
Emerging Therapeutic Targets and Drug Resistance Mechanisms in Immunotherapy of Hematological Malignancies


Abstract
:Simple Summary
Abstract
1. Introduction
2. Targets for Drugs in Cancer Immunotherapy
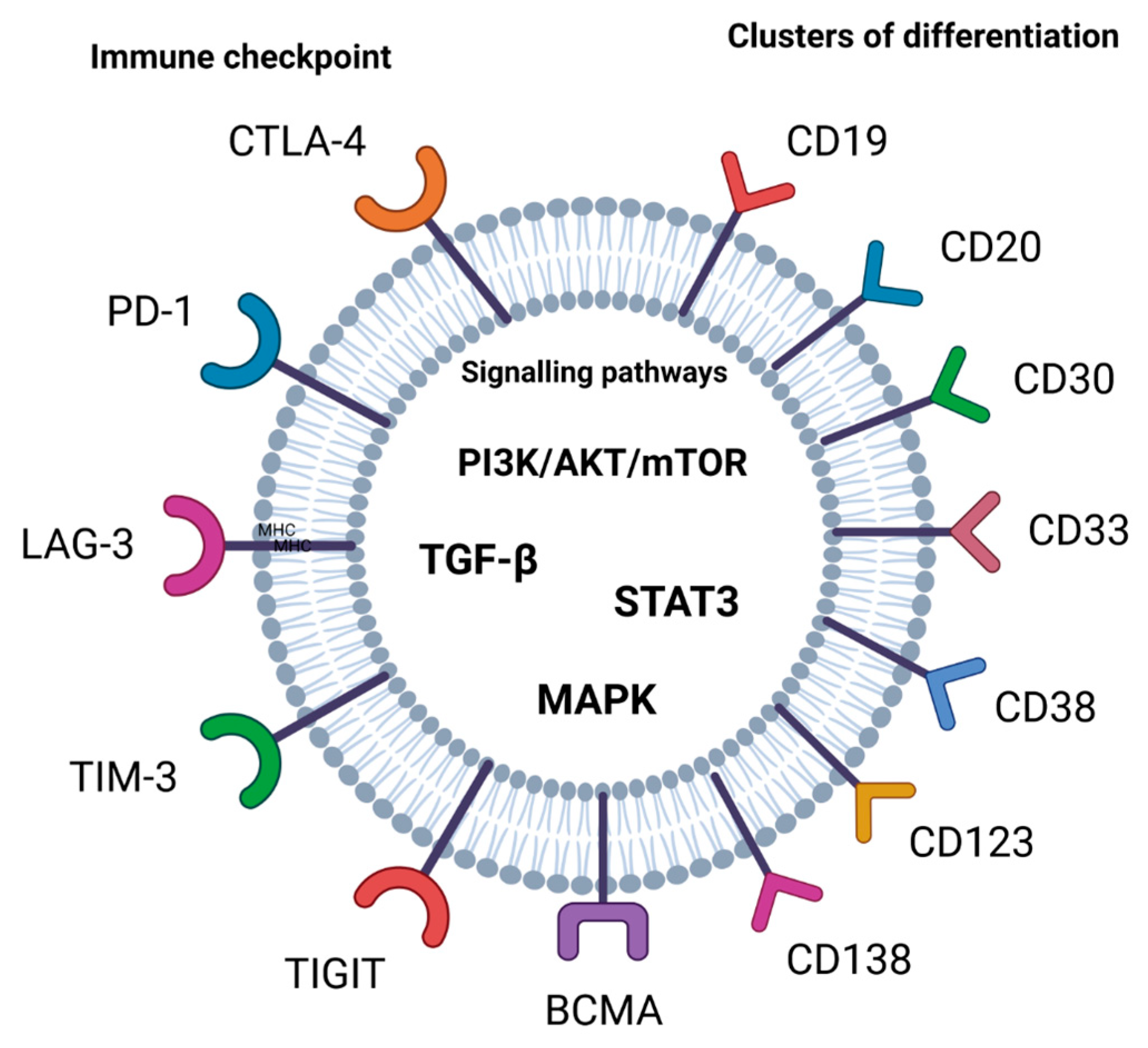
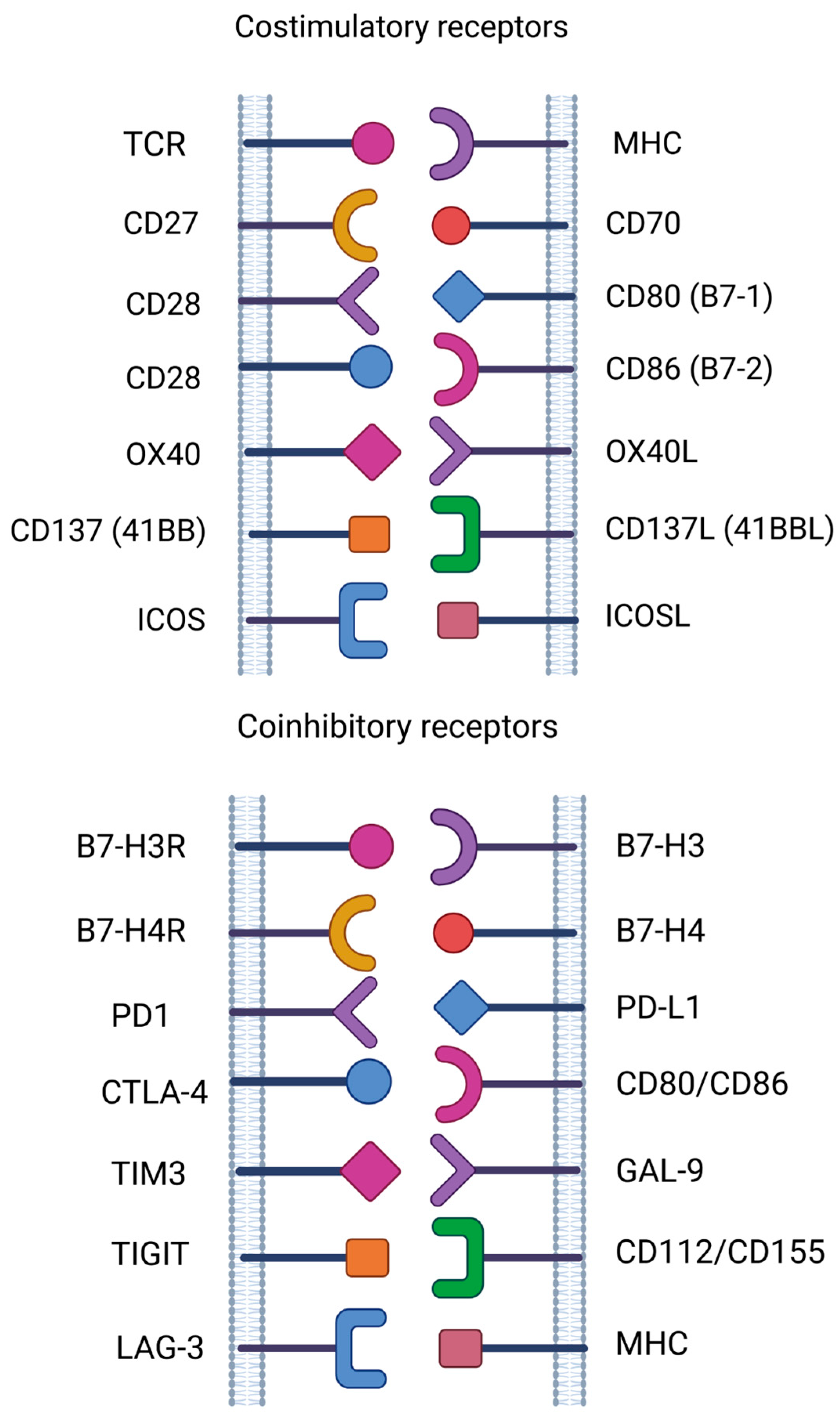
2.1. Immune Checkpoint
2.2. Clusters of Differentiation (CDs) and B-Cell Maturation Antigen (BCMA)
- CD20
- CD25
- CD30
- CD33
- CD38
- CD47
- CD123
- CD138
- BCMA
2.3. Signaling Pathways
2.3.1. Phosphatidylinositol 3-kinase (PI3K)/AKT/Mammalian Target of the Rapamycin (mTOR) Signaling Pathway
2.3.2. The Transforming Growth Factor (TGF)-β Signaling Pathway
2.3.3. Signal Transducer and Activator of Transcription 3 (STAT3)
2.3.4. Mitogen-Activated Protein Kinase (MAPK) Signaling Pathways
2.4. Exhaustion and Senescence
2.5. The Immunosuppressive Tumor Microenvironment (TME)
3. New Emerging Targets in Cancer Immunotherapy
- CD27 and CD70
- CD37
- CD80 and CD86
- B7-H3 and B7-H4
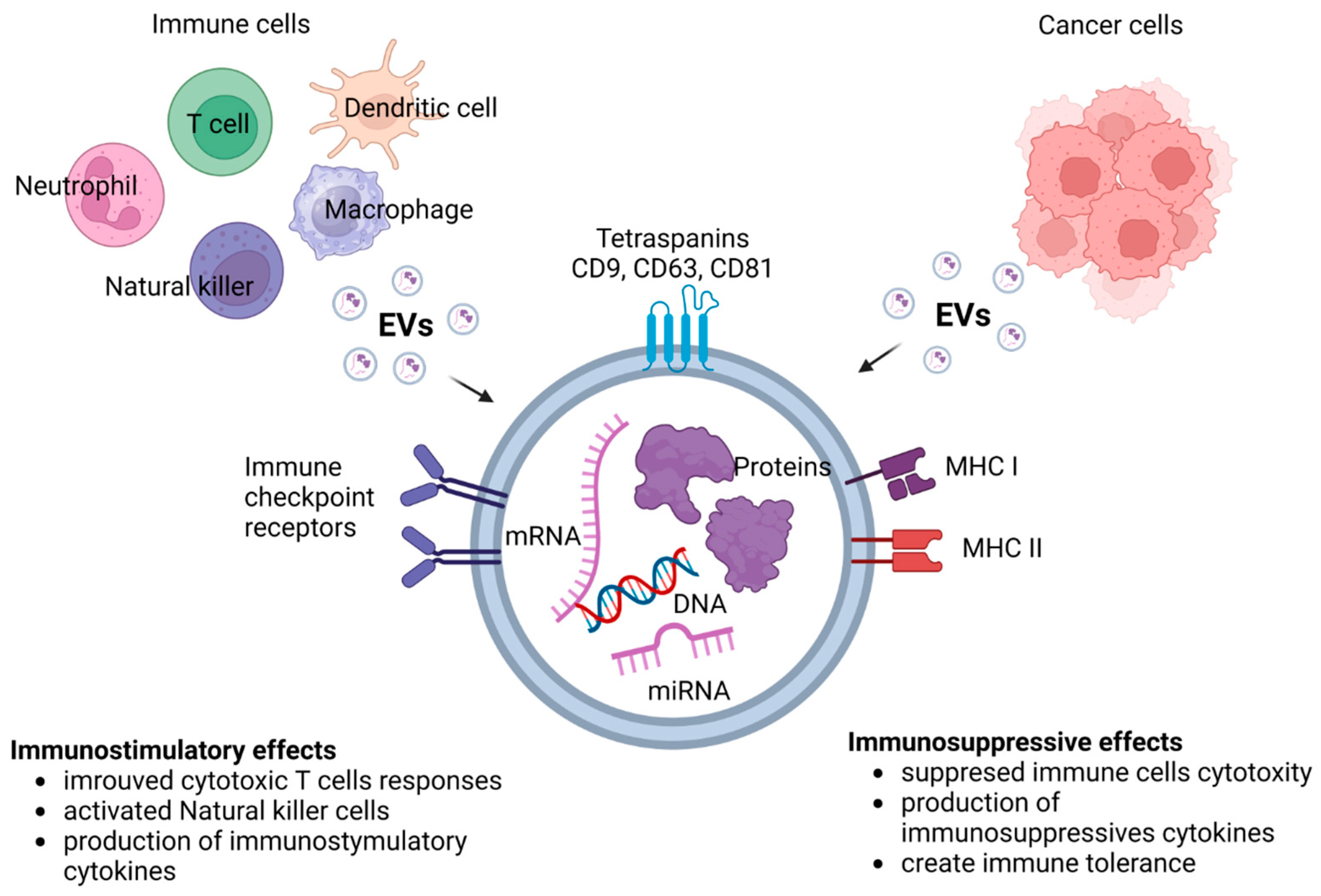
4. Extracellular Vesicles (EVs)
5. Mechanisms of Cancer Immune Resistance
5.1. Tumor Heterogeneity
5.2. Tumor Antigen and Major Histocompatibility Complex (MHC) Modulation
5.3. Immunosuppressive Cell Subsets and Factors in the Tumor Microenvironment (TME)
5.4. Anti-Apoptotic Pathways and T Cell Activation-Induced Cell Death (AICD)
5.5. Checkpoint Inhibitory Ligands
6. EVs in the Tumor Microenvironment as Mediators of Cancer Therapy Resistance
7. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, P.; Zhang, G.; Wan, X. Challenges and new technologies in adoptive cell therapy. J. Hematol. Oncol. 2023, 16, 97. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H. Precision medicine in acute lymphoblastic leukemia. Front. Med. 2020, 14, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhou, Y.; Zhang, M.; Ge, W.; Li, Y.; Yang, L.; Wei, G.; Han, L.; Wang, H.; Yu, S.; et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2021, 27, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.; Alati, C.; Canale, F.A.; Musuraca, G.; Martinelli, G.; Cerchione, C. A Review of Clinical Outcomes of CAR T-Cell Therapies for B-Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2021, 22, 2150. [Google Scholar] [CrossRef]
- Marofi, F.; Rahman, H.S.; Achmad, M.H.; Sergeevna, K.N.; Suksatan, W.; Abdelbasset, W.K.; Mikhailova, M.V.; Shomali, N.; Yazdanifar, M.; Hassanzadeh, A.; et al. A Deep Insight Into CAR-T Cell Therapy in Non-Hodgkin Lymphoma: Application, Opportunities, and Future Directions. Front. Immunol. 2021, 12, 681984. [Google Scholar] [CrossRef]
- Ghobadi, A. Chimeric antigen receptor T cell therapy for non-Hodgkin lymphoma. Curr. Res. Transl. Med. 2018, 66, 43–49. [Google Scholar] [CrossRef]
- Lin, H.; Cheng, J.; Mu, W.; Zhou, J.; Zhu, L. Advances in Universal CAR-T Cell Therapy. Front. Immunol. 2021, 12, 744823. [Google Scholar] [CrossRef]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Juarez-Avendano, G.; Mendez-Ramirez, N.; Luna-Silva, N.C.; Gomez-Almaguer, D.; Pelayo, R.; Balandran, J.C. Molecular and cellular markers for measurable residual disease in acute lymphoblastic leukemia. Bol. Med. Hosp. Infant. Mex. 2021, 78, 159–170. [Google Scholar] [CrossRef]
- Galimberti, S.; Genuardi, E.; Mazziotta, F.; Iovino, L.; Morabito, F.; Grassi, S.; Ciabatti, E.; Guerrini, F.; Petrini, M. The Minimal Residual Disease in Non-Hodgkin’s Lymphomas: From the Laboratory to the Clinical Practice. Front. Oncol. 2019, 9, 528. [Google Scholar] [CrossRef]
- Short, N.J.; Jabbour, E. Minimal Residual Disease in Acute Lymphoblastic Leukemia: How to Recognize and Treat It. Curr. Oncol. Rep. 2017, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Czyz, A.; Nagler, A. The Role of Measurable Residual Disease (MRD) in Hematopoietic Stem Cell Transplantation for Hematological Malignancies Focusing on Acute Leukemia. Int. J. Mol. Sci. 2019, 20, 5362. [Google Scholar] [CrossRef] [PubMed]
- Hopken, U.E.; Rehm, A. Targeting the Tumor Microenvironment of Leukemia and Lymphoma. Trends Cancer 2019, 5, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, D. Immune-checkpoint inhibitors in hematologic malignancies. Rinsho Ketsueki 2016, 57, 2381–2387. [Google Scholar] [CrossRef] [PubMed]
- Bryan, L.J.; Gordon, L.I. Blocking tumor escape in hematologic malignancies: The anti-PD-1 strategy. Blood Rev. 2015, 29, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.B.; Robert, C. Immune Checkpoint Inhibitors. Prog. Tumor Res. 2015, 42, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Kawashima, S.; Togashi, Y. Resistance to immune checkpoint inhibitors and the tumor microenvironment. Exp. Dermatol. 2023, 32, 240–249. [Google Scholar] [CrossRef]
- Liu, D. CAR-T “the living drugs”, immune checkpoint inhibitors, and precision medicine: A new era of cancer therapy. J. Hematol. Oncol. 2019, 12, 113. [Google Scholar] [CrossRef]
- Salik, B.; Smyth, M.J.; Nakamura, K. Targeting immune checkpoints in hematological malignancies. J. Hematol. Oncol. 2020, 13, 111. [Google Scholar] [CrossRef]
- Han, D.; Xu, Z.; Zhuang, Y.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Hematological Malignancies. J. Cancer 2021, 12, 326–334. [Google Scholar] [CrossRef]
- Li, B.; Chan, H.L.; Chen, P. Immune Checkpoint Inhibitors: Basics and Challenges. Curr. Med. Chem. 2019, 26, 3009–3025. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, X. Study and analysis of antitumor resistance mechanism of PD1/PD-L1 immune checkpoint blocker. Cancer Med. 2020, 9, 8086–8121. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Mehdizadeh, S.; Bayatipoor, H.; Pashangzadeh, S.; Jafarpour, R.; Shojaei, Z.; Motallebnezhad, M. Immune checkpoints and cancer development: Therapeutic implications and future directions. Pathol. Res. Pract. 2021, 223, 153485. [Google Scholar] [CrossRef] [PubMed]
- Burugu, S.; Dancsok, A.R.; Nielsen, T.O. Emerging targets in cancer immunotherapy. Semin. Cancer Biol. 2018, 52, 39–52. [Google Scholar] [CrossRef]
- Chihara, N.; Madi, A.; Kondo, T.; Zhang, H.; Acharya, N.; Singer, M.; Nyman, J.; Marjanovic, N.D.; Kowalczyk, M.S.; Wang, C.; et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 2018, 558, 454–459. [Google Scholar] [CrossRef]
- Watanabe, T. Approaches of the Innate Immune System to Ameliorate Adaptive Immunotherapy for B-Cell Non-Hodgkin Lymphoma in Their Microenvironment. Cancers 2021, 14, 141. [Google Scholar] [CrossRef]
- Chan, S.; Belmar, N.; Ho, S.; Rogers, B.; Stickler, M.; Graham, M.; Lee, E.; Tran, N.; Zhang, D.; Gupta, P.; et al. An anti-PD-1-GITR-L bispecific agonist induces GITR clustering-mediated T cell activation for cancer immunotherapy. Nat. Cancer 2022, 3, 337–354. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: Advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef]
- Yan, W.; Liu, Z.; Liu, J.; Xia, Y.; Hu, K.; Yu, J. Application of Chimeric Antigen Receptor T Cells in the Treatment of Hematological Malignancies. Biomed. Res. Int. 2020, 2020, 4241864. [Google Scholar] [CrossRef]
- Salles, G.; Barrett, M.; Foa, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef] [PubMed]
- Kansara, R.R.; Speziali, C. Immunotherapy in hematologic malignancies. Curr. Oncol. 2020, 27, S124–S131. [Google Scholar] [CrossRef] [PubMed]
- Fouda, G.E.; Bavbek, S. Rituximab Hypersensitivity: From Clinical Presentation to Management. Front. Pharmacol. 2020, 11, 572863. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Tao, Y.; Zhang, Y.; Wang, J.; Yang, J.; Wang, Y. CD25: A potential tumor therapeutic target. Int. J. Cancer 2023, 152, 1290–1303. [Google Scholar] [CrossRef]
- Zammarchi, F.; Havenith, K.; Bertelli, F.; Vijayakrishnan, B.; Chivers, S.; van Berkel, P.H. CD25-targeted antibody-drug conjugate depletes regulatory T cells and eliminates established syngeneic tumors via antitumor immunity. J. Immunother. Cancer 2020, 8, e000860. [Google Scholar] [CrossRef]
- Veyri, M.; Spano, J.P.; Le Bras, F.; Marcelin, A.G.; Todesco, E. CD30 as a therapeutic target in adult haematological malignancies: Where are we now? Br. J. Haematol. 2023, 201, 1033–1046. [Google Scholar] [CrossRef]
- Fabbri, A.; Cencini, E.; Gozzetti, A.; Schiattone, L.; Bocchia, M. Therapeutic Use of Brentuximab Vedotin in CD30+ Hematologic Malignancies. Anticancer. Agents Med. Chem. 2017, 17, 886–895. [Google Scholar] [CrossRef]
- Tschernia, N.P.; Heiling, H.; Deal, A.M.; Cheng, C.; Babinec, C.; Gonzalez, M.; Morrison, J.K.; Dittus, C.; Dotti, G.; Beaven, A.W.; et al. Patient-reported outcomes in CD30-directed CAR-T cells against relapsed/refractory CD30+ lymphomas. J. Immunother. Cancer 2023, 11, e006959. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Han, T.H.; Hunder, N.N.; Jang, G.; Zhao, B. Population Pharmacokinetics of Brentuximab Vedotin in Patients with CD30-Expressing Hematologic Malignancies. J. Clin. Pharmacol. 2017, 57, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Yang, L.; Chukinas, J.A.; Shah, N.; Tarun, S.; Pouzolles, M.; Chien, C.D.; Niswander, L.M.; Welch, A.R.; Taylor, N.; et al. Systematic preclinical evaluation of CD33-directed chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia defines optimized construct design. J. Immunother. Cancer 2021, 9, e003149. [Google Scholar] [CrossRef]
- Zugmaier, G.; Klinger, M.; Schmidt, M.; Subklewe, M. Clinical overview of anti-CD19 BiTE((R)) and ex vivo data from anti-CD33 BiTE((R)) as examples for retargeting T cells in hematologic malignancies. Mol. Immunol. 2015, 67, 58–66. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, R.; Chen, L.; Wu, S. CD38 as an immunomodulator in cancer. Future Oncol. 2020, 16, 2853–2861. [Google Scholar] [CrossRef]
- van de Donk, N.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef]
- Bonello, F.; D’Agostino, M.; Moscvin, M.; Cerrato, C.; Boccadoro, M.; Gay, F. CD38 as an immunotherapeutic target in multiple myeloma. Expert Opin. Biol. Ther. 2018, 18, 1209–1221. [Google Scholar] [CrossRef]
- van de Donk, N. Immunomodulatory effects of CD38-targeting antibodies. Immunol. Lett. 2018, 199, 16–22. [Google Scholar] [CrossRef]
- Yang, H.; Xun, Y.; You, H. The landscape overview of CD47-based immunotherapy for hematological malignancies. Biomark. Res. 2023, 11, 15. [Google Scholar] [CrossRef]
- Russ, A.; Hua, A.B.; Montfort, W.R.; Rahman, B.; Riaz, I.B.; Khalid, M.U.; Carew, J.S.; Nawrocki, S.T.; Persky, D.; Anwer, F. Blocking “don’t eat me” signal of CD47-SIRPalpha in hematological malignancies, an in-depth review. Blood Rev. 2018, 32, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, K. Cancer immunotherapy targeting the CD47/SIRPalpha axis. Eur. J. Cancer 2017, 76, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Beckett, A.N.; Chockley, P.; Pruett-Miller, S.M.; Nguyen, P.; Vogel, P.; Sheppard, H.; Krenciute, G.; Gottschalk, S.; DeRenzo, C. CD47 expression is critical for CAR T-cell survival in vivo. J. Immunother. Cancer 2023, 11, e005857. [Google Scholar] [CrossRef]
- Chen, H.; Yang, Y.; Deng, Y.; Wei, F.; Zhao, Q.; Liu, Y.; Liu, Z.; Yu, B.; Huang, Z. Delivery of CD47 blocker SIRPalpha-Fc by CAR-T cells enhances antitumor efficacy. J. Immunother. Cancer 2022, 10, e003737. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Castelli, G. CD123 as a Therapeutic Target in the Treatment of Hematological Malignancies. Cancers 2019, 11, 1358. [Google Scholar] [CrossRef]
- Bergaggio, E.; Riganti, C.; Garaffo, G.; Vitale, N.; Mereu, E.; Bandini, C.; Pellegrino, E.; Pullano, V.; Omede, P.; Todoerti, K.; et al. IDH2 inhibition enhances proteasome inhibitor responsiveness in hematological malignancies. Blood 2019, 133, 156–167. [Google Scholar] [CrossRef]
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef]
- Yu, B.; Jiang, T.; Liu, D. BCMA-targeted immunotherapy for multiple myeloma. J. Hematol. Oncol. 2020, 13, 125. [Google Scholar] [CrossRef]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin. Cancer Biol. 2022, 85, 69–94. [Google Scholar] [CrossRef]
- Barrett, D.; Brown, V.I.; Grupp, S.A.; Teachey, D.T. Targeting the PI3K/AKT/mTOR signaling axis in children with hematologic malignancies. Paediatr. Drugs 2012, 14, 299–316. [Google Scholar] [CrossRef]
- Yeh, Y.H.; Hsiao, H.F.; Yeh, Y.C.; Chen, T.W.; Li, T.K. Inflammatory interferon activates HIF-1alpha-mediated epithelial-to-mesenchymal transition via PI3K/AKT/mTOR pathway. J. Exp. Clin. Cancer Res. 2018, 37, 70. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.H.; Wang, S.W.; Yeh, Y.C.; Hsiao, H.F.; Li, T.K. Rhapontigenin inhibits TGF-beta-mediated epithelial—Mesenchymal transition via the PI3K/AKT/mTOR pathway and is not associated with HIF-1alpha degradation. Oncol. Rep. 2016, 35, 2887–2895. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, W.S.; Park, C. Interleukin-6 mediates resistance to PI3K-pathway-targeted therapy in lymphoma. BMC Cancer 2019, 19, 936. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Liao, R.; Lv, J.; Li, S.; Zheng, D.; Qin, L.; Wu, D.; Chen, S.; Long, Y.; Wu, Q.; et al. IL-6 trans-signaling promotes the expansion and anti-tumor activity of CAR T cells. Leukemia 2021, 35, 1380–1391. [Google Scholar] [CrossRef]
- Huynh, L.K.; Hipolito, C.J.; Ten Dijke, P. A Perspective on the Development of TGF-beta Inhibitors for Cancer Treatment. Biomolecules 2019, 9, 743. [Google Scholar] [CrossRef]
- Gu, S.; Feng, X.H. TGF-beta signaling in cancer. Acta Biochim. Biophys. Sin. 2018, 50, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Fabregat, I.; Fernando, J.; Mainez, J.; Sancho, P. TGF-beta signaling in cancer treatment. Curr. Pharm. Des. 2014, 20, 2934–2947. [Google Scholar] [CrossRef]
- Zhang, L.; Kuca, K.; You, L.; Zhao, Y.; Musilek, K.; Nepovimova, E.; Wu, Q.; Wu, W.; Adam, V. Signal transducer and activator of transcription 3 signaling in tumor immune evasion. Pharmacol. Ther. 2022, 230, 107969. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Gomez, H.J.; Thakkar, S.; Singh, S.P.; Parihar, A.S. Overcoming Acquired Drug Resistance to Cancer Therapies through Targeted STAT3 Inhibition. Int. J. Mol. Sci. 2023, 24, 4722. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef]
- Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.; Kempf, C.R.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080–1094. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef]
- Shelton, J.G.; Steelman, L.S.; Lee, J.T.; Knapp, S.L.; Blalock, W.L.; Moye, P.W.; Franklin, R.A.; Pohnert, S.C.; Mirza, A.M.; McMahon, M.; et al. Effects of the RAF/MEK/ERK and PI3K/AKT signal transduction pathways on the abrogation of cytokine-dependence and prevention of apoptosis in hematopoietic cells. Oncogene 2003, 22, 2478–2492. [Google Scholar] [CrossRef]
- Kurachi, M. CD8(+) T cell exhaustion. Semin. Immunopathol. 2019, 41, 327–337. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.T.; Pillai, R.N.; Ramalingam, S.S.; Araki, K.; Ahmed, R. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef]
- Verdon, D.J.; Mulazzani, M.; Jenkins, M.R. Cellular and Molecular Mechanisms of CD8(+) T Cell Differentiation, Dysfunction and Exhaustion. Int. J. Mol. Sci. 2020, 21, 7357. [Google Scholar] [CrossRef]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. Adv. Exp. Med. Biol. 2020, 1248, 33–59. [Google Scholar] [CrossRef]
- Dolina, J.S.; Van Braeckel-Budimir, N.; Thomas, G.D.; Salek-Ardakani, S. CD8(+) T Cell Exhaustion in Cancer. Front. Immunol. 2021, 12, 715234. [Google Scholar] [CrossRef] [PubMed]
- Ford, B.R.; Poholek, A.C. Regulation and Immunotherapeutic Targeting of the Epigenome in Exhausted CD8 T Cell Responses. J. Immunol. 2023, 210, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A. Activation of immunosuppressive network in the aging process. Ageing Res. Rev. 2020, 57, 100998. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Dupuis, G.; Witkowski, J.M.; Larbi, A. The Role of Immunosenescence in the Development of Age-Related Diseases. Rev. Investig. Clin. 2016, 68, 84–91. [Google Scholar]
- Thomas, R.; Wang, W.; Su, D.M. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun. Ageing 2020, 17, 2. [Google Scholar] [CrossRef]
- Regulski, M.J. Cellular Senescence: What, Why, and How. Wounds 2017, 29, 168–174. [Google Scholar]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat. Commun. 2019, 10, 4355. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. Int. J. Mol. Sci. 2019, 20, 805. [Google Scholar] [CrossRef]
- Sadowski, K.; Olejarz, W.; Basak, G. Modern Advances in CARs Therapy and Creating a New Approach to Future Treatment. Int. J. Mol. Sci. 2022, 23, 15006. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, H.; He, Q.; Tian, W.; Xia, A.; Lu, X.J. Exosomes derived from exhausted CD8+ T cells impaired the anticancer function of normal CD8+ T cells. J. Med. Genet. 2019, 56, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Elkord, E. Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression. Semin. Cancer Biol. 2020, 65, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Du, Y.; Lin, X.; Qian, Y.; Zhou, T.; Huang, Z. CD4+CD25+ regulatory T cells in tumor immunity. Int. Immunopharmacol. 2016, 34, 244–249. [Google Scholar] [CrossRef]
- Harris, R.J.; Willsmore, Z.; Laddach, R.; Crescioli, S.; Chauhan, J.; Cheung, A.; Black, A.; Geh, J.L.C.; MacKenzie Ross, A.D.; Healy, C.; et al. Enriched circulating and tumor-resident TGF-beta(+) regulatory B cells in patients with melanoma promote FOXP3(+) Tregs. Oncoimmunology 2022, 11, 2104426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Sun, H.X. Immune checkpoint molecules in pregnancy: Focus on regulatory T cells. Eur. J. Immunol. 2020, 50, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors in cancer therapy: A focus on T-regulatory cells. Immunol. Cell Biol. 2018, 96, 21–33. [Google Scholar] [CrossRef]
- Gunes, E.G.; Rosen, S.T.; Querfeld, C. The role of myeloid-derived suppressor cells in hematologic malignancies. Curr. Opin. Oncol. 2020, 32, 518–526. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, X.; Wu, S.; Cui, D.; Xu, Z. Myeloid-derived suppressor cells: Key immunosuppressive regulators and therapeutic targets in hematological malignancies. Biomark. Res. 2023, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Baldominos, P.; Barbera-Mourelle, A.; Barreiro, O.; Huang, Y.; Wight, A.; Cho, J.W.; Zhao, X.; Estivill, G.; Adam, I.; Sanchez, X.; et al. Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell 2022, 185, 1694–1708.e19. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V. CAR-T Cells Targeting Immune Checkpoint Pathway Players. Front. Biosci. (Landmark) 2022, 27, 121. [Google Scholar] [CrossRef] [PubMed]
- Leick, M.B.; Silva, H.; Scarfo, I.; Larson, R.; Choi, B.D.; Bouffard, A.A.; Gallagher, K.; Schmidts, A.; Bailey, S.R.; Kann, M.C.; et al. Non-cleavable hinge enhances avidity and expansion of CAR-T cells for acute myeloid leukemia. Cancer Cell 2022, 40, 494–508.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kaur, G.; Sankin, A.I.; Chen, F.; Guan, F.; Zang, X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J. Hematol. Oncol. 2019, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Starzer, A.M.; Berghoff, A.S. New emerging targets in cancer immunotherapy: CD27 (TNFRSF7). ESMO Open 2020, 4, e000629. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Flinn, I.; Taylor, M.H.; Sikic, B.I.; Brody, J.; Nemunaitis, J.; Feldman, A.; Hawthorne, T.R.; Rawls, T.; Keler, T.; et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, for hematologic malignancies. Blood Adv. 2020, 4, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Sauer, T.; Parikh, K.; Sharma, S.; Omer, B.; Sedloev, D.; Chen, Q.; Angenendt, L.; Schliemann, C.; Schmitt, M.; Muller-Tidow, C.; et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood 2021, 138, 318–330. [Google Scholar] [CrossRef]
- Payandeh, Z.; Noori, E.; Khalesi, B.; Mard-Soltani, M.; Abdolalizadeh, J.; Khalili, S. Anti-CD37 targeted immunotherapy of B-Cell malignancies. Biotechnol. Lett. 2018, 40, 1459–1466. [Google Scholar] [CrossRef]
- Witkowska, M.; Smolewski, P.; Robak, T. Investigational therapies targeting CD37 for the treatment of B-cell lymphoid malignancies. Expert Opin. Investig. Drugs 2018, 27, 171–177. [Google Scholar] [CrossRef]
- Deckert, J.; Park, P.U.; Chicklas, S.; Yi, Y.; Li, M.; Lai, K.C.; Mayo, M.F.; Carrigan, C.N.; Erickson, H.K.; Pinkas, J.; et al. A novel anti-CD37 antibody-drug conjugate with multiple anti-tumor mechanisms for the treatment of B-cell malignancies. Blood 2013, 122, 3500–3510. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, K.A.; Frissora, F.W.; Stefanovski, M.R.; Towns, W.H.; Cheney, C.; Mo, X.; Deckert, J.; Croce, C.M.; Flynn, J.M.; Andritsos, L.A.; et al. The CD37-targeted antibody-drug conjugate IMGN529 is highly active against human CLL and in a novel CD37 transgenic murine leukemia model. Leukemia 2014, 28, 1501–1510. [Google Scholar] [CrossRef]
- Heider, K.H.; Kiefer, K.; Zenz, T.; Volden, M.; Stilgenbauer, S.; Ostermann, E.; Baum, A.; Lamche, H.; Kupcu, Z.; Jacobi, A.; et al. A novel Fc-engineered monoclonal antibody to CD37 with enhanced ADCC and high proapoptotic activity for treatment of B-cell malignancies. Blood 2011, 118, 4159–4168. [Google Scholar] [CrossRef]
- Chen, R.; Ganesan, A.; Okoye, I.; Arutyunova, E.; Elahi, S.; Lemieux, M.J.; Barakat, K. Targeting B7-1 in immunotherapy. Med. Res. Rev. 2020, 40, 654–682. [Google Scholar] [CrossRef] [PubMed]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaav7431. [Google Scholar] [CrossRef] [PubMed]
- Podlesnykh, S.V.; Abramova, K.E.; Gordeeva, A.; Khlebnikov, A.I.; Chapoval, A.I. Peptide Blocking CTLA-4 and B7-1 Interaction. Molecules 2021, 26, 253. [Google Scholar] [CrossRef]
- Khan, M.; Arooj, S.; Wang, H. Soluble B7-CD28 Family Inhibitory Immune Checkpoint Proteins and Anti-Cancer Immunotherapy. Front. Immunol. 2021, 12, 651634. [Google Scholar] [CrossRef]
- Ni, L.; Dong, C. New checkpoints in cancer immunotherapy. Immunol. Rev. 2017, 276, 52–65. [Google Scholar] [CrossRef]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef]
- Wang, J.Y.; Wang, W.P. B7-H4, a promising target for immunotherapy. Cell Immunol. 2020, 347, 104008. [Google Scholar] [CrossRef]
- Wang, L.; Heng, X.; Lu, Y.; Cai, Z.; Yi, Q.; Che, F. Could B7-H4 serve as a target to activate anti-cancer immunity? Int. Immunopharmacol. 2016, 38, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Podojil, J.R.; Miller, S.D. Potential targeting of B7-H4 for the treatment of cancer. Immunol. Rev. 2017, 276, 40–51. [Google Scholar] [CrossRef]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 2020, 5, 145. [Google Scholar] [CrossRef] [PubMed]
- Olejarz, W.; Dominiak, A.; Zolnierzak, A.; Kubiak-Tomaszewska, G.; Lorenc, T. Tumor-Derived Exosomes in Immunosuppression and Immunotherapy. J. Immunol. Res. 2020, 2020, 6272498. [Google Scholar] [CrossRef] [PubMed]
- Olejarz, W.; Kubiak-Tomaszewska, G.; Chrzanowska, A.; Lorenc, T. Exosomes in Angiogenesis and Anti-angiogenic Therapy in Cancers. Int. J. Mol. Sci. 2020, 21, 5840. [Google Scholar] [CrossRef]
- Das, K.; Mukherjee, T.; Shankar, P. The Role of Extracellular Vesicles in the Pathogenesis of Hematological Malignancies: Interaction with Tumor Microenvironment; a Potential Biomarker and Targeted Therapy. Biomolecules 2023, 13, 897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ji, Q.; Yang, Y.; Li, Q.; Wang, Z. Exosome: Function and Role in Cancer Metastasis and Drug Resistance. Technol. Cancer Res. Treat. 2018, 17, 1533033818763450. [Google Scholar] [CrossRef]
- Raimondo, S.; Corrado, C.; Raimondi, L.; De Leo, G.; Alessandro, R. Role of Extracellular Vesicles in Hematological Malignancies. Biomed. Res. Int. 2015, 2015, 821613. [Google Scholar] [CrossRef]
- Xu, Z.; Zeng, S.; Gong, Z.; Yan, Y. Exosome-based immunotherapy: A promising approach for cancer treatment. Mol. Cancer 2020, 19, 160. [Google Scholar] [CrossRef]
- Wang, Y.; Hays, E.; Rama, M.; Bonavida, B. Cell-mediated immune resistance in cancer. Cancer Drug Resist. 2020, 3, 232–251. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational implications of tumor heterogeneity. Clin. Cancer Res. 2015, 21, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Holzel, M.; Bovier, A.; Tuting, T. Plasticity of tumour and immune cells: A source of heterogeneity and a cause for therapy resistance? Nat. Rev. Cancer 2013, 13, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef] [PubMed]
- Aptsiauri, N.; Ruiz-Cabello, F.; Garrido, F. The transition from HLA-I positive to HLA-I negative primary tumors: The road to escape from T-cell responses. Curr. Opin. Immunol. 2018, 51, 123–132. [Google Scholar] [CrossRef]
- Khong, H.T.; Wang, Q.J.; Rosenberg, S.A. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: Tumor escape by antigen loss and loss of MHC expression. J. Immunother. 2004, 27, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Rimsza, L.M.; Roberts, R.A.; Miller, T.P.; Unger, J.M.; LeBlanc, M.; Braziel, R.M.; Weisenberger, D.D.; Chan, W.C.; Muller-Hermelink, H.K.; Jaffe, E.S.; et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: A follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood 2004, 103, 4251–4258. [Google Scholar] [CrossRef] [PubMed]
- Jordanova, E.S.; Philippo, K.; Giphart, M.J.; Schuuring, E.; Kluin, P.M. Mutations in the HLA class II genes leading to loss of expression of HLA-DR and HLA-DQ in diffuse large B-cell lymphoma. Immunogenetics 2003, 55, 203–209. [Google Scholar] [CrossRef]
- Toes, R.E.; Ossendorp, F.; Offringa, R.; Melief, C.J. CD4 T cells and their role in antitumor immune responses. J. Exp. Med. 1999, 189, 753–756. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Gubin, M.M.; Vesely, M.D. Cancer Immunoediting in the Era of Immuno-oncology. Clin. Cancer Res. 2022, 28, 3917–3928. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Zhang, T.; Chen, L. Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu. Rev. Immunol. 2022, 40, 45–74. [Google Scholar] [CrossRef]
- Seliger, B.; Massa, C. Immune Therapy Resistance and Immune Escape of Tumors. Cancers 2021, 13, 551. [Google Scholar] [CrossRef] [PubMed]
- Erin, N.; Grahovac, J.; Brozovic, A.; Efferth, T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist. Updat. 2020, 53, 100715. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wolfel, T.; Holzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L. Immune regulation of cancer. J. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef]
- Khalaf, K.; Hana, D.; Chou, J.T.; Singh, C.; Mackiewicz, A.; Kaczmarek, M. Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Front. Immunol. 2021, 12, 656364. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Rashid, M.; Zadeh, L.R.; Baradaran, B.; Molavi, O.; Ghesmati, Z.; Sabzichi, M.; Ramezani, F. Up-down regulation of HIF-1alpha in cancer progression. Gene 2021, 798, 145796. [Google Scholar] [CrossRef]
- Lee, S.H.; Golinska, M.; Griffiths, J.R. HIF-1-Independent Mechanisms Regulating Metabolic Adaptation in Hypoxic Cancer Cells. Cells 2021, 10, 2371. [Google Scholar] [CrossRef]
- Tang, B.Z.; Zhao, F.Y.; Qu, Y.; Mu, D.Z. Hypoxia-inducible factor-1alpha: A promising target for tumor therapy. Ai Zheng = Aizheng = Chin. J. Cancer 2009, 28, 775–782. [Google Scholar] [CrossRef]
- Guo, H.; Yang, J.; Wang, H.; Liu, X.; Liu, Y.; Zhou, K. Reshaping the tumor microenvironment: The versatility of immunomodulatory drugs in B-cell neoplasms. Front. Immunol. 2022, 13, 1017990. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, N.; Jain, K.; Ramsay, A.G. Immunomodulatory Drugs for the Treatment of B Cell Malignancies. Int. J. Mol. Sci. 2021, 22, 8572. [Google Scholar] [CrossRef]
- Chaudhry, G.E.; Akim, A.M.; Sung, Y.Y.; Muhammad, T.S.T. Cancer and Apoptosis. Methods Mol. Biol. 2022, 2543, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef]
- Wu, H.; Medeiros, L.J.; Young, K.H. Apoptosis signaling and BCL-2 pathways provide opportunities for novel targeted therapeutic strategies in hematologic malignances. Blood Rev. 2018, 32, 8–28. [Google Scholar] [CrossRef]
- Valentin, R.; Grabow, S.; Davids, M.S. The rise of apoptosis: Targeting apoptosis in hematologic malignancies. Blood 2018, 132, 1248–1264. [Google Scholar] [CrossRef]
- Yalniz, F.F.; Wierda, W.G. Targeting BCL2 in Chronic Lymphocytic Leukemia and Other Hematologic Malignancies. Drugs 2019, 79, 1287–1304. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Walter, H.S.; Dyer, M.J.S. Targeting anti-apoptotic BCL2 family proteins in haematological malignancies—From pathogenesis to treatment. Br. J. Haematol. 2017, 178, 364–379. [Google Scholar] [CrossRef]
- Arakaki, R.; Yamada, A.; Kudo, Y.; Hayashi, Y.; Ishimaru, N. Mechanism of activation-induced cell death of T cells and regulation of FasL expression. Crit. Rev. Immunol. 2014, 34, 301–314. [Google Scholar] [CrossRef]
- Huan, T.; Chen, D.; Liu, G.; Zhang, H.; Wang, X.; Wu, Z.; Wu, Y.; Xu, Q.; Yu, F. Activation-induced cell death in CAR-T cell therapy. Hum. Cell 2022, 35, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E. Activation-induced and damage-induced cell death in aging human T cells. Mech. Ageing Dev. 2015, 151, 85–92. [Google Scholar] [CrossRef]
- Oberg, H.H.; Lengl-Janssen, B.; Kabelitz, D.; Janssen, O. Activation-induced T cell death: Resistance or susceptibility correlate with cell surface fas ligand expression and T helper phenotype. Cell Immunol. 1997, 181, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Kosti, P.; Maher, J.; Arnold, J.N. Perspectives on Chimeric Antigen Receptor T-Cell Immunotherapy for Solid Tumors. Front. Immunol. 2018, 9, 1104. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Strauss, G.; Meyer, E.; Debatin, K.M. Functional CD95 ligand and CD95 death-inducing signaling complex in activation-induced cell death and doxorubicin-induced apoptosis in leukemic T cells. Blood 2000, 95, 301–308. [Google Scholar] [CrossRef]
- Janssen, O.; Stocker, A.; Sanzenbacher, R.; Oberg, H.H.; Siddiqi, M.A.; Kabelitz, D. Differential regulation of activation-induced cell death in individual human T cell clones. Int. Arch. Allergy Immunol. 2000, 121, 183–193. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018, 7, e1466769. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Nogami, A.; Sasaki, K. Therapeutic Advances in Immunotherapies for Hematological Malignancies. Int. J. Mol. Sci. 2022, 23, 11526. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Dominik, P.K.; Stanfield, J.; Ding, S.; Yang, W.; Kurd, N.; Llewellyn, R.; Heyen, J.; Wang, C.; Melton, Z.; et al. Dual checkpoint blockade of CD47 and PD-L1 using an affinity-tuned bispecific antibody maximizes antitumor immunity. J. Immunother. Cancer 2021, 9, e003464. [Google Scholar] [CrossRef]
- Thieblemont, C.; Phillips, T.; Ghesquieres, H.; Cheah, C.Y.; Clausen, M.R.; Cunningham, D.; Do, Y.R.; Feldman, T.; Gasiorowski, R.; Jurczak, W.; et al. Epcoritamab, a Novel, Subcutaneous CD3xCD20 Bispecific T-Cell-Engaging Antibody, in Relapsed or Refractory Large B-Cell Lymphoma: Dose Expansion in a Phase I/II Trial. J. Clin. Oncol. 2023, 41, 2238–2247. [Google Scholar] [CrossRef]
- Srivastava, A.; Rathore, S.; Munshi, A.; Ramesh, R. Extracellular Vesicles in Oncology: From Immune Suppression to Immunotherapy. AAPS J. 2021, 23, 30. [Google Scholar] [CrossRef]
- Li, I.; Nabet, B.Y. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol. Cancer 2019, 18, 32. [Google Scholar] [CrossRef]
- Kugeratski, F.G.; Kalluri, R. Exosomes as mediators of immune regulation and immunotherapy in cancer. FEBS J. 2021, 288, 10–35. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.H.; Zheng, J.Q.; Ding, J.Y.; Wu, Y.F.; Liu, L.; Yu, Z.L.; Chen, G. Exosome-Mediated Immunosuppression in Tumor Microenvironments. Cells 2022, 11, 1946. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhu, Z.; Chen, X.; Zhang, H.; Huang, J.; Gu, S.; Zhao, X. The Importance of Exosomal PD-L1 in Cancer Progression and Its Potential as a Therapeutic Target. Cells 2021, 10, 3247. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Dong, Y.; Wei, M.; Gao, X.; Yang, G.; Zhang, J.; Liu, L. Exosomes in Tumor Immunotherapy: Mediator, Drug Carrier, and Prognostic Biomarker. Adv. Biosyst. 2020, 4, e2000061. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Savani, M.; Oluwole, O.; Dholaria, B. New targets for CAR T therapy in hematologic malignancies. Best Pract. Res. Clin. Haematol. 2021, 34, 101277. [Google Scholar] [CrossRef]
- Badura, S.; Tesanovic, T.; Pfeifer, H.; Wystub, S.; Nijmeijer, B.A.; Liebermann, M.; Falkenburg, J.H.; Ruthardt, M.; Ottmann, O.G. Differential effects of selective inhibitors targeting the PI3K/AKT/mTOR pathway in acute lymphoblastic leukemia. PLoS ONE 2013, 8, e80070. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets in Immunotherapy | Examples | Drugs | Mechanism of Action | References |
|---|---|---|---|---|
| Immune checkpoint |
| Ipilimumab, Nivolumab, Pembrolizumab, Avelumab, Cytarabine, Decitabine, Darubicin, MBG453, Tebotelimab (MGD013) | Reverses T-cell exhaustion. Enhances T-cell activation and effector functions. Broadens TCR repertoire. | [176] |
| CD and BCMA |
| Rituximab, Blinatumomab, Magrolimab (Hu5F9-G4), Epcoritamab, AMG330, Vixtimotamab (AMV564), Vibecotamab (XmAb14045), Flontetuzumab, AMG427, Brentuximab vedotin Belantamab | Activates effector cells. Upregulates proinflammatory cytokines. Increases genomic instability. Modulates metabolic response. | [176,177,178,179] |
| Signaling pathways |
| Rapalog, Rhapontigenin, Trastuzumab, Dabrafenib, Trametinib | Blocks signaling pathways. Decreases inhibitory cytokine production. Enhances T-cell effector function. | [60,61,62,73,74] |
| Immunosuppressive TME |
| Thalidomide, Lenalidomide, Pomalidomide, Avadomide | Depletes suppressive cells. Redirects cytotoxic effector cells to the TME. | [145,148,153,154] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olejarz, W.; Basak, G. Emerging Therapeutic Targets and Drug Resistance Mechanisms in Immunotherapy of Hematological Malignancies. Cancers 2023, 15, 5765. https://doi.org/10.3390/cancers15245765
Olejarz W, Basak G. Emerging Therapeutic Targets and Drug Resistance Mechanisms in Immunotherapy of Hematological Malignancies. Cancers. 2023; 15(24):5765. https://doi.org/10.3390/cancers15245765
Chicago/Turabian StyleOlejarz, Wioletta, and Grzegorz Basak. 2023. "Emerging Therapeutic Targets and Drug Resistance Mechanisms in Immunotherapy of Hematological Malignancies" Cancers 15, no. 24: 5765. https://doi.org/10.3390/cancers15245765
APA StyleOlejarz, W., & Basak, G. (2023). Emerging Therapeutic Targets and Drug Resistance Mechanisms in Immunotherapy of Hematological Malignancies. Cancers, 15(24), 5765. https://doi.org/10.3390/cancers15245765