
Functional and Molecular Heterogeneity in Glioma Stem Cells Derived from Multiregional Sampling

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Brain Tumor Biopsies and Cell Cultures
2.2. DNA and RNA Sequencing
2.3. Temozolomide Sensitivity Assays
2.4. Drug Sensitivity and Resistance Testing
2.5. Statistical Considerations
3. Results
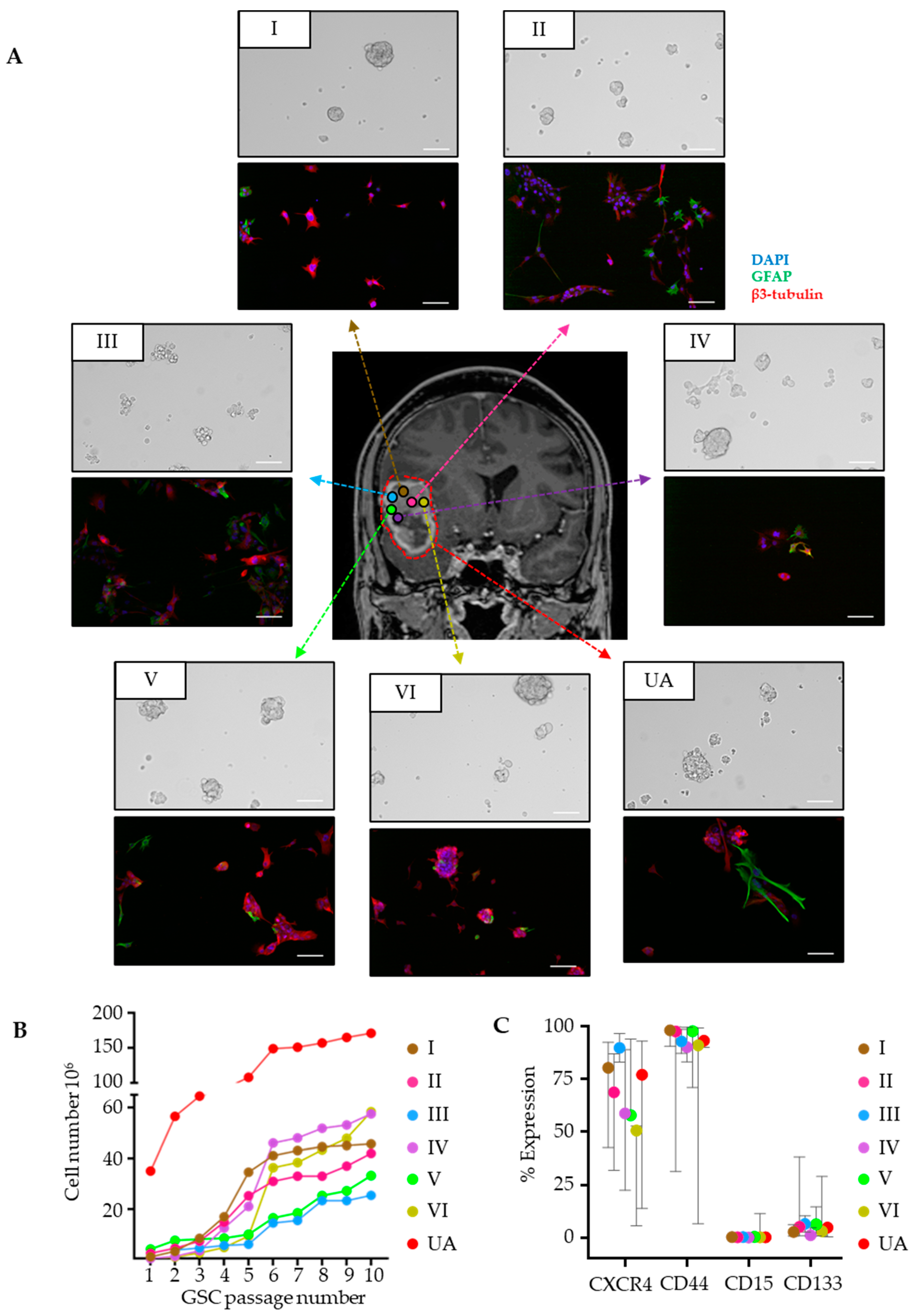
3.1. GSC Cultures Derived from Regionally Distant Biopsies Share Phenotypic Traits
3.2. ITH in Mutational and Gene Expression Profiles

3.3. ITH in Drug Sensitivity to Anticancer Drugs
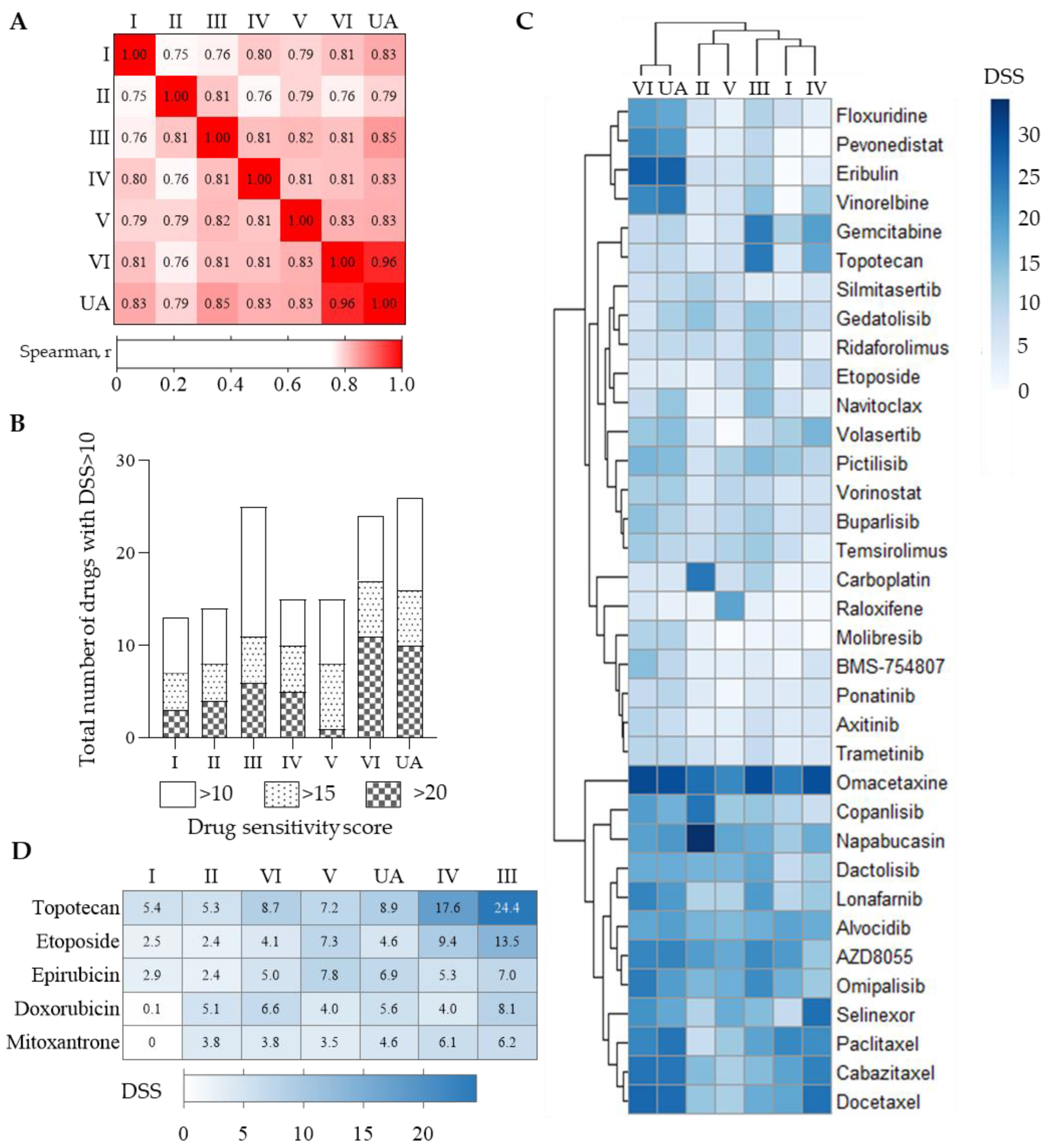
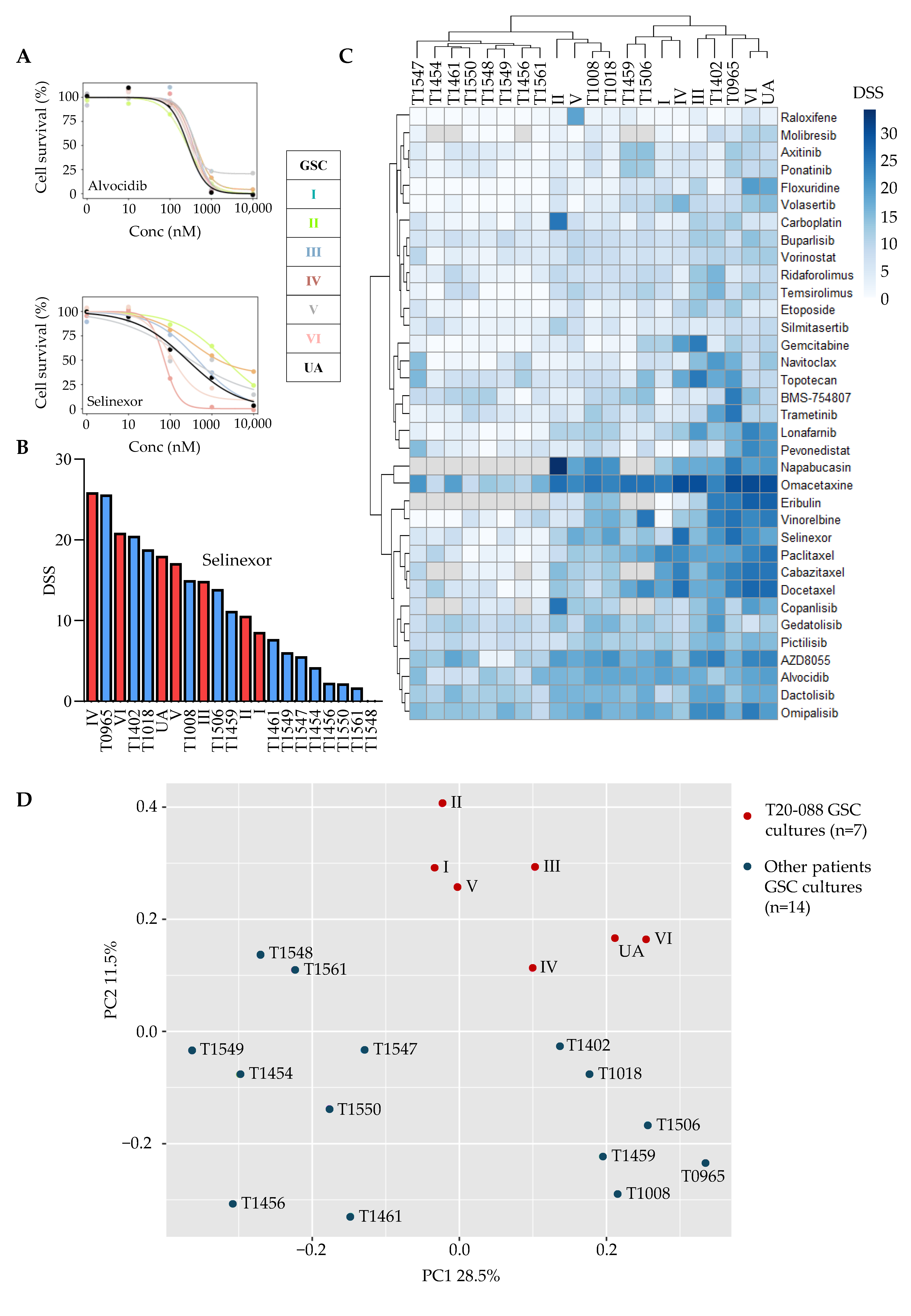
3.4. Tumor- and Culture-Specific Drug Sensitivity Patterns
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skaga, E.; Skretteberg, M.A.; Johannesen, T.B.; Brandal, P.; Vik-Mo, E.O.; Helseth, E.; Langmoen, I.A. Real-world validity of randomized controlled phase III trials in newly diagnosed glioblastoma: To whom do the results of the trials apply? Neuro-Oncol. Adv. 2021, 3, vdab008. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Skaga, E.; Kulesskiy, E.; Fayzullin, A.; Sandberg, C.J.; Potdar, S.; Kyttälä, A.; Langmoen, I.A.; Laakso, A.; Gaál-Paavola, E.; Perola, M.; et al. Intertumoral heterogeneity in patient-specific drug sensitivities in treatment-naïve glioblastoma. BMC Cancer 2019, 19, 628. [Google Scholar] [CrossRef]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.; Vega, S.F.; Kling, T.; Bontell, T.O.; Jakola, A.S.; Carén, H. Intratumor DNA methylation heterogeneity in glioblastoma: Implications for DNA methylation-based classification. Neuro. Oncol. 2019, 21, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.R.; Hudson, A.L.; Khong, P.; Parkinson, J.F.; Dwight, T.; Ikin, R.J.; Zhu, Y.; Cheng, Z.J.; Vafaee, F.; Chen, J.; et al. Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Sci. Rep. 2016, 6, 22477. [Google Scholar] [CrossRef]
- Johnson, K.C.; Anderson, K.J.; Courtois, E.T.; Gujar, A.D.; Barthel, F.P.; Varn, F.S.; Luo, D.; Seignon, M.; Yi, E.; Kim, H.; et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nat Genet. 2021, 53, 1456–1468. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Shawn, M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Meyer, M.; Reimand, J.; Lan, X.; Head, R.; Zhu, X.; Kushida, M.; Bayani, J.; Pressey, J.C.; Lionel, A.C.; Clarke, I.D.; et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl. Acad. Sci. USA 2015, 112, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergström, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sönmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Li, G.; Chang, S.D.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, C.J.; Altschuler, G.; Jeong, J.; Strømme, K.K.; Stangeland, B.; Murrell, W.; Grasmo-Wendler, U.H.; Myklebost, O.; Helseth, E.; Vik-Mo, E.O.; et al. Comparison of glioma stem cells to neural stem cells from the adult human brain identifies dysregulated Wnt-signaling and a fingerprint associated with clinical outcome. Exp. Cell Res. 2013, 319, 2230–2243. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Skaga, E.; Kulesskiy, E.; Potdar, S.; Panagopoulos, I.; Micci, F.; Langmoen, I.A.; Sandberg, C.J.; Vik-Mo, E.O. Functional temozolomide sensitivity testing of patient-specific glioblastoma stem cell cultures is predictive of clinical outcome. Transl. Oncol. 2022, 26, 101535. [Google Scholar] [CrossRef] [PubMed]
- Vik-Mo, E.O.; Sandberg, C.; Olstorn, H.; Varghese, M.; Brandal, P.; Ramm-Pettersen, J.; Murrell, W.; Langmoen, I.A. Brain tumor stem cells maintain overall phenotype and tumorigenicity after in vitro culturing in serum-free conditions. Neuro. Oncol. 2010, 12, 1220–1230. [Google Scholar] [CrossRef]
- Bhaduri, A.; Di Lullo, E.; Jung, D.; Müller, S.; Crouch, E.E.; Espinosa, C.S.; Ozawa, T.; Alvarado, B.; Spatazza, J.; Cadwell, C.R.; et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell 2020, 26, 48–63.e6. [Google Scholar] [CrossRef]
- Gulaia, V.; Shmelev, M.; Romanishin, A.; Shved, N.; Farniev, V.; Goncharov, N.; Biktimirov, A.; Vargas, I.L.; Khodosevich, K.; Kagansky, A.; et al. Single-nucleus transcriptomics of IDH1- and TP53-mutant glioma stem cells displays diversified commitment on invasive cancer progenitors. Sci. Rep. 2022, 12, 18975. [Google Scholar] [CrossRef]
- Duhamel, M.; Drelich, L.; Wisztorski, M.; Aboulouard, S.; Gimeno, J.P.; Ogrinc, N.; Devos, P.; Cardon, T.; Weller, M.; Escande, F.; et al. Spatial analysis of the glioblastoma proteome reveals specific molecular signatures and markers of survival. Nat. Commun. 2022, 13, 6665. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef]
- Vinel, C.; Rosser, G.; Guglielmi, L.; Constantinou, M.; Pomella, N.; Zhang, X.; Boot, J.R.; Jones, T.A.; Millner, T.O.; Dumas, A.A.; et al. Comparative epigenetic analysis of tumour initiating cells and syngeneic EPSC-derived neural stem cells in glioblastoma. Nat. Commun. 2021, 12, 6130. [Google Scholar] [CrossRef]
- Reinartz, R.; Wang, S.; Kebir, S.; Silver, D.J.; Wieland, A.; Zheng, T.; Kupper, M.; Rauschenbach, L.; Fimmers, R.; Shepherd, T.M.; et al. Functional Subclone profiling for prediction of treatment-induced intratumor population shifts and discovery of rational drug combinations in human glioblastoma. Clin. Cancer Res. 2017, 23, 562–574. [Google Scholar] [CrossRef]
- Letai, A.; Bhola, P.; Welm, A.L. Functional precision oncology: Testing tumors with drugs to identify vulnerabilities and novel combinations. Cancer Cell 2022, 40, 26–35. [Google Scholar] [CrossRef]
- Skaga, E.; Kulesskiy, E.; Brynjulvsen, M.; Sandberg, C.J.; Potdar, S.; Langmoen, I.A.; Laakso, A.; Gaál-Paavola, E.; Perola, M.; Wennerberg, K.; et al. Feasibility study of using high-throughput drug sensitivity testing to target recurrent glioblastoma stem cells for individualized treatment. Clin. Transl. Med. 2019, 8, 33. [Google Scholar] [CrossRef]
- Nakken, S.; Saveliev, V.; Hofmann, O.; Møller, P.; Myklebost, O.; Hovig, E. Cancer Predisposition Sequencing Reporter (CPSR): A flexible variant report engine for high-throughput germline screening in cancer. Int. J. Cancer 2021, 149, 1955–1960. [Google Scholar] [CrossRef]
- Nakken, S.; Fournous, G.; Vodák, D.; Aasheim, L.B.; Myklebost, O.; Hovig, E. Personal Cancer Genome Reporter: Variant interpretation report for precision oncology. Bioinformatics 2018, 34, 1778–1780. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, 452–457. [Google Scholar] [CrossRef]
- Yadav, B.; Pemovska, T.; Szwajda, A.; Kulesskiy, E.; Kontro, M.; Karjalainen, R.; Majumder, M.M.; Malani, D.; Murumägi, A.; Knowles, J.; et al. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci. Rep. 2014, 4, 5193. [Google Scholar] [CrossRef] [PubMed]
- Skaga, E. Drug Sensitivity and Resistance Testing of Brain Tumor Stem Cells for Individualized Functional Precision Medicine. Ph.D. Thesis, Faculty of Medicine, University of Oslo, Oslo, Norway, 2021. Available online: https://www.duo.uio.no/handle/10852/82455 (accessed on 6 November 2023).
- Potdar, S.; Ianevski, A.; Mpindi, J.P.; Bychkov, D.; Fiere, C.; Ianevski, P.; Yadav, B.; Wennerberg, K.; Aittokallio, T.; Kallioniemi, O.; et al. Breeze: An integrated quality control and data analysis application for high-throughput drug screening. Bioinformatics 2020, 36, 3602–3604. [Google Scholar] [CrossRef]
- Chen, Y.; He, L.; Ianevski, A.; Ayuda-durán, P.; Potdar, S.; Saarela, J.; Miettinen, J.J.; Kytölä, S.; Miettinen, S.; Manninen, M.; et al. Robust scoring of selective drug responses for patient-tailored therapy selection. Nat. Protoc. 2023, 401, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2014, 157, 753. [Google Scholar] [CrossRef]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Alenda, C.; Rojas, E.; Valor, L.M. FFPE samples from cavitational ultrasonic surgical aspirates are suitable for RNA profiling of gliomas. PLoS ONE 2021, 16, e0255168. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Delord, J.P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Marquart, J.; Chen, E.Y.; Prasad, V. Estimation of the percentage of US patients with cancer who benefit from genome-driven oncology. JAMA Oncol. 2018, 4, 1093–1098. [Google Scholar] [CrossRef]
- Pemovska, T.; Kontro, M.; Yadav, B.; Edgren, H.; Eldfors, S.; Szwajda, A.; Almusa, H.; Bespalov, M.M.; Ellonen, P.; Elonen, E.; et al. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 2013, 3, 1416–1429. [Google Scholar] [CrossRef]
- Malani, D.; Kumar, A.; Brück, O.; Kontro, M.; Yadav, B.; Hellesøy, M.; Kuusanmäki, H.; Dufva, O.; Kankainen, M.; Eldfors, S.; et al. Implementing a Functional Precision Medicine Tumor Board for Acute Myeloid Leukemia. Cancer Discov. 2022, 12, 388–401. [Google Scholar] [CrossRef]
- Rybin, M.J.; Ivan, M.E.; Ayad, N.G.; Zeier, Z. Organoid Models of Glioblastoma and Their Role in Drug Discovery. Front. Cell. Neurosci. 2021, 15, 605255. [Google Scholar] [CrossRef] [PubMed]
- Weth, F.R.; Peng, L.; Paterson, E.; Tan, S.T.; Gray, C. Utility of the Cerebral Organoid Glioma ‘GLICO’ Model for Screening Applications. Cells 2023, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Loong, H.H.F.; Wong, A.M.; Chan, D.T.M.; Cheung, M.S.H.; Chow, C.; Ding, X.; Chan, A.K.Y.; Johnston, P.A.; Lau, J.Y.W.; Poon, W.S.; et al. Patient-derived tumor organoid predicts drugs response in glioblastoma: A step forward in personalized cancer therapy? J. Clin. Neurosci. 2020, 78, 400–402. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brynjulvsen, M.; Solli, E.; Walewska, M.; Zucknick, M.; Djirackor, L.; Langmoen, I.A.; Mughal, A.A.; Skaga, E.; Vik-Mo, E.O.; Sandberg, C.J. Functional and Molecular Heterogeneity in Glioma Stem Cells Derived from Multiregional Sampling. Cancers 2023, 15, 5826. https://doi.org/10.3390/cancers15245826
Brynjulvsen M, Solli E, Walewska M, Zucknick M, Djirackor L, Langmoen IA, Mughal AA, Skaga E, Vik-Mo EO, Sandberg CJ. Functional and Molecular Heterogeneity in Glioma Stem Cells Derived from Multiregional Sampling. Cancers. 2023; 15(24):5826. https://doi.org/10.3390/cancers15245826
Chicago/Turabian StyleBrynjulvsen, Marit, Elise Solli, Maria Walewska, Manuela Zucknick, Luna Djirackor, Iver A. Langmoen, Awais Ahmad Mughal, Erlend Skaga, Einar O. Vik-Mo, and Cecilie J. Sandberg. 2023. "Functional and Molecular Heterogeneity in Glioma Stem Cells Derived from Multiregional Sampling" Cancers 15, no. 24: 5826. https://doi.org/10.3390/cancers15245826
APA StyleBrynjulvsen, M., Solli, E., Walewska, M., Zucknick, M., Djirackor, L., Langmoen, I. A., Mughal, A. A., Skaga, E., Vik-Mo, E. O., & Sandberg, C. J. (2023). Functional and Molecular Heterogeneity in Glioma Stem Cells Derived from Multiregional Sampling. Cancers, 15(24), 5826. https://doi.org/10.3390/cancers15245826