [PubMed]
- Ocean, A.J.; Starodub, A.N.; Bardia, A.; Vahdat, L.T.; Isakoff, S.J.; Guarino, M.; Messersmith, W.A.; Picozzi, V.J.; Mayer, I.A.; Wegener, W.A.; et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: Safety and pharmacokinetics. Cancer 2017, 123, 3843–3854. [Google Scholar] [CrossRef] [PubMed]
- FDA: US Prescribing Information: Sacituzumab Govitecan. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761115s009lbl.pdf (accessed on 2 April 2022).
- FDA: US Prescribing Information: Irinotecan. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020571s048lbl.pdf (accessed on 2 April 2022).
- Lee, A. Loncastuximab Tesirine: First Approval. Drugs 2021, 81, 1229–1233. [Google Scholar] [CrossRef]
- Zammarchi, F.; Corbett, S.; Adams, L.; Tyrer, P.C.; Kiakos, K.; Janghra, N.; Marafioti, T.; Britten, C.E.; Havenith, C.E.G.; Chivers, S.; et al. ADCT-402, a PBD dimer–containing antibody drug conjugate targeting CD19-expressing malignancies. Blood 2018, 131, 1094–1105. [Google Scholar] [CrossRef]
- Hamadani, M.; Radford, J.; Carlo-Stella, C.; Caimi, P.F.; Reid, E.; O’Connor, O.A.; Feingold, J.M.; Ardeshna, K.M.; Townsend, W.; Solh, M.; et al. Final results of a phase 1 study of loncastuximab tesirine in relapsed/refractory B-cell non-Hodgkin lymphoma. Blood 2021, 137, 2634–2645. [Google Scholar] [CrossRef]
- Caimi, P.F.; Ai, W.; Alderuccio, J.P.; Ardeshna, K.M.; Hamadani, M.; Hess, B.; Kahl, B.S.; Radford, J.; Solh, M.; Stathis, A.; et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 790–800. [Google Scholar] [CrossRef] [PubMed]
- FDA: US Prescribing Information: Loncastuximab Tesirine. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761196s000lbl.pdf (accessed on 2 April 2022).
- Elgersma, R.C.; Coumans, R.G.E.; Huijbregts, T.; Menge, W.M.P.B.; Joosten, J.A.F.; Spijker, H.J.; de Groot, F.M.H.; van der Lee, M.M.C.; Ubink, R.; van den Dobbelsteen, D.J.; et al. Design, Synthesis, and Evaluation of Linker-Duocarmycin Payloads: Toward Selection of HER2-Targeting Antibody–Drug Conjugate SYD985. Mol. Pharm. 2015, 12, 1813–1835. [Google Scholar] [CrossRef] [PubMed]
- Saura Manich, C.; O’Shaughnessy, J.; Aftimos, P.G.; van den Tweel, E.; Oesterholt, M.; Escrivá-de-Romaní, S.I.; Quenel Tueux, N.; Tan, T.J.; Lim, J.S.; Ladoire, S.; et al. LBA15 Primary outcome of the phase III SYD985.002/TULIP trial comparing [vic-]trastuzumab duocarmazine to physician’s choice treatment in patients with pre-treated HER2-positive locally advanced or metastatic breast cancer. Ann. Oncol. 2021, 32, S1288. [Google Scholar] [CrossRef]
- Jiang, J.; Li, S.; Shan, X.; Wang, L.; Ma, J.; Huang, M.; Dong, L.; Chen, F. Preclinical safety profile of disitamab vedotin: A novel anti-HER2 antibody conjugated with MMAE. Toxicol. Lett. 2020, 324, 30–37. [Google Scholar] [CrossRef]
- Deeks, E.D. Disitamab Vedotin: First Approval. Drugs 2021, 81, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Shen, L.; Wang, W.; Fang, J. Safety, pharmacokinetics and efficacy of RC48-ADC in a phase I study in patients with HER2-overexpression advanced solid cancer. J. Clin. Oncol. 2018, 36, e16059. [Google Scholar] [CrossRef]
- Actor, J.K. 2-Cells and Organs of the Immune System. In Elsevier’s Integrated Review Immunology and Microbiology, 2nd ed.; Actor, J.K., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2012; pp. 7–16. [Google Scholar]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef]
- Bekkering, S. Another look at the life of a neutrophil. World J. Hematol. 2013, 2, 44. [Google Scholar] [CrossRef]
- Lahoz-Beneytez, J.; Elemans, M.; Zhang, Y.; Ahmed, R.; Salam, A.; Block, M.; Niederalt, C.; Asquith, B.; Macallan, D. Human neutrophil kinetics: Modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood 2016, 127, 3431–3438. [Google Scholar] [CrossRef]
- Zhao, H.; Gulesserian, S.; Malinao, M.C.; Ganesan, S.K.; Song, J.; Chang, M.S.; Williams, M.M.; Zeng, Z.; Mattie, M.; Mendelsohn, B.A.; et al. A Potential Mechanism for ADC-Induced Neutropenia: Role of Neutrophils in Their Own Demise. Mol. Cancer Ther. 2017, 16, 1866. [Google Scholar] [CrossRef]
- Kung Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. SGN-CD33A: A novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sutherland, M.K.; Yu, C.; Walter, R.B.; Westendorf, L.; Valliere-Douglass, J.; Pan, L.; Cronkite, A.; Sussman, D.; Klussman, K.; et al. Characterization of SGN-CD123A, A Potent CD123-Directed Antibody–Drug Conjugate for Acute Myeloid Leukemia. Mol. Cancer Ther. 2018, 17, 554–564. [Google Scholar] [CrossRef]
- Zheng, B.; Yu, S.-F.; del Rosario, G.; Leong, S.R.; Lee, G.Y.; Vij, R.; Chiu, C.; Liang, W.-C.; Wu, Y.; Chalouni, C.; et al. An Anti–CLL-1 Antibody–Drug Conjugate for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 1358–1368. [Google Scholar] [CrossRef]
- Haubner, S.; Perna, F.; Köhnke, T.; Schmidt, C.; Berman, S.; Augsberger, C.; Schnorfeil, F.M.; Krupka, C.; Lichtenegger, F.S.; Liu, X.; et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 2019, 33, 64–74. [Google Scholar] [CrossRef]
- Grisold, W.; Cavaletti, G.; Windebank, A.J. Peripheral neuropathies from chemotherapeutics and targeted agents: Diagnosis, treatment, and prevention. Neuro Oncol. 2012, 14, iv45–iv54. [Google Scholar] [CrossRef] [PubMed]
- Galsky, M.D.; Eisenberger, M.; Moore-Cooper, S.; Kelly, W.K.; Slovin, S.F.; Delacruz, A.; Lee, Y.; Webb, I.J.; Scher, H.I. Phase I Trial of the Prostate-Specific Membrane Antigen–Directed Immunoconjugate MLN2704 in Patients with Progressive Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2008, 26, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Milowsky, M.I.; Galsky, M.D.; Morris, M.J.; Crona, D.J.; George, D.J.; Dreicer, R.; Tse, K.; Petruck, J.; Webb, I.J.; Bander, N.H.; et al. Phase 1/2 multiple ascending dose trial of the prostate-specific membrane antigen-targeted antibody drug conjugate MLN2704 in metastatic castration-resistant prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2016, 34, 530.e15–530.e21. [Google Scholar] [CrossRef]
- Mita, M.M.; Ricart, A.D.; Mita, A.C.; Patnaik, A.; Sarantopoulos, J.; Sankhala, K.; Fram, R.J.; Qin, A.; Watermill, J.; Tolcher, A.W.; et al. A phase I study of a CanAg-targeted immunoconjugate, huC242-DM4, in patients with Can Ag-expressing solid tumors. J. Clin. Oncol. 2007, 25, 3062. [Google Scholar] [CrossRef]
- Borghaei, H.; O’Malley, D.M.; Seward, S.M.; Bauer, T.M.; Perez, R.P.; Oza, A.M.; Jeong, W.; Michenzie, M.F.; Kirby, M.W.; Chandorkar, G.; et al. Phase 1 study of IMGN853, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC) in patients (Pts) with epithelial ovarian cancer (EOC) and other FRA-positive solid tumors. J. Clin. Oncol. 2015, 33, 5558. [Google Scholar] [CrossRef]
- Younes, A.; Kim, S.; Romaguera, J.; Copeland, A.; Farial Sde, C.; Kwak, L.W.; Fayad, L.; Hagemeister, F.; Fanale, M.; Neelapu, S.; et al. Phase I multidose-escalation study of the anti-CD19 maytansinoid immunoconjugate SAR3419 administered by intravenous infusion every 3 weeks to patients with relapsed/refractory B-cell lymphoma. J. Clin. Oncol. 2012, 30, 2776–2782. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Motzer, R.; Molina, A.M.; Choueiri, T.K.; Heath, E.I.; Kollmannsberger, C.K.; Redman, B.G.; Sangha, R.S.; Ernst, D.S.; Pili, R.; et al. Phase I studies of anti-ENPP3 antibody drug conjugates (ADCs) in advanced refractory renal cell carcinomas (RRCC). J. Clin. Oncol. 2015, 33, 2503. [Google Scholar] [CrossRef]
- Tannir, N.M.; Forero-Torres, A.; Ramchandren, R.; Pal, S.K.; Ansell, S.M.; Infante, J.R.; de Vos, S.; Hamlin, P.A.; Kim, S.K.; Whiting, N.C.; et al. Phase I dose-escalation study of SGN-75 in patients with CD70-positive relapsed/refractory non-Hodgkin lymphoma or metastatic renal cell carcinoma. Investig. New Drugs 2014, 32, 1246–1257. [Google Scholar] [CrossRef]
- Eaton, J.S.; Miller, P.E.; Mannis, M.J.; Murphy, C.J. Ocular Adverse Events Associated with Antibody–Drug Conjugates in Human Clinical Trials. J. Ocul. Pharmacol. 2015, 31, 589–604. [Google Scholar] [CrossRef]
- Hong, D.S.; Concin, N.; Vergote, I.; de Bono, J.S.; Slomovitz, B.M.; Drew, Y.; Arkenau, H.-T.; Machiels, J.-P.; Spicer, J.F.; Jones, R.; et al. Tisotumab Vedotin in Previously Treated Recurrent or Metastatic Cervical Cancer. Clin. Cancer Res. 2020, 26, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- De Goeij, B.E.; Lambert, J.M. New developments for antibody-drug conjugate-based therapeutic approaches. Curr. Opin. Immunol. 2016, 40, 14–23. [Google Scholar] [CrossRef]
- Kim, M.T.; Chen, Y.; Marhoul, J.; Jacobson, F. Statistical Modeling of the Drug Load Distribution on Trastuzumab Emtansine (Kadcyla), a Lysine-Linked Antibody Drug Conjugate. Bioconjugate Chem. 2014, 25, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef]
- Matsuda, Y.; Mendelsohn, B.A. Recent Advances in Drug–Antibody Ratio Determination of Antibody–Drug Conjugates. Chem. Pharm. Bull. 2021, 69, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Herrera, A.F.; Patel, M.R.; Burke, J.M.; Advani, R.H.; Cheson, B.D.; Sharman, J.P.; Penuel, E.; Polson, A.G.; Leng, N.; Li, C.; et al. A Phase I Study of the Anti-CD79b THIOMABTM-Drug Conjugate DCDS0780A in Patients (pts) with Relapsed or Refractory B-Cell Non-Hodgkin’s Lymphoma (B-NHL). Blood 2017, 130, 4129. [Google Scholar] [CrossRef]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde Tag Coupled with HIPS Chemistry Enables the Production of ADCs Conjugated Site-Specifically to Different Antibody Regions with Distinct in Vivo Efficacy and PK Outcomes. Bioconjugate Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bertozzi, C.R. Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjugate Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-selective modification strategies in antibody–drug conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef]
- Schumacher, D.; Hackenberger, C.P.R.; Leonhardt, H.; Helma, J. Current Status: Site-Specific Antibody Drug Conjugates. J. Clin. Immunol. 2016, 36, 100–107. [Google Scholar] [CrossRef]
- Behrens, C.R.; Liu, B. Methods for site-specific drug conjugation to antibodies. mAbs 2014, 6, 46–53. [Google Scholar] [CrossRef]
- Matsuda, Y.; Mendelsohn, B.A. An overview of process development for antibody-drug conjugates produced by chemical conjugation technology. Expert Opin. Biol. Ther. 2021, 21, 963–975. [Google Scholar] [CrossRef]
- Hussain, A.F.; Grimm, A.; Sheng, W.; Zhang, C.; Al-Rawe, M.; Bräutigam, K.; Abu Mraheil, M.; Zeppernick, F.; Meinhold-Heerlein, I. Toward Homogenous Antibody Drug Conjugates Using Enzyme-Based Conjugation Approaches. Pharmaceuticals 2021, 14, 343. [Google Scholar] [CrossRef]
- Burke, P.J.; Hamilton, J.Z.; Jeffrey, S.C.; Hunter, J.H.; Doronina, S.O.; Okeley, N.M.; Miyamoto, J.B.; Anderson, M.E.; Stone, I.J.; Ulrich, M.L.; et al. Optimization of a PEGylated Glucuronide-Monomethylauristatin E Linker for Antibody–Drug Conjugates. Mol. Cancer Ther. 2017, 16, 116–123. [Google Scholar] [CrossRef]
- Simmons, J.K.; Burke, P.J.; Cochran, J.H.; Pittman, P.G.; Lyon, R.P. Reducing the antigen-independent toxicity of antibody-drug conjugates by minimizing their non-specific clearance through PEGylation. Toxicol. Appl. Pharmacol. 2020, 392, 114932. [Google Scholar] [CrossRef]
- Zhao, R.Y.; Wilhelm, S.D.; Audette, C.; Jones, G.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Singh, R.; Kovtun, Y.; Widdison, W.C.; et al. Synthesis and Evaluation of Hydrophilic Linkers for Antibody–Maytansinoid Conjugates. J. Med. Chem. 2011, 54, 3606–3623. [Google Scholar] [CrossRef]
- Hartimath, S.V.; Alizadeh, E.; Solomon, V.R.; Chekol, R.; Bernhard, W.; Hill, W.; Parada, A.C.; Barreto, K.; Geyer, C.R.; Fonge, H.; et al. Preclinical Evaluation of111In-Labeled PEGylated Maytansine Nimotuzumab Drug Conjugates in EGFR-Positive Cancer Models. J. Nucl. Med. 2019, 60, 1103–1110. [Google Scholar] [CrossRef]
- Shao, S.; Tsai, M.-H.; Lu, J.; Yu, T.; Jin, J.; Xiao, D.; Jiang, H.; Han, M.; Wang, M.; Wang, J. Site-specific and hydrophilic ADCs through disulfide-bridged linker and branched PEG. Bioorganic Med. Chem. Lett. 2018, 28, 1363–1370. [Google Scholar] [CrossRef]
- Cheng, X.; Li, J.; Tanaka, K.; Majumder, U.; Milinichik, A.Z.; Verdi, A.C.; Maddage, C.J.; Rybinski, K.A.; Fernando, S.; Fernando, D.; et al. MORAb-202, an Antibody–Drug Conjugate Utilizing Humanized Anti-human FRα Farletuzumab and the Microtubule-targeting Agent Eribulin, has Potent Antitumor Activity. Mol. Cancer Ther. 2018, 17, 2665–2675. [Google Scholar] [CrossRef]
- Kovtun, Y.; Noordhuis, P.; Whiteman, K.R.; Watkins, K.; Jones, G.E.; Harvey, L.; Lai, K.C.; Portwood, S.; Adams, S.; Sloss, C.M.; et al. IMGN779, a Novel CD33-Targeting Antibody–Drug Conjugate with DNA-Alkylating Activity, Exhibits Potent Antitumor Activity in Models of AML. Mol. Cancer Ther. 2018, 17, 1271–1279. [Google Scholar] [CrossRef]
- Tiberghien, A.C.; Levy, J.-N.; Masterson, L.A.; Patel, N.V.; Adams, L.R.; Corbett, S.; Williams, D.G.; Hartley, J.A.; Howard, P.W. Design and Synthesis of Tesirine, a Clinical Antibody–Drug Conjugate Pyrrolobenzodiazepine Dimer Payload. ACS Med. Chem. Lett. 2016, 7, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Flynn, M.J.; Zammarchi, F.; Tyrer, P.C.; Akarca, A.U.; Janghra, N.; Britten, C.E.; Havenith, C.E.G.; Levy, J.-N.; Tiberghien, A.; Masterson, L.A.; et al. ADCT-301, a Pyrrolobenzodiazepine (PBD) Dimer–Containing Antibody–Drug Conjugate (ADC) Targeting CD25-Expressing Hematological Malignancies. Mol. Cancer Ther. 2016, 15, 2709–2721. [Google Scholar] [CrossRef] [PubMed]
- Sandall, S.L.; Mason, M.; Olson, D.; Mazahreh, R.; Pires, T.; Sahetya, D.; Westendorf, L.; Leiske, C.; Schimpf, B.; Nguyen, L.; et al. SGN-CD228A: A novel humanized anti-CD228 antibody-drug conjugate for the treatment of solid tumors. Cancer Res. 2019, 79, 2688. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sacituzumab Govitecan: First Approval. Drugs 2020, 80, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Carl, P.L.; Chakravarty, P.K.; Katzenellenbogen, J.A. A novel connector linkage applicable in prodrug design. J. Med. Chem. 1981, 24, 479–480. [Google Scholar] [CrossRef]
- Edupuganti, V.V.S.R.; Tyndall, J.D.A.; Gamble, A.B. Self-immolative Linkers in Prodrugs and Antibody Drug Conjugates in Cancer Treatment. Recent Pat. Anti-Cancer Drug Discov. 2021, 16, 479–497. [Google Scholar] [CrossRef]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody-Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Pillow, T.H.; Schutten, M.; Yu, S.-F.; Ohri, R.; Sadowsky, J.; Poon, K.A.; Solis, W.; Zhong, F.; del Rosario, G.; Go, M.A.T.; et al. Modulating Therapeutic Activity and Toxicity of Pyrrolobenzodiazepine Antibody–Drug Conjugates with Self-Immolative Disulfide Linkers. Mol. Cancer Ther. 2017, 16, 871–878. [Google Scholar] [CrossRef]
- Chuprakov, S.; Ogunkoya, A.O.; Barfield, R.M.; Bauzon, M.; Hickle, C.; Kim, Y.C.; Yeo, D.; Zhang, F.; Rabuka, D.; Drake, P.M. Tandem-Cleavage Linkers Improve the In Vivo Stability and Tolerability of Antibody–Drug Conjugates. Bioconjugate Chem. 2021, 32, 746–754. [Google Scholar] [CrossRef]
- Anami, Y.; Yamazaki, C.M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid–valine–citrulline linkers ensure stability and efficacy of antibody–drug conjugates in mice. Nat. Commun. 2018, 9, 2512. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.Y.Y.; Anami, Y.; Yamazaki, C.M.; Xiong, W.; Haase, C.M.; Olson, S.D.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; et al. An Enzymatically Cleavable Tripeptide Linker for Maximizing the Therapeutic Index of Antibody–Drug Conjugates. Mol. Cancer Ther. 2022, 21, 1449–1461. [Google Scholar] [CrossRef]
- Miller, M.L.; Fishkin, N.E.; Li, W.; Whiteman, K.R.; Kovtun, Y.; Reid, E.E.; Archer, K.E.; Maloney, E.K.; Audette, C.A.; Mayo, M.F.; et al. A New Class of Antibody-Drug Conjugates with Potent DNA Alkylating Activity. Mol. Cancer Ther. 2016, 15, 1870–1878. [Google Scholar] [CrossRef]
- Singh, R.; Reid, E.E.; Harris, L.; Salomon, P.L.; Miller, M.L.; Chari, R.V.J.; Keating, T.A. Antibody–Drug Conjugates with Indolinobenzodiazepine Dimer Payloads: DNA-Binding Mechanism of Indolinobenzodiazepine Dimer Catabolites in Target Cancer Cells. Mol. Pharmaceutics 2020, 17, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.L.; Shizuka, M.; Wilhelm, A.; Salomon, P.; Reid, E.E.; Lanieri, L.; Sikka, S.; Maloney, E.K.; Harvey, L.; Qiu, Q.; et al. A DNA-Interacting Payload Designed to Eliminate Cross-Linking Improves the Therapeutic Index of Antibody–Drug Conjugates (ADCs). Mol. Cancer Ther. 2018, 17, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Damelin, M.; Zhong, W.; Myers, J.; Sapra, P. Evolving Strategies for Target Selection for Antibody-Drug Conjugates. Pharm. Res. 2015, 32, 3494–3507. [Google Scholar] [CrossRef]
- Lin, J.; Sagert, J. Targeting Drug Conjugates to the Tumor Microenvironment: Probody Drug Conjugates. In Innovations for Next-Generation Antibody-Drug Conjugates; Damelin, M., Ed.; Springer International Publishing: Cham, Germany, 2018; pp. 281–298. [Google Scholar]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Sevenich, L.; Joyce, J.A. Pericellular proteolysis in cancer. Genes Dev. 2014, 28, 2331–2347. [Google Scholar] [CrossRef]
- Fujimori, K.; Covell, D.G.; Fletcher, J.E.; Weinstein, J.N. A modeling analysis of monoclonal antibody percolation through tumors: A binding-site barrier. J. Nucl. Med. 1990, 31, 1191–1198. [Google Scholar]
- Bordeau, B.M.; Yang, Y.; Balthasar, J.P. Transient Competitive Inhibition Bypasses the Binding Site Barrier to Improve Tumor Penetration of Trastuzumab and Enhance T-DM1 Efficacy. Cancer Res. 2021, 81, 4145–4154. [Google Scholar] [CrossRef]
- Bordeau, B.M.; Abuqayyas, L.; Nguyen, T.D.; Chen, P.; Balthasar, J.P. Development and Evaluation of Competitive Inhibitors of Trastuzumab-HER2 Binding to Bypass the Binding-Site Barrier. Front. Pharm. 2022, 13, 837744. [Google Scholar] [CrossRef]
- Chen, P.; Bordeau, B.M.; Zhang, Y.; Balthasar, J.P. Transient Inhibition of Trastuzumab-Tumor Binding to Overcome the “Binding-Site Barrier” and Improve the Efficacy of a Trastuzumab-Gelonin Immunotoxin. Mol. Cancer 2022, 21, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Sagert, J.G.; West, J.W.; Wong, C.; Desnoyers, L.R.; Vasiljeva, O.; Richardson, J.H.; Polu, K.R.; Lowman, H.B. Abstract 2665: Transforming Notch ligands into tumor-antigen targets: A Probody-Drug Conjugate (PDC) targeting Jagged 1 and Jagged 2. Cancer Res. 2014, 74, 2665. [Google Scholar] [CrossRef]
- Autio, K.A.; Boni, V.; Humphrey, R.W.; Naing, A. Probody Therapeutics: An Emerging Class of Therapies Designed to Enhance On-Target Effects with Reduced Off-Tumor Toxicity for Use in Immuno-Oncology. Clin. Cancer Res. 2020, 26, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Corbacho, J.; Spira, A.; Boni, V.; Feliu, J.; Middleton, M.; Burris, H.; Weaver, A.; Will, M.; Harding, J.; Meric-Bernstam, F.; et al. 422TiPPROCLAIM-CX-2009: A first-in-human trial to evaluate CX-2009 in adults with metastatic or locally advanced unresectable solid tumors. Ann. Oncol. 2017, 28, V140. [Google Scholar] [CrossRef]
- Johnson, M.; El-Khoueiry, A.; Hafez, N.; Lakhani, N.; Mamdani, H.; Rodon, J.; Sanborn, R.E.; Garcia-Corbacho, J.; Boni, V.; Stroh, M.; et al. Phase I, First-in-Human Study of the Probody Therapeutic CX-2029 in Adults with Advanced Solid Tumor Malignancies. Clin. Cancer Res. 2021, 27, 4521–4530. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Shen, J.; Yu, W.; Jing, C.; Saha, S.; Rangan, V.; Richardson, J.; Liu, Y.; Boule, S.; Lucas, J.; et al. CX-2043, an EpCAM-targeting probody drug conjugate, demonstrates anti-tumor activity with a favorable safety profile in preclinical models. Eur. J. Cancer 2020, 138, S15. [Google Scholar] [CrossRef]
- Comer, F.; Gao, C.; Coats, S. Bispecific and Biparatopic Antibody Drug Conjugates. In Innovations for Next-Generation Antibody-Drug Conjugates; Damelin, M., Ed.; Springer International Publishing: Cham, Germany, 2018; pp. 267–280. [Google Scholar]
- May, C.; Sapra, P.; Gerber, H.-P. Advances in bispecific biotherapeutics for the treatment of cancer. Biochem. Pharmacol. 2012, 84, 1105–1112. [Google Scholar] [CrossRef]
- Mazor, Y.; Oganesyan, V.; Yang, C.; Hansen, A.; Wang, J.; Liu, H.; Sachsenmeier, K.; Carlson, M.; Gadre, D.V.; Borrok, M.J.; et al. Improving target cell specificity using a novel monovalent bispecific IgG design. mAbs 2015, 7, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Mazor, Y.; Hansen, A.; Yang, C.; Chowdhury, P.S.; Wang, J.; Stephens, G.; Wu, H.; Dall’Acqua, W.F. Insights into the molecular basis of a bispecific antibody’s target selectivity. mAbs 2015, 7, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Mazor, Y.; Sachsenmeier, K.F.; Yang, C.; Hansen, A.; Filderman, J.; Mulgrew, K.; Wu, H.; Dall’Acqua, W.F. Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci. Rep. 2017, 7, 40098. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, C.; Doerner, A.; Knuehl, C.; Rasche, N.; Sood, V.; Krah, S.; Rhiel, L.; Messemer, A.; Wesolowski, J.; Schuette, M.; et al. Balancing Selectivity and Efficacy of Bispecific Epidermal Growth Factor Receptor (EGFR) × c-MET Antibodies and Antibody-Drug Conjugates. J. Biol. Chem. 2016, 291, 25106–25119. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.; Gregorc, V.; Belli, C.; Roca, E.; Lazzari, C.; Viganò, M.G.; Serafico, A.; Villa, E. Clinical Significance of Skin Toxicity due to EGFR-Targeted Therapies. J. Oncol. 2009, 2009, 849051. [Google Scholar] [CrossRef] [PubMed]
- Holcmann, M.; Sibilia, M. Mechanisms underlying skin disorders induced by EGFR inhibitors. Mol. Cell. Oncol. 2015, 2, e1004969. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Anami, Y.; Ha, S.Y.Y.; Roeder, T.J.; Xiong, W.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; Tsuchikama, K. Chemical generation of small molecule-based bispecific antibody-drug conjugates for broadening the target scope. Bioorganic Med. Chem. 2021, 32, 116013. [Google Scholar] [CrossRef] [PubMed]
- De Goeij, B.E.C.G.; Vink, T.; Ten Napel, H.; Breij, E.C.W.; Satijn, D.; Wubbolts, R.; Miao, D.; Parren, P.W.H.I. Efficient Payload Delivery by a Bispecific Antibody–Drug Conjugate Targeting HER2 and CD63. Mol. Cancer Ther. 2016, 15, 2688–2697. [Google Scholar] [CrossRef]
- Andreev, J.; Thambi, N.; Perez Bay, A.E.; Delfino, F.; Martin, J.; Kelly, M.P.; Kirshner, J.R.; Rafique, A.; Kunz, A.; Nittoli, T.; et al. Bispecific Antibodies and Antibody–Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol. Cancer Ther. 2017, 16, 681–693. [Google Scholar] [CrossRef]
- Sapra, P.; Betts, A.; Boni, J. Preclinical and clinical pharmacokinetic/pharmacodynamic considerations for antibody-drug conjugates. Expert Rev. Clin. Pharm. 2013, 6, 541–555. [Google Scholar] [CrossRef]
- Smith, T.W.; Haber, E.; Yeatman, L.; Butler, V.P., Jr. Reversal of advanced digoxin intoxication with Fab fragments of digoxin-specific antibodies. N. Engl. J. Med. 1976, 294, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Pollack, C.V.; Reilly, P.A.; Eikelboom, J.; Glund, S.; Verhamme, P.; Bernstein, R.A.; Dubiel, R.; Huisman, M.V.; Hylek, E.M.; Kamphuisen, P.W.; et al. Idarucizumab for Dabigatran Reversal. N. Engl. J. Med. 2015, 373, 511–520. [Google Scholar] [CrossRef]
- Balthasar, J.; Fung, H.L. Utilization of antidrug antibody fragments for the optimization of intraperitoneal drug therapy: Studies using digoxin as a model drug. J. Pharmacol. Exp. Ther. 1994, 268, 734–739. [Google Scholar]
- Balthasar, J.P.; Fung, H.L. Inverse targeting of peritoneal tumors: Selective alteration of the disposition of methotrexate through the use of anti-methotrexate antibodies and antibody fragments. J. Pharm. Sci. 1996, 85, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E.D.; Soda, D.M.; Balthasar, J.P. Application of pharmacokinetic-pharmacodynamic modeling to predict the kinetic and dynamic effects of anti-methotrexate antibodies in mice. J. Pharm. Sci. 2003, 92, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E.D.; Balthasar, J.P. Application of anti-methotrexate Fab fragments for the optimization of intraperitoneal methotrexate therapy in a murine model of peritoneal cancer. J. Pharm. Sci. 2005, 94, 1957–1964. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Balthasar, J.P. PK/TD modeling for prediction of the effects of 8C2, an anti-topotecan mAb, on topotecan-induced toxicity in mice. Int. J. Pharm. 2014, 465, 228–238. [Google Scholar] [CrossRef]
- Bordeau, B.M.; Nguyen, T.D.; Polli, R.; Chen, P.; Balthasar, J.P. Payload-binding fab fragments increase the therapeutic index of MMAE antibody-drug conjugates. Mol. Cancer Ther. 2023, accepted. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Use of Payload Binding Selectivity Enhancers to Improve Therapeutic Index of Maytansinoid-Antibody-Drug Conjugates. Mol. Cancer Ther. 2023, submitted.


{kind=link}
| Approved ADCs | Target | Indications | Approved Year/ R&D Organization | Common Adverse Events (Any Grades) | Common Grade ≥ 3 Adverse Events |
|---|---|---|---|---|---|
| Gemtuzumab Ozogamicin (Mylotarg®) | CD-33 | Acute myeloid leukemia | 2001 Pfizer | Thrombocytopenia, fatigue, neutropenia, pyrexia, nausea, infection, chills, hemorrhage, vomiting, headache, stomatitis, diarrhea, and abdominal pain | Neutropenia, thrombocytopenia, increased AST/ALT levels, and sepsis |
| Inotuzumab Ozogamicin (Besponsa®) | CD-22 | B-cell precursor acute lymphoblastic leukemia | 2017 Pfizer | Neutropenia, thrombocytopenia, infection, anemia, leukopenia, febrile neutropenia, and nausea | Neutropenia, thrombocytopenia, leukopenia, febrile neutropenia, anemia, and lymphopenia |
| Brentuximab Vedotin (Adcetris®) | CD-30 | Hodgkin lymphoma, systemic anaplastic large-cell lymphoma, T-cell lymphoma | 2011 Seattle Genetics | Peripheral sensory neuropathy, nausea, fatigue, neutropenia, diarrhea, pyrexia, vomiting, arthralgia, pruritus, myalgia, peripheral motor neuropathy, and alopecia | Neutropenia, peripheral sensory neuropathy, thrombocytopenia, and anemia |
| Polatuzumab Vedotin (Polivy®) | CD-79b | Diffuse large B-cell lymphoma | 2019 Genentech | Neutropenia, anemia, and peripheral neuropathy | Neutropenia, anemia, and peripheral sensory neuropathy |
| Enfortumab Vedotin (Padcev®) | Nectin-4 | Urothelial cancer | 2019 Astellas | Fatigue, alopecia, decreased appetite, dysgeusia, nausea, peripheral sensory neuropathy, pruritus, diarrhea, and maculopapular rash | Rash, neutropenia, anemia, and fatigue |
| Tisotumab Vedotin (Tivdak®) | Tissue factor | Cervical cancer | 2021 Genmab | Epistaxis, fatigue, nausea, alopecia, conjunctivitis, decreased appetite, constipation, diarrhea, vomiting, peripheral neuropathy, dry eye, and abdominal pain | Fatigue, anemia, abdominal pain, hypokalemia, conjunctivitis, hyponatremia, peripheral neuropathy, and vomiting |
| Belantamab Mafodotin (Blenrep®) | B-cell maturation antigen | Multiple myeloma | 2020 GSK | Keratopathy, thrombocytopenia, anemia, nausea, pyrexia, blurred vision, increased aspartate aminotransferase | Keratopathy, thrombocytopenia, anemia |
| Trastuzumab Emtansine (Kadcyla®) | HER-2 | Breast cancer | 2013 Genentech | Thrombocytopenia, elevated transaminases, fatigue, anemia, and nausea | Thrombocytopenia, increased aspartate aminotransferase levels, and anemia |
| Mirvetuximab Soravtansine (Elahere®) | Folate receptor α | Ovarian cancer | 2022 Immunogen | Nausea, blurred vision, keratopathy, diarrhea, fatigue, peripheral neuropathy, dry eye, and decreased visual acuity | Blurred vision, peripheral neuropathy, and diarrhea |
| Trastuzumab Deruxtecan (Enhertu®) | HER-2 | Breast cancer | 2019 Daiichi Sankyo | Nausea, fatigue, alopecia, vomiting, neutropenia, constipation, anemia, decreased appetite, diarrhea, leukopenia, and thrombocytopenia | Neutropenia, anemia, nausea, leukopenia, lymphopenia, and fatigue |
| Sacituzumab Govitecan (Trodelvy®) | Trop-2 | Breast cancer, urothelial cancer | 2020 Gilead Sciences | Nausea, diarrhea, neutropenia, fatigue, vomiting, anemia, alopecia, and constipation | Neutropenia, anemia, diarrhea, and leukopenia |
| Loncastuximab Tesirine (Zynlonta®) | CD-19 | Large B-cell lymphoma | 2021 ADC Therapeutics | Neutropenia, thrombocytopenia, anemia, fatigue, and gamma-glutamyl transferase increase | Thrombocytopenia, neutropenia, anemia, gamma-glutamyl transferase increased, leukopenia, lymphopenia, and hypophosphatemia |
| Approved/Late-Stage ADCs | Linker Type | Key Toxicities | |
|---|---|---|---|
| Tubulin inhibitors | |||
| MMAE | Brentuximab Vedotin, Polatuzumab Vedotin, Enfortumab Vedotin, Tisotumab Vedotin, Disitamab Vedotin | Cleavable | Neutropenia, peripheral neuropathy, anemia, skin toxicity |
| MMAF | Belantamab Mafodotin | Non-cleavable | Thrombocytopenia, ocular toxicity, hepatic toxicity |
| DM1 | Trastuzumab Emtansine | Non-cleavable | Thrombocytopenia, hepatic toxicity |
| DM4 | Mirvetuximab Soravtansine | Cleavable | Neutropenia, anemia, peripheral neuropathy, ocular toxicity |
| DNA-crosslinkers/ DNA-alkylators | |||
| Calicheamicin | Gemtuzumab Ozogamicin, Inotuzumab Ozogamicin | Cleavable | Neutropenia, thrombocytopenia, hepatic toxicity |
| PBD | Loncastuximab Tesirine | Cleavable | Neutropenia, thrombocytopenia, anemia, serosal effusion, nephron toxicity, skin toxicity |
| Duocarmycin | Trastuzumab Duocarmazine | Cleavable | Neutropenia, thrombocytopenia, serosal effusion |
| Topoisomerase inhibitors | |||
| SN-38 | Sacituzumab Govitecan | Cleavable | Neutropenia, gastrointestinal toxicity |
| Deruxtecan | Trastuzumab Deruxtecan | Cleavable | Neutropenia, gastrointestinal toxicity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers 2023, 15, 713. https://doi.org/10.3390/cancers15030713
Nguyen TD, Bordeau BM, Balthasar JP. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers. 2023; 15(3):713. https://doi.org/10.3390/cancers15030713
Chicago/Turabian StyleNguyen, Toan D., Brandon M. Bordeau, and Joseph P. Balthasar. 2023. "Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability" Cancers 15, no. 3: 713. https://doi.org/10.3390/cancers15030713
APA StyleNguyen, T. D., Bordeau, B. M., & Balthasar, J. P. (2023). Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers, 15(3), 713. https://doi.org/10.3390/cancers15030713