
Role of Vitamin C in Targeting Cancer Stem Cells and Cellular Plasticity


Abstract
:Simple Summary
Abstract
1. Introduction
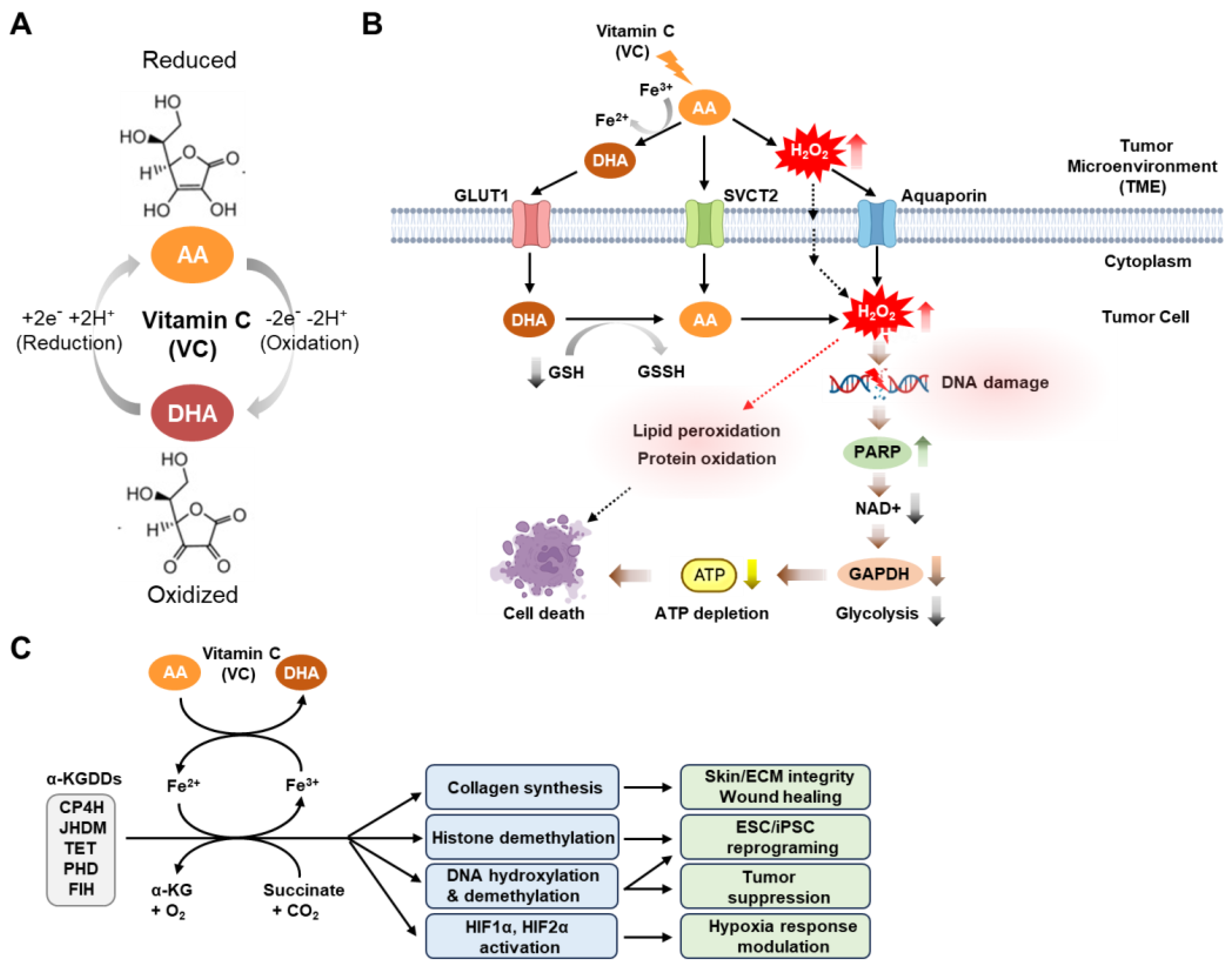
2. Physiological and Anti-Tumor Activities of Vitamin C
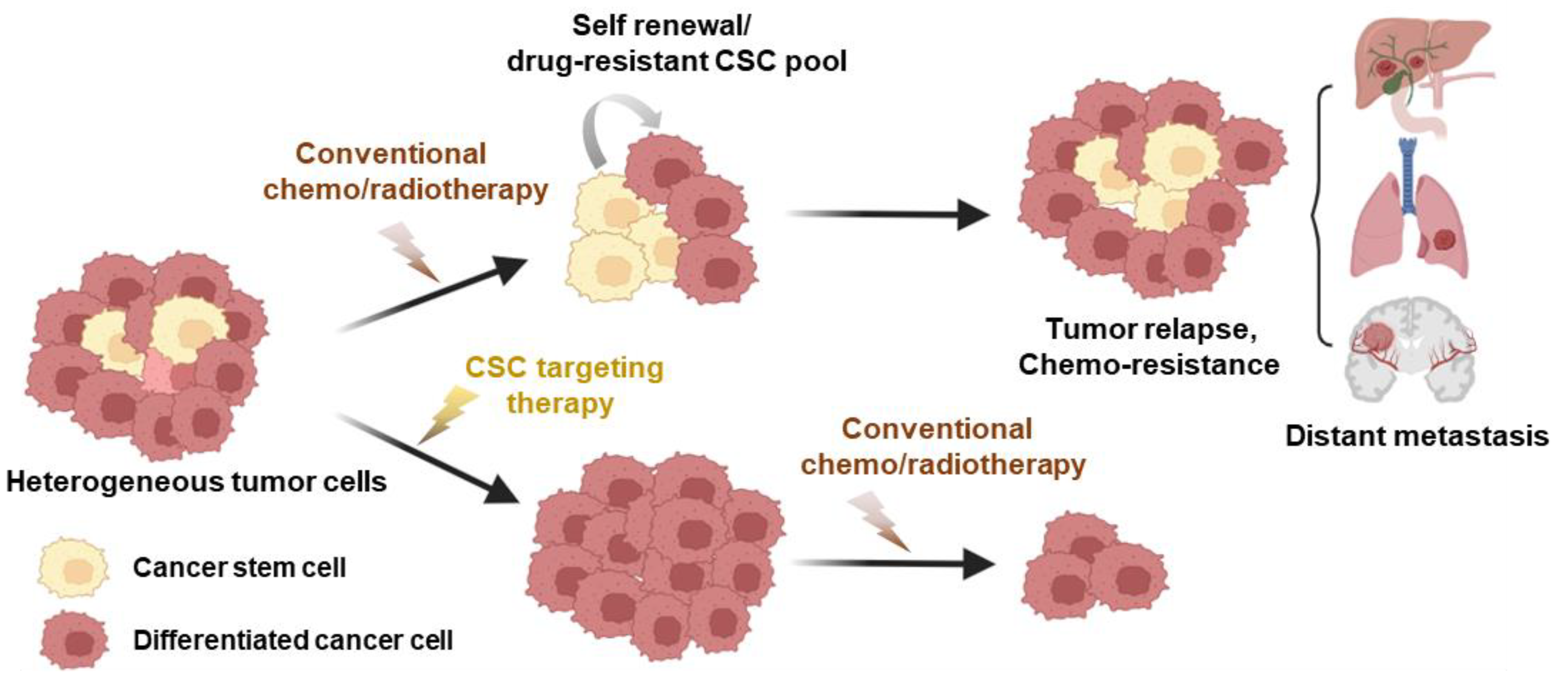
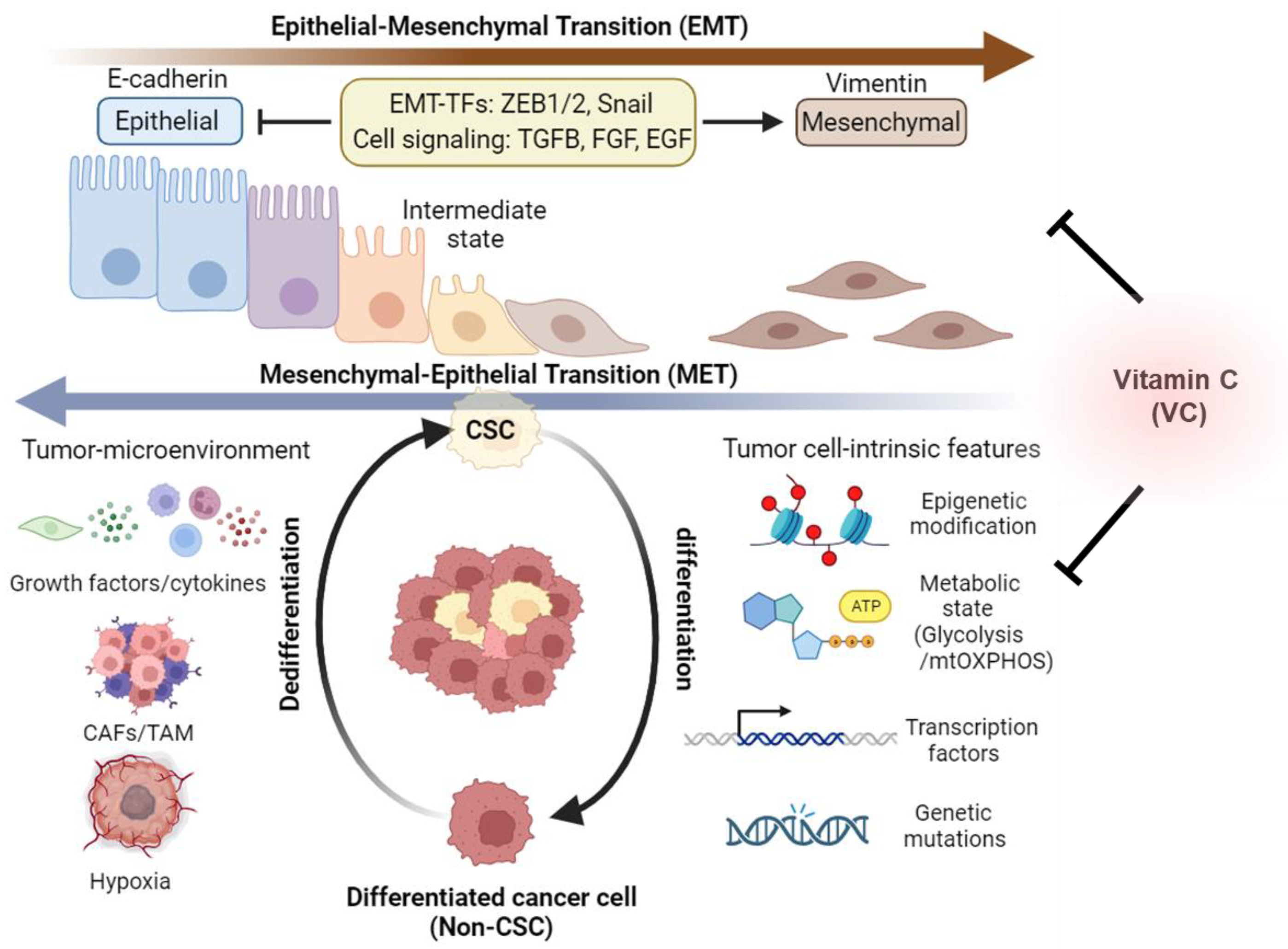
3. Cancer Stem Cell Phenotypes and Plasticity
4. Metabolic Plasticity of Cancer Stem Cells
5. Anti-Cancer Mechanism of Vitamin C in Targeting Cancer Stem Cells
5.1. Targeting Leukemic Stem Cells with Vitamin C
5.2. Targeting Liver Cancer Stem Cells with Vitamin C
5.3. Targeting Breast Cancer Stem Cells with Vitamin C
5.4. Targeting Metabolic Plasticity in Pancreatic Cancer with Vitamin C
5.5. Targeting Cancer Stem Cells with Vitamin C in Combination Therapy
6. Conclusions
Funding
Conflicts of Interest
References
- Figueroa-Méndez, R.; Rivas-Arancibia, S. Vitamin C in Health and Disease: Its Role in the Metabolism of Cells and Redox State in the Brain. Front. Physiol. 2015, 6, 397. [Google Scholar] [CrossRef] [PubMed]
- Pullar, J.M.; Carr, A.C.; Vissers, M.C.M. The Roles of Vitamin C in Skin Health. Nutrients 2017, 9, 866. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Maggini, S. Vitamin C and Immune Function. Nutrients 2017, 9, 1211. [Google Scholar] [CrossRef]
- Chen, H.Y.; Almonte-Loya, A.; Lay, F.Y.; Hsu, M.; Johnson, E.; González-Avalos, E.; Yin, J.; Bruno, R.S.; Ma, Q.; Ghoneim, H.E.; et al. Epigenetic remodeling by vitamin C potentiates plasma cell differentiation. Elife 2022, 11, e73754. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak-Barańska, J.; Boguszewska, K.; Adamus-Grabicka, A.; Karwowski, B.T. Two Faces of Vitamin C—Antioxidative and Pro-Oxidative Agent. Nutrients 2020, 12, 1501. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed]
- Villagran, M.; Ferreira, J.; Martorell, M.; Mardones, L. The Role of Vitamin C in Cancer Prevention and Therapy: A Literature Review. Antioxidants 2021, 10, 1894. [Google Scholar] [CrossRef]
- Zhitkovich, A. Nuclear and Cytoplasmic Functions of Vitamin C. Chem. Res. Toxicol. 2020, 33, 2515–2526. [Google Scholar] [CrossRef]
- Brabson, J.P.; Leesang, T.; Mohammad, S.; Cimmino, L. Epigenetic Regulation of Genomic Stability by Vitamin C. Front. Genet. 2021, 12, 675780. [Google Scholar] [CrossRef]
- Cenigaonandia-Campillo, A.; Serna-Blasco, R.; Gómez-Ocabo, L.; Solanes-Casado, S.; Baños-Herraiz, N.; Puerto-Nevado, L.D.; Cañas, J.A.; Aceñero, M.J.; García-Foncillas, J.; Aguilera, Ó. Vitamin C activates pyruvate dehydrogenase (PDH) targeting the mitochondrial tricarboxylic acid (TCA) cycle in hypoxic. Theranostics 2021, 11, 3595–3606. [Google Scholar] [CrossRef]
- Szarka, A.; Kapuy, O.; Lőrincz, T.; Bánhegyi, G. Vitamin C and Cell Death. Antioxid. Redox Signal 2021, 34, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Böttger, F.; Vallés-Martí, A.; Cahn, L.; Jimenez, C.R. High-dose intravenous vitamin C, a promising multi-targeting agent in the treatment of cancer. J. Exp. Clin. Cancer Res. 2021, 40, 343. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Idris, R.A.M.; Ahmed, N.; Ahmad, S.; Murtadha, A.H.; Din, T.A.D.A.A.T.; Yean, C.Y.; Rahman, W.F.W.A.; Lazim, N.M.; Uskoković, V.; et al. High-Dose Vitamin C for Cancer Therapy. Pharmaceuticals 2022, 15, 711. [Google Scholar] [CrossRef] [PubMed]
- Di Tano, M.; Raucci, F.; Vernieri, C.; Caffa, I.; Buono, R.; Fanti, M.; Brandhorst, S.; Curigliano, G.; Nencioni, A.; de Braud, F.; et al. Synergistic effect of fasting-mimicking diet and vitamin C against KRAS mutated cancers. Nat. Commun. 2020, 11, 2332. [Google Scholar] [CrossRef] [PubMed]
- Magrì, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chilà, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay8707. [Google Scholar] [CrossRef] [PubMed]
- Bedhiafi, T.; Inchakalody, V.P.; Fernandes, Q.; Mestiri, S.; Billa, N.; Uddin, S.; Merhi, M.; Dermime, S. The potential role of vitamin C in empowering cancer immunotherapy. Biomed. Pharmacother. 2021, 146, 112553. [Google Scholar] [CrossRef]
- Li, W.-N.; Zhang, S.-J.; Feng, J.-Q.; Jin, W.-L. Repurposing Vitamin C for Cancer Treatment: Focus on Targeting the Tumor Microenvironment. Cancers 2022, 14, 2608. [Google Scholar] [CrossRef]
- Das, P.K.; Pillai, S.; Rakib, M.A.; Khanam, J.A.; Gopalan, V.; Lam, A.K.Y.; Islam, F. Plasticity of Cancer Stem Cell: Origin and Role in Disease Progression and Therapy Resistance. Stem Cell Rev. Rep. 2020, 16, 397–412. [Google Scholar] [CrossRef]
- Tanabe, S.; Quader, S.; Cabral, H.; Ono, R. Interplay of EMT and CSC in Cancer and the Potential Therapeutic Strategies. Front. Pharmacol. 2020, 11, 904. [Google Scholar] [CrossRef]
- Kumar, V.E.; Nambiar, R.; De Souza, C.; Nguyen, A.; Chien, J.; Lam, K.S. Targeting Epigenetic Modifiers of Tumor Plasticity and Cancer Stem Cell Behavior. Cells 2022, 11, 1403. [Google Scholar] [CrossRef]
- Yadav, U.P.; Singh, T.; Kumar, P.; Sharma, P.; Kaur, H.; Sharma, S.; Singh, S.; Kumar, S.; Mehta, K. Metabolic Adaptations in Cancer Stem Cells. Front. Oncol. 2020, 10, 1010. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Gallo, C.; Pajardi, G.; Noonan, D.M.; Dallaglio, K. Cancer stem cells and the tumor microenvironment: Interplay in tumor heterogeneity. Connect. Tissue Res. 2015, 56, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Shi, X.; Jiang, M.; Liu, H. Cross-talk between cancer stem cells and immune cells: Potential therapeutic targets in the tumor immune microenvironment. Mol. Cancer 2023, 22, 38. [Google Scholar] [CrossRef] [PubMed]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Kim, J.H. Cancer stem cell metabolism: Target for cancer therapy. BMB Rep. 2018, 51, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629 . [Google Scholar] [CrossRef]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef]
- Du, F.Y.; Zhou, Q.F.; Sun, W.J.; Chen, G.L. Targeting cancer stem cells in drug discovery: Current state and future perspectives. World J. Stem Cells 2019, 11, 398–420. [Google Scholar] [CrossRef]
- Lee, Y.; Tanggono, A.S. Potential Role of the Circadian Clock in the Regulation of Cancer Stem Cells and Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 14181. [Google Scholar] [CrossRef] [PubMed]
- Basak, D.; Arrighi, S.; Darwiche, Y.; Deb, S. Comparison of Anticancer Drug Toxicities: Paradigm Shift in Adverse Effect Profile. Life 2021, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Meerson, A.; Khatib, S.; Mahajna, J. Natural Products Targeting Cancer Stem Cells for Augmenting Cancer Therapeutics. Int. J. Mol. Sci. 2021, 22, 13044. [Google Scholar] [CrossRef]
- Telang, N.T.; Nair, H.B.; Wong, G.Y.C. Growth Inhibitory Efficacy of Chinese Herbs in a Cellular Model for Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 1318. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e1020. [Google Scholar] [CrossRef] [PubMed]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Wang, C.; Fang, T.; Li, T.; Lv, G.; Han, Q.; Yang, W.; Wang, H. Vitamin C preferentially kills cancer stem cells in hepatocellular carcinoma via SVCT-2. NPJ Precis. Oncol. 2018, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Zhou, J.; Fu, L.; Li, Y.; Zeng, H.; Xu, X.; Lv, C.; Jin, H. Ascorbic Acid Inhibits Liver Cancer Growth and Metastasis. Front. Pharmacol. 2021, 12, 726015. [Google Scholar] [CrossRef]
- Bonuccelli, G.; De Francesco, E.M.; de Boer, R.; Tanowitz, H.B.; Lisanti, M.P. NADH autofluorescence, a new metabolic biomarker for cancer stem cells: Identification of Vitamin C and CAPE as natural products targeting “stemness”. Oncotarget 2017, 8, 20667–20678. [Google Scholar] [CrossRef]
- Sen, U.; Chaudhury, D.; Shenoy, P.S.; Bose, B. Differential sensitivities of triple-negative breast cancer stem cell towards various doses of vitamin C: An insight into the internal antioxidant systems. J. Cell Biochem. 2021, 122, 349–366. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, J.; Mao, J.; Han, W.; Fan, Y.; Luo, T.; Xia, J.; Lee, M.J.; Lin, W. Pharmacological ascorbate potentiates combination nanomedicines and reduces cancer cell stemness to prevent post-surgery recurrence and systemic metastasis. Biomaterials 2023, 295, 122037. [Google Scholar] [CrossRef] [PubMed]
- El Banna, N.; Hatem, E.; Heneman-Masurel, A.; Léger, T.; Baïlle, D.; Vernis, L.; Garcia, C.; Martineau, S.; Dupuy, C.; Vagner, S.; et al. Redox modifications of cysteine-containing proteins, cell cycle arrest and translation inhibition: Involvement in vitamin C-induced breast cancer cell death. Redox Biol. 2019, 26, 101290. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.; Chen, P.; Wilkins, H.M.; Wang, T.; Swerdlow, R.H.; Chen, Q. Pharmacologic ascorbate induces neuroblastoma cell death by hydrogen peroxide mediated DNA damage and reduction in cancer cell glycolysis. Free Radic. Biol. Med. 2017, 113, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Neel, B.G.; Aifantis, I. Vitamin C in Stem Cell Reprogramming and Cancer. Trends Cell Biol. 2018, 28, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Maurya, V.K.; Shakya, A.; McClements, D.J.; Srinivasan, R.; Bashir, K.; Ramesh, T.; Lee, J.; Sathiyamoorthi, E. Vitamin C fortification: Need and recent trends in encapsulation technologies. Front. Nutr. 2023, 10, 1229243. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, O.; Keith, M.O. Bioavailability of synthetic and natural ascorbic acid. J. Am. Diet. Assoc. 1974, 64, 271–275. [Google Scholar] [CrossRef]
- Carr, A.C.; Vissers, M.C. Synthetic or food-derived vitamin C—Are they equally bioavailable? Nutrients 2013, 5, 4284–4304. [Google Scholar] [CrossRef]
- Carr, A.C.; Bozonet, S.M.; Vissers, M.C. A randomised cross-over pharmacokinetic bioavailability study of synthetic versus kiwifruit-derived vitamin C. Nutrients 2013, 5, 4451–4461. [Google Scholar] [CrossRef]
- Michels, A.J.; Hagen, T.M.; Frei, B. Human genetic variation influences vitamin C homeostasis by altering vitamin C transport and antioxidant enzyme function. Annu. Rev. Nutr. 2013, 33, 45–70. [Google Scholar] [CrossRef]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef]
- Wang, H.; Dutta, B.; Huang, W.; Devoe, L.D.; Leibach, F.H.; Ganapathy, V.; Prasad, P.D. Human Na(+)-dependent vitamin C transporter 1 (hSVCT1): Primary structure, functional characteristics and evidence for a non-functional splice variant. Biochim. Biophys. Acta 1999, 1461, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Daruwala, R.; Song, J.; Koh, W.S.; Rumsey, S.C.; Levine, M. Cloning and functional characterization of the human sodium-dependent vitamin C transporters hSVCT1 and hSVCT2. FEBS Lett. 1999, 460, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mackenzie, B.; Tsukaguchi, H.; Weremowicz, S.; Morton, C.C.; Hediger, M.A. Human vitamin C (L-ascorbic acid) transporter SVCT1. Biochem. Biophys. Res. Commun. 2000, 267, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Pierce, M.R.; Raj, A.; Betke, K.M.; Zeidan, L.N.; Matthies, H.J.; May, J.M. Sodium-dependent vitamin C transporter-2 mediates vitamin C transport at the cortical nerve terminal. J. Neurosci. Res. 2015, 93, 1881–1890. [Google Scholar] [CrossRef] [PubMed]
- Salazar, K.; Espinoza, F.; Cerda-Gallardo, G.; Ferrada, L.; Magdalena, R.; Ramírez, E.; Ulloa, V.; Saldivia, N.; Troncoso, N.; Oviedo, M.J.; et al. SVCT2 Overexpression and Ascorbic Acid Uptake Increase Cortical Neuron Differentiation, Which Is Dependent on Vitamin C Recycling between Neurons and Astrocytes. Antioxidants 2021, 10, 1413. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Lee, S.H.; Moon, J.H.; Hwang, J.J.; Kim, D.E.; Ko, E.; Kim, H.S.; Cho, I.J.; Kang, J.S.; Kim, D.J.; et al. SVCT-2 in breast cancer acts as an indicator for L-ascorbate treatment. Oncogene 2013, 32, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 1997, 272, 18982–18989. [Google Scholar] [CrossRef]
- Rumsey, S.C.; Daruwala, R.; Al-Hasani, H.; Zarnowski, M.J.; Simpson, I.A.; Levine, M. Dehydroascorbic acid transport by GLUT4 in Xenopus oocytes and isolated rat adipocytes. J. Biol. Chem. 2000, 275, 28246–28253. [Google Scholar] [CrossRef]
- Corpe, C.P.; Eck, P.; Wang, J.; Al-Hasani, H.; Levine, M. Intestinal dehydroascorbic acid (DHA) transport mediated by the facilitative sugar transporters, GLUT2 and GLUT8. J. Biol. Chem. 2013, 288, 9092–9101. [Google Scholar] [CrossRef]
- Vera, J.C.; Rivas, C.I.; Fischbarg, J.; Golde, D.W. Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature 1993, 364, 79–82. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Griffiths, P.T.; Campbell, S.J.; Utinger, B.; Kalberer, M.; Paulson, S.E. Ascorbate oxidation by iron, copper and reactive oxygen species: Review, model development, and derivation of key rate constants. Sci. Rep. 2021, 11, 7417. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wagner, B.A.; Buettner, G.R.; Cullen, J.J. Role of labile iron in the toxicity of pharmacological ascorbate. Free Radic. Biol. Med. 2015, 84, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Carosio, R.; Zuccari, G.; Orienti, I.; Mangraviti, S.; Montaldo, P.G. Sodium ascorbate induces apoptosis in neuroblastoma cell lines by interfering with iron uptake. Mol. Cancer 2007, 6, 55. [Google Scholar] [CrossRef]
- Tsuma-Kaneko, M.; Sawanobori, M.; Kawakami, S.; Uno, T.; Nakamura, Y.; Onizuka, M.; Ando, K.; Kawada, H. Iron removal enhances vitamin C-induced apoptosis and growth inhibition of K-562 leukemic cells. Sci. Rep. 2018, 8, 17377. [Google Scholar] [CrossRef]
- Mojić, M.; Bogdanović Pristov, J.; Maksimović-Ivanić, D.; Jones, D.R.; Stanić, M.; Mijatović, S.; Spasojević, I. Extracellular iron diminishes anticancer effects of vitamin C: An in vitro study. Sci. Rep. 2014, 4, 5955. [Google Scholar] [CrossRef]
- Zhong, B.; Zhao, L.; Yu, J.; Hou, Y.; Ai, N.; Lu, J.J.; Ge, W.; Chen, X. Exogenous iron impairs the anti-cancer effect of ascorbic acid both in vitro and in vivo. J. Adv. Res. 2023, 46, 149–158. [Google Scholar] [CrossRef]
- Kim, J.E.; Kang, J.S.; Lee, W.J. Vitamin C Induces Apoptosis in Human Colon Cancer Cell Line, HCT-8 Via the Modulation of Calcium Influx in Endoplasmic Reticulum and the Dissociation of Bad from 14-3-3β. Immune Netw. 2012, 12, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Nam, A.; Song, K.H.; Lee, K.; Rebhun, R.B.; Seo, K.W. Anticancer effects of high-dose ascorbate on canine melanoma cell lines. Vet. Comp. Oncol. 2018, 16, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Chen, Y.; Qu, C.J.; Pan, Z.H.; Qin, Y.; Zhang, X.; Liu, W.J.; Li, D.F.; Zheng, Q. Vitamin C induces human melanoma A375 cell apoptosis via Bax- and Bcl-2-mediated mitochondrial pathways. Oncol. Lett. 2019, 18, 3880–3886. [Google Scholar] [CrossRef]
- Baek, M.W.; Cho, H.S.; Kim, S.H.; Kim, W.J.; Jung, J.Y. Ascorbic Acid Induces Necrosis in Human Laryngeal Squamous Cell Carcinoma via ROS, PKC, and Calcium Signaling. J. Cell Physiol. 2017, 232, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Hwang, S.; Lee, J.H.; Im, S.S.; Son, J. Vitamin C Suppresses Pancreatic Carcinogenesis through the Inhibition of Both Glucose Metabolism and Wnt Signaling. Int. J. Mol. Sci. 2022, 23, 12249. [Google Scholar] [CrossRef] [PubMed]
- Greco, G.; Catanzaro, E.; Fimognari, C. Natural Products as Inducers of Non-Canonical Cell Death: A Weapon against Cancer. Cancers 2021, 13, 304. [Google Scholar] [CrossRef]
- Chen, P.; Yu, J.; Chalmers, B.; Drisko, J.; Yang, J.; Li, B.; Chen, Q. Pharmacological ascorbate induces cytotoxicity in prostate cancer cells through ATP depletion and induction of autophagy. Anticancer Drugs 2012, 23, 437–444. [Google Scholar] [CrossRef]
- Buranasudja, V.; Doskey, C.M.; Gibson, A.R.; Wagner, B.A.; Du, J.; Gordon, D.J.; Koppenhafer, S.L.; Cullen, J.J.; Buettner, G.R. Pharmacologic Ascorbate Primes Pancreatic Cancer Cells for Death by Rewiring Cellular Energetics and Inducing DNA Damage. Mol. Cancer Res. 2019, 17, 2102–2114. [Google Scholar] [CrossRef]
- Zhang, X.; Li, S.; He, J.; Jin, Y.; Zhang, R.; Dong, W.; Lin, M.; Yang, Y.; Tian, T.; Zhou, Y.; et al. TET2 Suppresses VHL Deficiency-Driven Clear Cell Renal Cell Carcinoma by Inhibiting HIF Signaling. Cancer Res. 2022, 82, 2097–2109. [Google Scholar] [CrossRef]
- Gustafson, C.B.; Yang, C.; Dickson, K.M.; Shao, H.; Van Booven, D.; Harbour, J.W.; Liu, Z.J.; Wang, G. Epigenetic reprogramming of melanoma cells by vitamin C treatment. Clin. Epigenet. 2015, 7, 51. [Google Scholar] [CrossRef]
- Miles, S.L.; Fischer, A.P.; Joshi, S.J.; Niles, R.M. Ascorbic acid and ascorbate-2-phosphate decrease HIF activity and malignant properties of human melanoma cells. BMC Cancer 2015, 15, 867. [Google Scholar] [CrossRef] [PubMed]
- Brabson, J.P.; Leesang, T.; Yap, Y.S.; Wang, J.; Lam, M.Q.; Fang, B.; Dolgalev, I.; Barbieri, D.A.; Strippoli, V.; Bañuelos, C.P.; et al. Oxidized mC modulates synthetic lethality to PARP inhibitors for the treatment of leukemia. Cell Rep. 2023, 42, 112027. [Google Scholar] [CrossRef] [PubMed]
- Zasowska-Nowak, A.; Nowak, P.J.; Ciałkowska-Rysz, A. High-Dose Vitamin C in Advanced-Stage Cancer Patients. Nutrients 2021, 13, 735. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Sun, A.Y.; Chen, Q.; Espey, M.G.; Drisko, J.; Levine, M. Vitamin C: Intravenous use by complementary and alternative medicine practitioners and adverse effects. PLoS ONE 2010, 5, e11414. [Google Scholar] [CrossRef] [PubMed]
- Doseděl, M.; Jirkovský, E.; Macáková, K.; Krčmová, L.K.; Javorská, L.; Pourová, J.; Mercolini, L.; Remião, F.; Nováková, L.; Mladěnka, P.; et al. Vitamin C-Sources, Physiological Role, Kinetics, Deficiency, Use, Toxicity, and Determination. Nutrients 2021, 13, 615. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, W.; Dobrzyński, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef]
- Telang, N. Stem Cell Models for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 7055. [Google Scholar] [CrossRef]
- Telang, N. Drug-Resistant Stem Cells: Novel Approach for Colon Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 2519. [Google Scholar] [CrossRef]
- Telang, N. Isolation and Characterization of Chemo-Resistant Stem Cells from a Mouse Model of Hereditary Non-Polyposis Colon Cancer. Stem Cells Cloning 2021, 14, 19–25. [Google Scholar] [CrossRef]
- Uckun, F.M.; Sather, H.; Reaman, G.; Shuster, J.; Land, V.; Trigg, M.; Gunther, R.; Chelstrom, L.; Bleyer, A.; Gaynon, P. Leukemic cell growth in SCID mice as a predictor of relapse in high-risk B-lineage acute lymphoblastic leukemia. Blood 1995, 85, 873–878. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef]
- Doherty, M.R.; Smigiel, J.M.; Junk, D.J.; Jackson, M.W. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.L.; Zhao, Z.Q.; Li, J.C.; Liang, Y.; Yin, J.Q.; Zou, C.Y.; Xie, X.B.; Zeng, Y.X.; Shen, J.N.; Kang, T.; et al. Salinomycin inhibits osteosarcoma by targeting its tumor stem cells. Cancer Lett. 2011, 311, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, S.; Wang, Y.; Dai, W.; Zou, H.; Wang, S.; Zhang, J.; Pan, J. Salinomycin effectively eliminates cancer stem-like cells and obviates hepatic metastasis in uveal melanoma. Mol. Cancer 2019, 18, 159. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, S.; Serttas, R.; Turkekul, K.; Dibirdik, I. The synergistic anticancer effect of salinomycin combined with cabazitaxel in CD44+ prostate cancer cells by downregulating wnt, NF-κB and AKT signaling. Mol. Biol. Rep. 2022, 49, 4873–4884. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.; Liu, Y.; Li, J.; Huang, J.H.; Hu, X.; Wu, E. Salinomycin as a potent anticancer stem cell agent: State of the art and future directions. Med. Res. Rev. 2022, 42, 1037–1063. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, G.N.; Naccarato, A.G.; Scatena, C. Recent Advances in Cancer Plasticity: Cellular Mechanisms, Surveillance Strategies, and Therapeutic Optimization. Front. Oncol. 2020, 10, 569. [Google Scholar] [CrossRef]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef]
- Zheng, X.; Dai, F.; Feng, L.; Zou, H.; Xu, M. Communication Between Epithelial-Mesenchymal Plasticity and Cancer Stem Cells: New Insights Into Cancer Progression. Front. Oncol. 2021, 11, 617597. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; Hausen, A.Z.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial–Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Cell 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Jeanes, A.; Gottardi, C.; Yap, A. Cadherins and cancer: How does cadherin dysfunction promote tumor progression? Oncogene 2008, 27, 6920–6929. [Google Scholar] [CrossRef]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; Hogenesch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar] [CrossRef]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of their Normal Counterparts. Stem Cell Rep. 2013, 2, 78–91. [Google Scholar] [CrossRef]
- Beerling, E.; Seinstra, D.; Wit, E.D.; Kester, L.; Velden, D.v.D.; Maynard, C.; Schäfer, R.; Diest, P.V.; Voest, E.; Oudenaarden, A.V.; et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell 2016, 14, 2281–2288. [Google Scholar] [CrossRef]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef]
- van Denderen, B.J.; Thompson, E.W. Cancer: The to and fro of tumour spread. Nature 2013, 493, 487–488. [Google Scholar] [CrossRef]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, O.H.; Córcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Akhmetkaliyev, A.; Alibrahim, N.; Shafiee, D.; Tulchinsky, E. EMT/MET plasticity in cancer and Go-or-Grow decisions in quiescence: The two sides of the same coin? Mol. Cancer 2023, 22, 90. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Inguva, A.; Jordan, C.T. Targeting Energy Metabolism in Cancer Stem Cells: Progress and Challenges in Leukemia and Solid Tumors. Cell Stem Cell 2021, 28, 378–393. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Liu, C.; Jin, Y.; Fan, Z. The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 2021, 11, 698023. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cao, X.; Li, B.; Zhao, Z.; Chen, S.; Lai, S.W.T.; Muend, S.A.; Nossa, G.K.; Wang, L.; Guo, W.; et al. Warburg Effect Is a Cancer Immune Evasion Mechanism Against Macrophage Immunosurveillance. Front. Immunol. 2020, 11, 621757. [Google Scholar] [CrossRef] [PubMed]
- Facucho-Oliveira, J.M.; St John, J.C. The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev. Rep. 2009, 5, 140–158. [Google Scholar] [CrossRef]
- Panopoulos, A.D.; Yanes, O.; Ruiz, S.; Kida, Y.S.; Diep, D.; Tautenhahn, R.; Herrerías, A.; Batchelder, E.M.; Plongthongkum, N.; Lutz, M.; et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012, 22, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Liao, J.; Tang, Z.J.; Wu, W.J.; Yang, J.; Zeng, Z.L.; Hu, Y.; Wang, P.; Ju, H.Q.; Xu, R.H.; et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014, 21, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic alterations in highly tumorigenic glioblastoma cells: Preference for hypoxia and high dependency on glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Qian, F.; Tchabo, N.; Mhawech-Fauceglia, P.; Beck, A.; Qian, Z.; Wang, X.; Huss, W.J.; Lele, S.B.; Morrison, C.D.; et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS ONE 2014, 9, e84941. [Google Scholar] [CrossRef] [PubMed]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’Agostino, D.; et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef]
- Emmink, B.L.; Verheem, A.; Van Houdt, W.J.; Steller, E.J.; Govaert, K.M.; Pham, T.V.; Piersma, S.R.; Borel Rinkes, I.H.; Jimenez, C.R.; Kranenburg, O. The secretome of colon cancer stem cells contains drug-metabolizing enzymes. J. Proteom. 2013, 91, 84–96. [Google Scholar] [CrossRef]
- Palorini, R.; Votta, G.; Balestrieri, C.; Monestiroli, A.; Olivieri, S.; Vento, R.; Chiaradonna, F. Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J. Cell Biochem. 2014, 115, 368–379. [Google Scholar] [CrossRef]
- Tamada, M.; Nagano, O.; Tateyama, S.; Ohmura, M.; Yae, T.; Ishimoto, T.; Sugihara, E.; Onishi, N.; Yamamoto, T.; Yanagawa, H.; et al. Modulation of glucose metabolism by CD44 contributes to antioxidant status and drug resistance in cancer cells. Cancer Res. 2012, 72, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Q.; Fu, Y.L.; Zhang, J.; Zhang, K.Y.; Ma, J.; Tang, J.Y.; Zhang, Z.W.; Zhou, Z.Y. Targeting glycolysis in non-small cell lung cancer: Promises and challenges. Front. Pharmacol. 2022, 13, 1037341. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 126. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 2015, 6, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Deshpande, K.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Cancer stem cell metabolism: A potential target for cancer therapy. Mol. Cancer 2016, 15, 69. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The Role of PGC1α in Cancer Metabolism and its Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Fiorillo, M.; Peiris-Pagès, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Uthaya Kumar, D.B.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e637. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef]
- Li, J.; Eu, J.Q.; Kong, L.R.; Wang, L.; Lim, Y.C.; Goh, B.C.; Wong, A.L.A. Targeting Metabolism in Cancer Cells and the Tumour Microenvironment for Cancer Therapy. Molecules 2020, 25, 4831. [Google Scholar] [CrossRef] [PubMed]
- Satheesh, N.J.; Samuel, S.M.; Büsselberg, D. Combination Therapy with Vitamin C Could Eradicate Cancer Stem Cells. Biomolecules 2020, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Lee Chong, T.; Ahearn, E.L.; Cimmino, L. Reprogramming the Epigenome With Vitamin C. Front. Cell Dev. Biol. 2019, 7, 128. [Google Scholar] [CrossRef]
- Chung, T.L.; Brena, R.M.; Kolle, G.; Grimmond, S.M.; Berman, B.P.; Laird, P.W.; Pera, M.F.; Wolvetang, E.J. Vitamin C promotes widespread yet specific DNA demethylation of the epigenome in human embryonic stem cells. Stem Cells 2010, 28, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, K.; Zeng, X.; Yang, J.; Wu, Y.; Shi, X.; Qin, B.; Zeng, L.; Esteban, M.A.; Pan, G.; et al. The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell 2011, 9, 575–587. [Google Scholar] [CrossRef]
- Doege, C.A.; Inoue, K.; Yamashita, T.; Rhee, D.B.; Travis, S.; Fujita, R.; Guarnieri, P.; Bhagat, G.; Vanti, W.B.; Shih, A.; et al. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature 2012, 488, 652–655. [Google Scholar] [CrossRef]
- Costa, Y.; Ding, J.; Theunissen, T.W.; Faiola, F.; Hore, T.A.; Shliaha, P.V.; Fidalgo, M.; Saunders, A.; Lawrence, M.; Dietmann, S.; et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 2013, 495, 370–374. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, J.; Li, K.; Wu, T.; Huang, B.; Liu, W.; Kou, X.; Zhang, Y.; Huang, H.; Jiang, Y.; et al. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell 2013, 12, 453–469. [Google Scholar] [CrossRef]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674. [Google Scholar] [CrossRef] [PubMed]
- Guillamot, M.; Cimmino, L.; Aifantis, I. The Impact of DNA Methylation in Hematopoietic Malignancies. Trends Cancer 2016, 2, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Tefferi, A.; Lim, K.H.; Levine, R. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 361. author reply 1117–1118. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, L.; Dawlaty, M.M.; Pan, F.; Weeks, O.; Zhou, Y.; Cao, Z.; Shi, H.; Wang, J.; Lin, L.; et al. Combined Loss of Tet1 and Tet2 Promotes B Cell, but Not Myeloid Malignancies, in Mice. Cell Rep. 2015, 13, 1692–1704. [Google Scholar] [CrossRef]
- Cimmino, L.; Dawlaty, M.M.; Ndiaye-Lobry, D.; Yap, Y.S.; Bakogianni, S.; Yu, Y.; Bhattacharyya, S.; Shaknovich, R.; Geng, H.; Lobry, C.; et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat. Immunol. 2015, 16, 653–662. [Google Scholar] [CrossRef]
- Ko, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Thompson, E.C.; Hastie, R.; Tsangaratou, A.; Rajewsky, K.; Koralov, S.B.; Rao, A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 14566–14571. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef]
- Maeda, N.; Hagihara, H.; Nakata, Y.; Hiller, S.; Wilder, J.; Reddick, R. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc. Natl. Acad. Sci. USA 2000, 97, 841–846. [Google Scholar] [CrossRef]
- Tanabe, A.; Sahara, H. The Metabolic Heterogeneity and Flexibility of Cancer Stem Cells. Cancers 2020, 12, 2780. [Google Scholar] [CrossRef]
- Ambrose, J.M.; Veeraraghavan, V.P.; Vennila, R.; Rupert, S.; Sathyanesan, J.; Meenakshisundaram, R.; Selvaraj, S.; Malayaperumal, S.; Kullappan, M.; Dorairaj, S.; et al. Comparison of mammosphere formation from stem-like cells of normal breast, malignant primary breast tumors, and MCF-7 cell line. J. Egypt. Natl. Cancer Inst. 2022, 34, 51. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.H.; Wang, Q.M.; Feng, L.Y.; Ke, Y.D.; Xu, Q.Z.; Wei, A.Y.; Zhang, C.; Ying, R.B. High-dose vitamin C suppresses the invasion and metastasis of breast cancer cells via inhibiting epithelial-mesenchymal transition. Onco Targets Ther. 2019, 12, 7405–7413. [Google Scholar] [CrossRef] [PubMed]
- Cieslak, J.A.; Cullen, J.J. Treatment of Pancreatic Cancer with Pharmacological Ascorbate. Curr. Pharm. Biotechnol. 2015, 16, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Cancer Stem Cell Type/Origin | Methods | Results | Ref. |
|---|---|---|---|
| Hematopoietic stem cells (HSCs) purified from the bone marrow of mice (Gulo−/−, Tet2fl/fl, Flt3ITD, Slc23a2−/−) or bone marrow aspirates collected from patients, aged 34–85, who were being assessed for lymphoma | Human hematopoietic cell purification Bone marrow reconstitution assays HSC culture Metabolomics to measure 5 hmC, 5 mC, and C by LC–MS/MS RNA-sequencing (RNA-seq) analysis | HSCs have high Vitamin C (VC) levels and ascorbate depletion increases HSC frequency. VC depletion reduces Tet2 activity in HSCs and progenitors in vivo. Low VC levels cooperate with Flt3ITD to promote myelopoiesis, in part, by reducing TET2 function, and cell-autonomously promote HSC function. Low VC levels accelerate leukemogenesis. | Agathocleous, et al. [36] |
| Primary mouse hematopoietic progenitor cells or bone marrow cells from TRE-TurboGFP-shTet2 and TRE-TurboGFP-shTet3 transgenic mice; Vav-tTA, Rosa-M2rtTA, and TRE-GFP-Ren mice; C57BL/6 B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice; and germ-line Tet2-deficient mice Human leukemia cell lines: HL60, MOLM13, K562, KG1, THP1, and KASUMI1 Diagnostic bone marrow aspirates obtained from acute myeloid leukemia (AML) patients | Primary AML colony formation and liquid differentiation assays Bone marrow competitive transplantation Global DNA methylation quantitation, RNS sequencing, bisulfite sequencing analysis 5-hydroxymethylcytosine DNA immunoprecipitation (5 hmeDIP), sequencing, and analysis | TET2 restoration reverses aberrant self-renewal of Tet2-deficient cells. TET2 restoration promotes DNA demethylation, differentiation, and cell death. VC treatment mimics TET2 restoration to block leukemia progression. VC treatment enhances leukemia cell sensitivity to PARP inhibition. | Cimmino, et al. [35] |
| Human hepatocellular carcinoma (HCC) and mouse liver cancer cells Patient-derived xenograft (PDX) liver tumors from human patients | Colony formation assays with HCC cells (HCC-LM3 and HuH-7 cells) and liver CSCs Cell viability and cell invasion assays Knockdown of SVCT-2 via shSVCT-2 plasmid transfection SVCT-2 immunohistochemistry staining in HCC tumors Microarrays In vivo xenograft assays using the HCC PDX model and PDXs | SVCT-2 is highly expressed in liver CSCs and is required for the maintenance of liver CSCs. SVCT-2 determines the differential susceptibility to pharmacological VC-induced cell death. Pharmacological VC (10 mM) preferentially eradicates liver CSCs in vitro. SVCT-2-dependent mechanisms of pharmacological VC-induced cell death. Pharmacological VC (4 g/kg) impairs tumor growth and eradicates liver CSCs in vivo. | Lv, et al. [37] |
| Huh7 and Hep3B HCC cell lines | 3D sphere formation and colony formation assays Cell viability analysis RT-qPCR H2O2 Assays In vivo xenograft assays | VC (0.5~1 mM) selectively inhibits the viability of liver cancer cells and liver CSCs in vitro. VC inhibits sphere formation and colony formation in liver cancer cells. VC (4 g/kg) prevents HCC xenograft tumor growth and metastasis in vivo. | Wan, et al. [38] |
| MCF7 human breast cancer cell line | CSC identification with a mitochondrial metabolism reporter (mPGC1α-eGFP-Puro-R) and NADH auto-fluorescence analysis 3D mammosphere formation assays Mitochondrial ROS/H2O2 detection assays Cell migration: in vitro scratch assays Metabolic flux analysis (MFA) | Mitochondrial biogenesis indicated by PGC1α reporter activity correlates with stemness. Increased mitochondrial ROS levels and H2O2 production contribute to stemness. Increased NAD(P)H levels directly correlate with stemness. VC (1~2 mM) blocks mammosphere formation. | Bonuccelli, et al. [39] |
| MDA-MB-231 and MDA-MB-468 Triple-negative breast cancer (TNBC) stem cells | Fluorescence-activated cell sorting (FACS) of CSC populations (CD44+/24−) Population doubling time (PDT)/cell proliferation assays Detection of ROS generation in CD44+/24− CSCs via fluorescence microscopy and nitroblue tetrazolium (NBT) assays Mitotracker staining assays and JC-1 staining for qualitative assessments of mitochondrial integrity and membrane potential (ΔΨm) | Breast CSC yields are ~80% from TNBC cell lines with different morphologies and similar doubling times. Treatment with VC (10~20 mM) leads to changes in morphology followed by proliferation inhibition in breast CSCs. VC-induced ROS production and mitochondrial damage in sorted breast CSCs occurs in a dose dependent manner, with pronounced effects on MDA-MB-231 CSCs compared to MDA-MB-468 CSCs. The antioxidant activities/redox alterations that occur upon VC treatment are correlated with the VC sensitivities of the CSCs. | Sen, et al. [40] |
| CT26, MC38, and 4T1 murine carcinoma cells | Synthesis and characterization of nanocarrier particles (NCPs): Carboplatin (Carb)/Docetaxel (DTX) and Oxaliplatin (OX)/SN-38 (active metabolite of irinotecan) Flow cytometry analysis of pluripotency factors (SOX2, OCT4, and NANOG) 3D sphere formation assays Metabolic flux analysis with fluorescence lifetime imaging microscopy (FLIM)/GAPDH activity assays/mitochondrial morphology and membrane potential assessments In vitro apoptosis/cytotoxicity assays In vivo orthotopic xenografts and measurements of tumor growth/metastasis | VC (5 mM) enhances the cytotoxicity of NCPs against CSCs in vitro. VC transitions CSCs from glycolysis to mtOXPHOS and inhibits CSC self-renewal. VC (4 g/kg) potentiates the antitumor efficacy of NCPs and reduces tumor cell stemness in vivo. VC and NCPs in combination treatments prevent post-surgery relapse and inhibit systemic metastasis. | Jiang, et al. [41] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y. Role of Vitamin C in Targeting Cancer Stem Cells and Cellular Plasticity. Cancers 2023, 15, 5657. https://doi.org/10.3390/cancers15235657
Lee Y. Role of Vitamin C in Targeting Cancer Stem Cells and Cellular Plasticity. Cancers. 2023; 15(23):5657. https://doi.org/10.3390/cancers15235657
Chicago/Turabian StyleLee, Yool. 2023. "Role of Vitamin C in Targeting Cancer Stem Cells and Cellular Plasticity" Cancers 15, no. 23: 5657. https://doi.org/10.3390/cancers15235657
APA StyleLee, Y. (2023). Role of Vitamin C in Targeting Cancer Stem Cells and Cellular Plasticity. Cancers, 15(23), 5657. https://doi.org/10.3390/cancers15235657