Cellular Plasticity in Mammary Gland Development and Breast Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Cellular Plasticity
2. The Evolving Model of a Mammary Epithelial Hierarchy
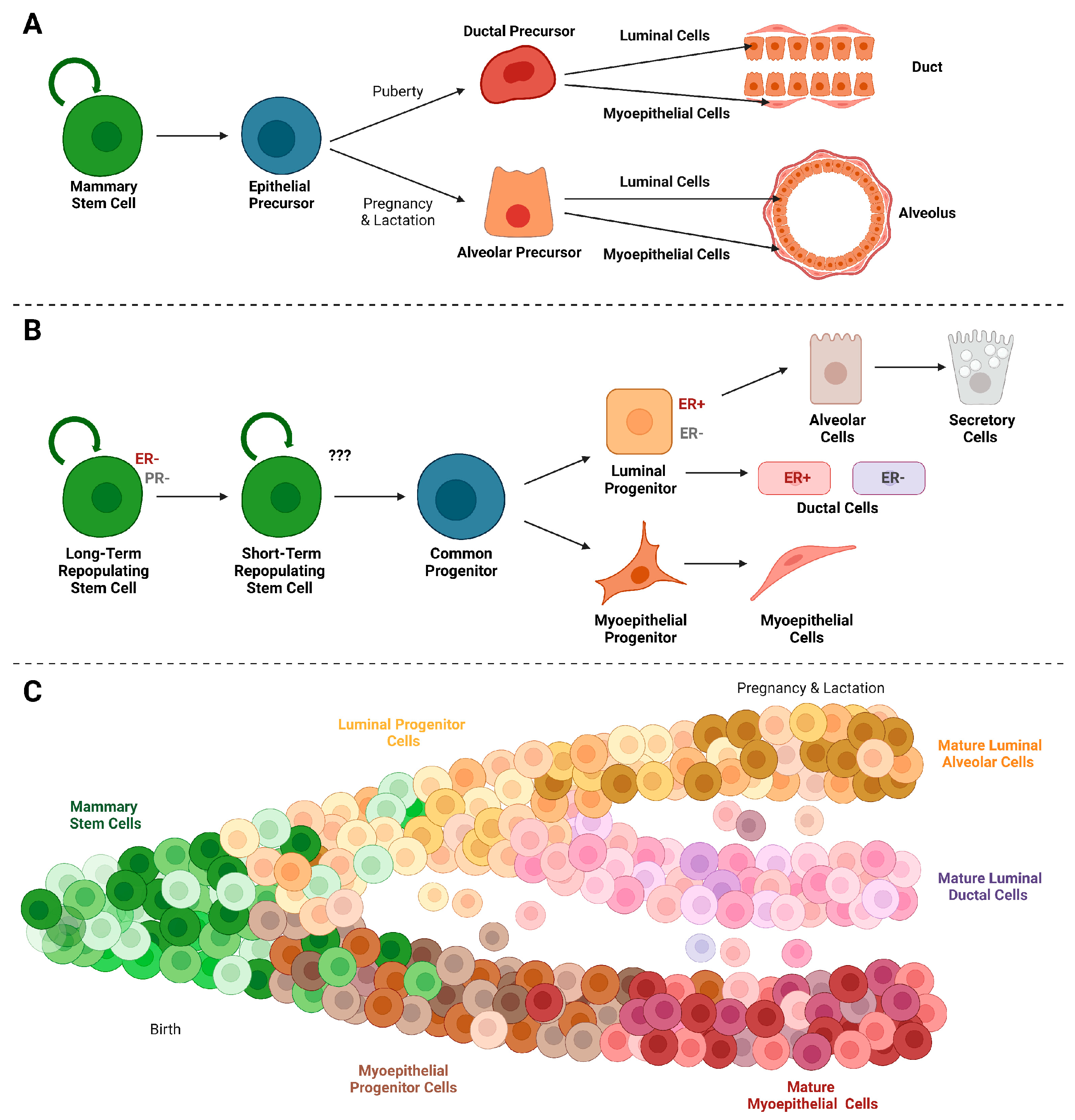
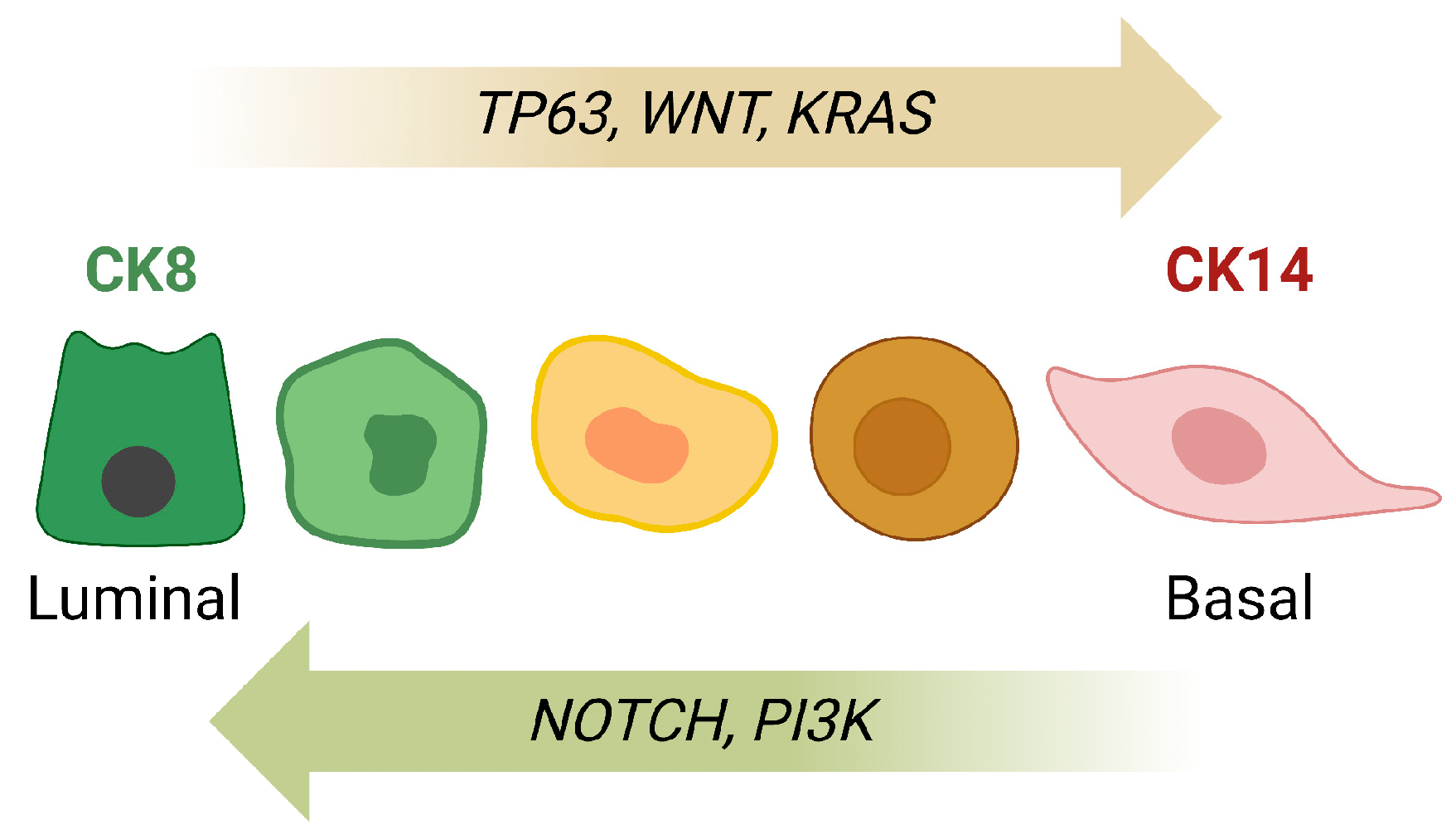
3. Cellular Plasticity in Normal Mammary Gland Development
4. Cellular and Molecular Mechanisms That Promote Plasticity in the Mammary Gland
5. Cellular Plasticity during Early Stages of Mammary Tumorigenesis
6. Cellular Plasticity during Breast Cancer Progression
7. Selection of Experimental Models to Study Cellular Plasticity in Tumor Initiation and Progression
8. Summary and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADM | Acinar-to-ductal metaplasia |
| ALDH1A1 | Aldehyde dehydrogenase 1 family member A1 |
| BRCA1 | Breast and ovarian cancer susceptibility gene 1, early onset |
| CD24 | Cluster of differentiation 24, cell adhesion molecule |
| CD29 | Integrin beta 1 (ITGB1) |
| CD44 | Homing cell adhesion molecule, HCAM |
| CD49f | Integrin alpha 6 (ITGA6) |
| CD61 | Integrin beta 3 (ITGB3) |
| CDH2 | Neural cadherin (N-cadherin) |
| CK14 | Cytokeratin 14 (KRT14) |
| CK18 | Cytokeratin 18 (KRT18) |
| CK6 | Cytokeratin 6 (KRT6A) |
| CK8 | Cytokeratin 8 (KRT8) |
| CLBC | Claudin-low breast cancer |
| Cre | Cre recombinase from the P1 bacteriophage |
| CTNNB1 | Catenin beta 1, beta-catenin |
| ECM | Extracellular matrix |
| EF1 | Eef1a1 gene promoter |
| EMT | Epithelial-to-mesenchymal transition |
| EpCAM | Epithelial cellular adhesion molecule, CD326 |
| ER | Estrogen receptor |
| FLP, FRT | Flp recombinase from yeast |
| FRT | FLP recombination target site |
| GFP | Green fluorescent protein |
| H2B | Histone H2B |
| HER2 | Human epidermal growth factor receptor 2 |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LacZ | Lactose operon, beta-galactosidase |
| LGR5 | Leucine-rich-repeat-containing G-protein-coupled receptor 5 |
| loxP | Locus of crossing over, recognition site of Cre recombinase |
| MMTV | Mouse mammary tumor virus |
| NOTCH | Family of conserved receptors discovered in Drosophila melanogaster |
| PIK3ca | Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit |
| PI-MECs | Parity-induced mammary epithelial cells |
| PR | Progesterone receptor |
| PyMT | Polyoma virus middle T antigen |
| Rosa26 | Ubiquitously active locus on chromosome 6 in mice |
| SCA1 | Lymphocyte antigen 6 family member A (Ly6a) |
| TAg | Simian virus 40 large T antigen |
| TEB | Terminal end buds |
| TetO | Tetracycline-controlled operator/promoter |
| TNBC | Triple-negative breast cancer |
| tTA | Tetracycline-controlled transactivator |
| TP63 | Transformation-related protein 63, p63 |
| VIM | Vimentin, type III intermediate filament protein |
| WAP | Whey acidic protein, milk protein |
| WNT | Wingless, integration site 1 (int1), signal transduction pathway |
References
- Merrell, A.J.; Stanger, B.Z. Adult cell plasticity in vivo: De-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol. 2016, 17, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Scheibner, K.; Schirge, S.; Burtscher, I.; Büttner, M.; Sterr, M.; Yang, D.; Böttcher, A.; Ansarullah; Irmler, M.; Beckers, J.; et al. Epithelial cell plasticity drives endoderm formation during gastrulation. Nat. Cell Biol. 2021, 23, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Spatz, L.B.; Jin, R.U.; Mills, J.C. Cellular plasticity at the nexus of development and disease. Development 2021, 148, dev197392. [Google Scholar] [CrossRef] [PubMed]
- Tata, A.; Chow, R.D.; Tata, P.R. Epithelial cell plasticity: Breaking boundaries and changing landscapes. EMBO Rep. 2021, 22, e51921. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Dennaoui, R.; Shrestha, H.; Wagner, K.U. Models of pancreatic ductal adenocarcinoma. Cancer Metastasis Rev. 2021, 40, 803–818. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef]
- Lee, E.; Piranlioglu, R.; Wicha, M.S.; Korkaya, H. Plasticity and Potency of Mammary Stem Cell Subsets During Mammary Gland Development. Int. J. Mol. Sci. 2019, 20, 2357. [Google Scholar] [CrossRef]
- Kong, D.; Hughes, C.J.; Ford, H.L. Cellular Plasticity in Breast Cancer Progression and Therapy. Front. Mol. Biosci. 2020, 7, 72. [Google Scholar] [CrossRef]
- DeOme, K.B.; Faulkin, L.J.; Bern, H.A.; Blair, P.E. Development of mammary tumors from hyperplastic alveolar nodules transplanted into gland-free mammary fat pads of female C3H mice. Cancer Res. 1959, 19, 515–520. [Google Scholar]
- Young, L.J.; Medina, D.; DeOme, K.B.; Daniel, C.W. The influence of host and tissue age on life span and growth rate of serially transplanted mouse mammary gland. Exp. Gerontol. 1971, 6, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.W.; De Ome, K.B.; Young, J.T.; Blair, P.B.; Faulkin, L.J., Jr. The in vivo life span of normal and preneoplastic mouse mammary glands: A serial transplantation study. Proc. Natl. Acad. Sci. USA 1968, 61, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.W.; Young, L.J.; Medina, D.; DeOme, K.B. The influence of mammogenic hormones on serially transplanted mouse mammary gland. Exp. Gerontol. 1971, 6, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Kordon, E.C.; Smith, G.H. An entire functional mammary gland may comprise the progeny from a single cell. Development 1998, 125, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.H. Experimental mammary epithelial morphogenesis in an in vivo model: Evidence for distinct cellular progenitors of the ductal and lobular phenotype. Breast Cancer Res. Treat. 1996, 39, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Hennighausen, L.; Robinson, G.W. Information networks in the mammary gland. Nat. Rev. Mol. Cell Biol. 2005, 6, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Stingl, J.; Eirew, P.; Ricketson, I.; Shackleton, M.; Vaillant, F.; Choi, D.; Li, H.I.; Eaves, C.J. Purification and unique properties of mammary epithelial stem cells. Nature 2006, 439, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, M.; Vaillant, F.; Simpson, K.J.; Stingl, J.; Smyth, G.K.; Asselin-Labat, M.L.; Wu, L.; Lindeman, G.J.; Visvader, J.E. Generation of a functional mammary gland from a single stem cell. Nature 2006, 439, 84–88. [Google Scholar] [CrossRef]
- Sleeman, K.E.; Kendrick, H.; Ashworth, A.; Isacke, C.M.; Smalley, M.J. CD24 staining of mouse mammary gland cells defines luminal epithelial, myoepithelial/basal and non-epithelial cells. Breast Cancer Res. 2006, 8, R7. [Google Scholar] [CrossRef]
- Shehata, M.; Teschendorff, A.; Sharp, G.; Novcic, N.; Russell, I.A.; Avril, S.; Prater, M.; Eirew, P.; Caldas, C.; Watson, C.J.; et al. Phenotypic and functional characterisation of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. BCR 2012, 14, R134. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Mammary stem cells and mammopoiesis. Cancer Res. 2006, 66, 9798–9801. [Google Scholar] [CrossRef] [PubMed]
- George, A.L.; Smith, G.H. Mammary Epithelial Cell Lineage Analysis via the Lyon’s Hypothesis. Int. J. Stem Cell Res. Ther. 2016, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Rocha, A.S.; Ousset, M.; Beck, B.; Bouvencourt, G.; Rock, J.; Sharma, N.; Dekoninck, S.; Blanpain, C. Distinct stem cells contribute to mammary gland development and maintenance. Nature 2011, 479, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Wuidart, A.; Ousset, M.; Rulands, S.; Simons, B.D.; Van Keymeulen, A.; Blanpain, C. Quantitative lineage tracing strategies to resolve multipotency in tissue-specific stem cells. Genes. Dev. 2016, 30, 1261–1277. [Google Scholar] [CrossRef] [PubMed]
- Wuidart, A.; Sifrim, A.; Fioramonti, M.; Matsumura, S.; Brisebarre, A.; Brown, D.; Centonze, A.; Dannau, A.; Dubois, C.; Van Keymeulen, A.; et al. Early lineage segregation of multipotent embryonic mammary gland progenitors. Nat. Cell Biol. 2018, 20, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Giraddi, R.R.; Chung, C.Y.; Heinz, R.E.; Balcioglu, O.; Novotny, M.; Trejo, C.L.; Dravis, C.; Hagos, B.M.; Mehrabad, E.M.; Rodewald, L.W.; et al. Single-Cell Transcriptomes Distinguish Stem Cell State Changes and Lineage Specification Programs in Early Mammary Gland Development. Cell Rep. 2018, 24, 1653–1666.e7. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.H.; Medina, D. Does the Mouse Mammary Gland Arise from Unipotent or Multipotent Mammary Stem/Progenitor Cells? J. Mammary Gland Biol. Neoplasia 2018, 23, 1–3. [Google Scholar] [CrossRef]
- Ragle, L.E.; Bruno, R.D.; Boulanger, C.A.; Smith, G.H. Long-label-retaining mammary epithelial cells are created early in ductal development and distributed throughout the branching ducts. Mech. Dev. 2019, 159, 103565. [Google Scholar] [CrossRef]
- Chhabra, S.N.; Booth, B.W. Asymmetric cell division of mammary stem cells. Cell Div. 2021, 16, 5. [Google Scholar] [CrossRef]
- Rodilla, V.; Fre, S. Cellular Plasticity of Mammary Epithelial Cells Underlies Heterogeneity of Breast Cancer. Biomedicines 2018, 6, 103. [Google Scholar] [CrossRef]
- Anstine, L.J.; Keri, R. A new view of the mammary epithelial hierarchy and its implications for breast cancer initiation and metastasis. J. Cancer Metastasis Treat. 2019, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Pal, B.; Chen, Y.; Vaillant, F.; Capaldo, B.D.; Joyce, R.; Song, X.; Bryant, V.L.; Penington, J.S.; Di Stefano, L.; Tubau Ribera, N.; et al. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. 2021, 40, e107333. [Google Scholar] [CrossRef]
- Sun, P.; Yuan, Y.; Li, A.; Li, B.; Dai, X. Cytokeratin expression during mouse embryonic and early postnatal mammary gland development. Histochem. Cell Biol. 2010, 133, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Miao, Z.; Zhang, X.; Chan, U.I.; Su, S.M.; Guo, S.; Wong, C.K.H.; Xu, X.; Deng, C.X. Single-cell RNA-Seq reveals cell heterogeneity and hierarchy within mouse mammary epithelia. J. Biol. Chem. 2018, 293, 8315–8329. [Google Scholar] [CrossRef] [PubMed]
- Bach, K.; Pensa, S.; Grzelak, M.; Hadfield, J.; Adams, D.J.; Marioni, J.C.; Khaled, W.T. Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing. Nat. Commun. 2017, 8, 2128. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.; Nee, K.; Wei, R.; He, S.; Nguyen, Q.H.; Bai, S.; Blake, K.; Pein, M.; Gong, Y.; Sei, E.; et al. A spatially resolved single-cell genomic atlas of the adult human breast. Nature 2023, 620, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Gray, G.K.; Li, C.M.; Rosenbluth, J.M.; Selfors, L.M.; Girnius, N.; Lin, J.R.; Schackmann, R.C.J.; Goh, W.L.; Moore, K.; Shapiro, H.K.; et al. A human breast atlas integrating single-cell proteomics and transcriptomics. Dev. Cell 2022, 57, 1400–1420.e7. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Campbell, K.H.; McWhir, J.; Ritchie, W.A.; Wilmut, I. Sheep cloned by nuclear transfer from a cultured cell line. Nature 1996, 380, 64–66. [Google Scholar] [CrossRef]
- Burdon, T.; Sankaran, L.; Wall, R.J.; Spencer, M.; Hennighausen, L. Expression of a whey acidic protein transgene during mammary development. Evidence for different mechanisms of regulation during pregnancy and lactation. J. Biol. Chem. 1991, 266, 6909–6914. [Google Scholar] [CrossRef]
- Triplett, A.A.; Sakamoto, K.; Matulka, L.A.; Shen, L.; Smith, G.H.; Wagner, K.U. Expression of the whey acidic protein (Wap) is necessary for adequate nourishment of the offspring but not functional differentiation of mammary epithelial cells. Genesis 2005, 43, 1–11. [Google Scholar] [CrossRef]
- Robinson, G.W.; McKnight, R.A.; Smith, G.H.; Hennighausen, L. Mammary epithelial cells undergo secretory differentiation in cycling virgins but require pregnancy for the establishment of terminal differentiation. Development 1995, 121, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.U.; Wall, R.J.; St-Onge, L.; Gruss, P.; Wynshaw-Boris, A.; Garrett, L.; Li, M.; Furth, P.A.; Hennighausen, L. Cre-mediated gene deletion in the mammary gland. Nucleic. Acids. Res. 1997, 25, 4323–4330. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat.Genet. 1999, 21, 70–71. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.U.; Boulanger, C.A.; Henry, M.D.; Sgagias, M.; Hennighausen, L.; Smith, G.H. An adjunct mammary epithelial cell population in parous females: Its role in functional adaptation and tissue renewal. Development 2002, 129, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.A.; Wagner, K.U.; Smith, G.H. Parity-induced mouse mammary epithelial cells are pluripotent, self-renewing and sensitive to TGF-beta1 expression. Oncogene 2005, 24, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Matulka, L.A.; Triplett, A.A.; Wagner, K.U. Parity-induced mammary epithelial cells are multipotent and express cell surface markers associated with stem cells. Dev. Biol. 2007, 303, 29–44. [Google Scholar] [CrossRef]
- Henry, M.D.; Triplett, A.A.; Oh, K.B.; Smith, G.H.; Wagner, K.U. Parity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic mice. Oncogene 2004, 23, 6980–6985. [Google Scholar] [CrossRef]
- Ahmed, F.; Wyckoff, J.; Lin, E.Y.; Wang, W.; Wang, Y.; Hennighausen, L.; Miyazaki, J.; Jones, J.; Pollard, J.W.; Condeelis, J.S.; et al. GFP expression in the mammary gland for imaging of mammary tumor cells in transgenic mice. Cancer Res. 2002, 62, 7166–7169. [Google Scholar]
- Creamer, B.A.; Triplett, A.A.; Wagner, K.U. Longitudinal analysis of mammogenesis using a novel tetracycline-inducible mouse model and in vivo imaging. Genesis 2009, 47, 234–245. [Google Scholar] [CrossRef]
- Booth, B.W.; Smith, G.H. Estrogen receptor-alpha and progesterone receptor are expressed in label-retaining mammary epithelial cells that divide asymmetrically and retain their template DNA strands. Breast Cancer Res. 2006, 8, R49. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.U.; Smith, G.H. Pregnancy and stem cell behavior. J. Mammary Gland. Biol. Neoplasia 2005, 10, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Kritikou, E.A.; Sharkey, A.; Abell, K.; Came, P.J.; Anderson, E.; Clarkson, R.W.; Watson, C.J. A dual, non-redundant, role for LIF as a regulator of development and STAT3-mediated cell death in mammary gland. Development 2003, 130, 3459–3468. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.S.; Lourenco, P.C.; Tonner, E.; Flint, D.J.; Selbert, S.; Takeda, K.; Akira, S.; Clarke, A.R.; Watson, C.J. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes. Dev. 1999, 13, 2604–2616. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, R.C.; Bierie, B.; Zhao, L.; Raz, R.; Levy, D.; Hennighausen, L. Deletion of Stat3 blocks mammary gland involution and extends functional competence of the secretory epithelium in the absence of lactogenic stimuli. Endocrinology 2002, 143, 3641–3650. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Wehde, B.L.; Yoo, K.H.; Kim, T.; Rajbhandari, N.; Shin, H.Y.; Triplett, A.A.; Radler, P.D.; Schuler, F.; Villunger, A.; et al. Janus Kinase 1 Is Essential for Inflammatory Cytokine Signaling and Mammary Gland Remodeling. Mol. Cell. Biol. 2016, 36, 1673–1690. [Google Scholar] [CrossRef] [PubMed]
- Schedin, P.; Mitrenga, T.; McDaniel, S.; Kaeck, M. Mammary ECM composition and function are altered by reproductive state. Mol. Carcinog. 2004, 41, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.D.; Fleming, J.M.; George, A.L.; Boulanger, C.A.; Schedin, P.; Smith, G.H. Mammary extracellular matrix directs differentiation of testicular and embryonic stem cells to form functional mammary glands in vivo. Sci. Rep. 2017, 7, 40196. [Google Scholar] [CrossRef]
- Booth, B.W.; Mack, D.L.; Androutsellis-Theotokis, A.; McKay, R.D.; Boulanger, C.A.; Smith, G.H. The mammary microenvironment alters the differentiation repertoire of neural stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 14891–14896. [Google Scholar] [CrossRef]
- Boulanger, C.A.; Bruno, R.D.; Rosu-Myles, M.; Smith, G.H. The mouse mammary microenvironment redirects mesoderm-derived bone marrow cells to a mammary epithelial progenitor cell fate. Stem Cells Dev. 2012, 21, 948–954. [Google Scholar] [CrossRef]
- Boulanger, C.A.; Mack, D.L.; Booth, B.W.; Smith, G.H. Interaction with the mammary microenvironment redirects spermatogenic cell fate in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 3871–3876. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, T.; Nishizuka, Y.; Dawe, C.J. Mesenchyme-dependent morphogenesis and epithelium-specific cytodifferentiation in mouse mammary gland. Science 1976, 194, 1439–1441. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R.; Young, P.; Christov, K.; Guzman, R.; Nandi, S.; Talamantes, F.; Thordarson, G. Mammary phenotypic expression induced in epidermal cells by embryonic mammary mesenchyme. Acta Anat. 1995, 152, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Bouras, T.; Pal, B.; Vaillant, F.; Harburg, G.; Asselin-Labat, M.L.; Oakes, S.R.; Lindeman, G.J.; Visvader, J.E. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell 2008, 3, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. BCR 2004, 6, R605–R615. [Google Scholar] [CrossRef] [PubMed]
- Buono, K.D.; Robinson, G.W.; Martin, C.; Shi, S.; Stanley, P.; Tanigaki, K.; Honjo, T.; Hennighausen, L. The canonical Notch/RBP-J signaling pathway controls the balance of cell lineages in mammary epithelium during pregnancy. Dev. Biol. 2006, 293, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Yalcin-Ozuysal, O.; Fiche, M.; Guitierrez, M.; Wagner, K.U.; Raffoul, W.; Brisken, C. Antagonistic roles of Notch and p63 in controlling mammary epithelial cell fates. Cell Death.Differ. 2010, 17, 1600–1612. [Google Scholar] [CrossRef]
- Gu, B.; Watanabe, K.; Sun, P.; Fallahi, M.; Dai, X. Chromatin effector Pygo2 mediates Wnt-notch crosstalk to suppress luminal/alveolar potential of mammary stem and basal cells. Cell Stem Cell 2013, 13, 48–61. [Google Scholar] [CrossRef]
- Holliday, H.; Baker, L.A.; Junankar, S.R.; Clark, S.J.; Swarbrick, A. Epigenomics of mammary gland development. Breast Cancer Res. BCR 2018, 20, 100. [Google Scholar] [CrossRef]
- Pal, B.; Bouras, T.; Shi, W.; Vaillant, F.; Sheridan, J.M.; Fu, N.; Breslin, K.; Jiang, K.; Ritchie, M.E.; Young, M.; et al. Global changes in the mammary epigenome are induced by hormonal cues and coordinated by Ezh2. Cell Rep. 2013, 3, 411–426. [Google Scholar] [CrossRef]
- Rädler, P.D.; Wehde, B.L.; Triplett, A.A.; Shrestha, H.; Shepherd, J.H.; Pfefferle, A.D.; Rui, H.; Cardiff, R.D.; Perou, C.M.; Wagner, K.U. Highly metastatic claudin-low mammary cancers can originate from luminal epithelial cells. Nat. Commun. 2021, 12, 3742. [Google Scholar] [CrossRef] [PubMed]
- Rädler, P.D.; Vistisen, K.; Triplett, A.A.; Dennaoui, R.; Li, Y.; Shrestha, H.; Ferraiuolo, R.-M.; Thangasamy, A.; Saur, D.; Wagner, K.-U. Dual recombinase action in the normal and neoplastic mammary gland epithelium. Sci. Rep. 2021, 11, 20775. [Google Scholar] [CrossRef] [PubMed]
- Best, S.A.; Hutt, K.J.; Fu, N.Y.; Vaillant, F.; Liew, S.H.; Hartley, L.; Scott, C.L.; Lindeman, G.J.; Visvader, J.E. Dual roles for Id4 in the regulation of estrogen signaling in the mammary gland and ovary. Development 2014, 141, 3159–3164. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, D.; Na, R.; Feuermann, Y.; Pechhold, S.; Chen, W.; Robinson, G.W.; Hennighausen, L. Development of mammary luminal progenitor cells is controlled by the transcription factor STAT5A. Genes. Dev. 2009, 23, 2382–2387. [Google Scholar] [CrossRef] [PubMed]
- Chiche, A.; Moumen, M.; Petit, V.; Jonkers, J.; Medina, D.; Deugnier, M.A.; Faraldo, M.M.; Glukhova, M.A. Somatic loss of p53 leads to stem/progenitor cell amplification in both mammary epithelial compartments, basal and luminal. Stem Cells 2013, 31, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Xiang, D.; Xie, Y.; Bronson, R.T.; Li, Z. Induced p53 loss in mouse luminal cells causes clonal expansion and development of mammary tumours. Nat. Commun. 2017, 8, 14431. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.R.; Naylor, M.J.; Asselin-Labat, M.L.; Blazek, K.D.; Gardiner-Garden, M.; Hilton, H.N.; Kazlauskas, M.; Pritchard, M.A.; Chodosh, L.A.; Pfeffer, P.L.; et al. The Ets transcription factor Elf5 specifies mammary alveolar cell fate. Genes. Dev. 2008, 22, 581–586. [Google Scholar] [CrossRef]
- Schaefer, F.V.; Custer, R.P.; Sorof, S. Squamous Metaplasia in Human Breast Culture: Induction by Cyclic Adenine Nucleotide and Prostaglandins, and Influence of Menstrual Cycle1. Cancer Res. 1983, 43, 279–286. [Google Scholar]
- Carter, B.A.; Page, D.L.; Schuyler, P.; Parl, F.F.; Simpson, J.F.; Jensen, R.A.; Dupont, W.D. No elevation in long-term breast carcinoma risk for women with fibroadenomas that contain atypical hyperplasia. Cancer 2001, 92, 30–36. [Google Scholar] [CrossRef]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef]
- Molyneux, G.; Geyer, F.C.; Magnay, F.A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; Mackay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010, 7, 403–417. [Google Scholar] [CrossRef]
- Smart, C.E.; Clarke, C.; Brooks, K.M.; Raghavendra, A.; Brewster, B.L.; French, J.D.; Hetherington, R.; Fleming, J.S.; Rothnagel, J.A.; Wainwright, B.; et al. Targeted disruption of Brca1 in restricted compartments of the mouse mammary epithelia. Breast Cancer Res. Treat. 2008, 112, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wagner, K.U.; Larson, D.; Weaver, Z.; Li, C.; Ried, T.; Hennighausen, L.; Wynshaw-Boris, A.; Deng, C.X. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat. Genet. 1999, 22, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Smith, M.D.; Chan, H.L.; Pei, X.H. Germline mutation of Brca1 alters the fate of mammary luminal cells and causes luminal-to-basal mammary tumor transformation. Oncogene 2013, 32, 2715–2725. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Zheng, C.; Liu, X.; Chan, H.L.; Liu, S.; Ma, J.; Ren, S.; Zhu, W.-G.; Pei, X.-H. Loss of function of GATA3 induces basal-like mammary tumors. Theranostics 2022, 12, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Reavie, L.; Couto, J.P.; De Silva, D.; Stadler, M.B.; Roloff, T.; Britschgi, A.; Eichlisberger, T.; Kohler, H.; Aina, O.; et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 2015, 525, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Lee, M.Y.; Ousset, M.; Brohée, S.; Rorive, S.; Giraddi, R.R.; Wuidart, A.; Bouvencourt, G.; Dubois, C.; Salmon, I.; et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015, 525, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Schachter, N.F.; Adams, J.R.; Skowron, P.; Kozma, K.J.; Lee, C.A.; Raghuram, N.; Yang, J.; Loch, A.J.; Wang, W.; Kucharczuk, A.; et al. Single allele loss-of-function mutations select and sculpt conditional cooperative networks in breast cancer. Nat. Commun. 2021, 12, 5238. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef]
- Pérez-González, A.; Bévant, K.; Blanpain, C. Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat. Cancer 2023, 4, 1063–1082. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Grasset, E.M.; Dunworth, M.; Sharma, G.; Loth, M.; Tandurella, J.; Cimino-Mathews, A.; Gentz, M.; Bracht, S.; Haynes, M.; Fertig, E.J.; et al. Triple-negative breast cancer metastasis involves complex epithelial-mesenchymal transition dynamics and requires vimentin. Sci. Transl. Med. 2022, 14, eabn7571. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef] [PubMed]
- Lüönd, F.; Sugiyama, N.; Bill, R.; Bornes, L.; Hager, C.; Tang, F.; Santacroce, N.; Beisel, C.; Ivanek, R.; Bürglin, T.; et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev. Cell 2021, 56, 3203–3221.e11. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lv, Z.; Zhang, S.; Wang, Z.; He, L.; Tang, M.; Pu, W.; Zhao, H.; Zhang, Z.; Shi, Q.; et al. Genetic Fate Mapping of Transient Cell Fate Reveals N-Cadherin Activity and Function in Tumor Metastasis. Dev. Cell 2020, 54, 593–607.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-Y.; Jiang, Z.; Ben-David, Y.; Woodgett, J.R.; Zacksenhaus, E. Molecular stratification within triple-negative breast cancer subtypes. Sci. Rep. 2019, 9, 19107. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. BCR 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- Fougner, C.; Bergholtz, H.; Norum, J.H.; Sorlie, T. Re-definition of claudin-low as a breast cancer phenotype. Nat. Commun. 2020, 11, 1787. [Google Scholar] [CrossRef]
- Cejalvo, J.M.; Martinez de Duenas, E.; Galvan, P.; Garcia-Recio, S.; Burgues Gasion, O.; Pare, L.; Antolin, S.; Martinello, R.; Blancas, I.; Adamo, B.; et al. Intrinsic Subtypes and Gene Expression Profiles in Primary and Metastatic Breast Cancer. Cancer Res. 2017, 77, 2213–2221. [Google Scholar] [CrossRef]
- Priedigkeit, N.; Hartmaier, R.J.; Chen, Y.; Vareslija, D.; Basudan, A.; Watters, R.J.; Thomas, R.; Leone, J.P.; Lucas, P.C.; Bhargava, R.; et al. Intrinsic Subtype Switching and Acquired ERBB2/HER2 Amplifications and Mutations in Breast Cancer Brain Metastases. JAMA Oncol. 2017, 3, 666–671. [Google Scholar] [CrossRef]
- Hulsbergen, A.F.C.; Claes, A.; Kavouridis, V.K.; Ansaripour, A.; Nogarede, C.; Hughes, M.E.; Smith, T.R.; Brastianos, P.K.; Verhoeff, J.J.C.; Lin, N.U.; et al. Subtype switching in breast cancer brain metastases: A multicenter analysis. Neuro-oncol. 2020, 22, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Recio, S.; Thennavan, A.; East, M.P.; Parker, J.S.; Cejalvo, J.M.; Garay, J.P.; Hollern, D.P.; He, X.; Mott, K.R.; Galván, P.; et al. FGFR4 regulates tumor subtype differentiation in luminal breast cancer and metastatic disease. J. Clin. Investig. 2020, 130, 4871–4887. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A.; Tan, A.R. Triple-negative breast cancer: Molecular subtypes and new targets for therapy. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, e31–e39. [Google Scholar] [CrossRef] [PubMed]
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; Rasmussen, K.E.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S.; et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007, 8, R76. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, A.; Kuperwasser, C. The origin of breast tumor heterogeneity. Oncogene 2015, 34, 5309–5316. [Google Scholar] [CrossRef] [PubMed]
- Gross, K.; Wronski, A.; Skibinski, A.; Phillips, S.; Kuperwasser, C. Cell Fate Decisions During Breast Cancer Development. J. Dev. Biol. 2016, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Pommier, R.M.; Sanlaville, A.; Tonon, L.; Kielbassa, J.; Thomas, E.; Ferrari, A.; Sertier, A.S.; Hollande, F.; Martinez, P.; Tissier, A.; et al. Comprehensive characterization of claudin-low breast tumors reflects the impact of the cell-of-origin on cancer evolution. Nat. Commun. 2020, 11, 3431. [Google Scholar] [CrossRef]
- Fougner, C.; Bergholtz, H.; Kuiper, R.; Norum, J.H.; Sorlie, T. Claudin-low-like mouse mammary tumors show distinct transcriptomic patterns uncoupled from genomic drivers. Breast Cancer Res. BCR 2019, 21, 85. [Google Scholar] [CrossRef]
- Wagner, K.U. Know thy cells: Commonly used triple-negative human breast cancer cell lines carry mutations in RAS and effectors. Breast Cancer Res. BCR 2022, 24, 44. [Google Scholar] [CrossRef]
- Matulka, L.A.; Wagner, K.U. Models of breast cancer. Drug Discov. Today Dis. Models 2005, 2, 1–6. [Google Scholar] [CrossRef]
- Sakamoto, K.; Schmidt, J.W.; Wagner, K.U. Mouse models of breast cancer. Methods Mol. Biol. 2015, 1267, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Attalla, S.; Taifour, T.; Bui, T.; Muller, W. Insights from transgenic mouse models of PyMT-induced breast cancer: Recapitulating human breast cancer progression in vivo. Oncogene 2021, 40, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, A.D.; Herschkowitz, J.I.; Usary, J.; Harrell, J.C.; Spike, B.T.; Adams, J.R.; Torres-Arzayus, M.I.; Brown, M.; Egan, S.E.; Wahl, G.M.; et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 2013, 14, R125. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, G.A.; Mahmood, S.; Ognjenovic, N.B.; Lee, M.K.; Wilkins, O.M.; Christensen, B.C.; Muller, K.E.; Pattabiraman, D.R. Lineage plasticity enables low-ER luminal tumors to evolve and gain basal-like traits. Breast Cancer Res. BCR 2023, 25, 23. [Google Scholar] [CrossRef] [PubMed]
- Podsypanina, K.; Du, Y.C.; Jechlinger, M.; Beverly, L.J.; Hambardzumyan, D.; Varmus, H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science 2008, 321, 1841–1844. [Google Scholar] [CrossRef]
- Moody, S.E.; Sarkisian, C.J.; Hahn, K.T.; Gunther, E.J.; Pickup, S.; Dugan, K.D.; Innocent, N.; Cardiff, R.D.; Schnall, M.D.; Chodosh, L.A. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell 2002, 2, 451–461. [Google Scholar] [CrossRef]
- Turpin, J.; Ling, C.; Crosby, E.J.; Hartman, Z.C.; Simond, A.M.; Chodosh, L.A.; Rennhack, J.P.; Andrechek, E.R.; Ozcelik, J.; Hallett, M.; et al. The ErbB2DeltaEx16 splice variant is a major oncogenic driver in breast cancer that promotes a pro-metastatic tumor microenvironment. Oncogene 2016, 35, 6053–6064. [Google Scholar] [CrossRef]
- Podsypanina, K.; Politi, K.; Beverly, L.J.; Varmus, H.E. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc. Natl. Acad. Sci. USA 2008, 105, 5242–5247. [Google Scholar] [CrossRef]
- Andrechek, E.R.; Hardy, W.R.; Siegel, P.M.; Rudnicki, M.A.; Cardiff, R.D.; Muller, W.J. Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 3444–3449. [Google Scholar] [CrossRef]
- Li, Z.; Tognon, C.E.; Godinho, F.J.; Yasaitis, L.; Hock, H.; Herschkowitz, J.I.; Lannon, C.L.; Cho, E.; Kim, S.J.; Bronson, R.T.; et al. ETV6-NTRK3 fusion oncogene initiates breast cancer from committed mammary progenitors via activation of AP1 complex. Cancer Cell 2007, 12, 542–558. [Google Scholar] [CrossRef]
- Klinakis, A.; Szabolcs, M.; Chen, G.; Xuan, S.; Hibshoosh, H.; Efstratiadis, A. Igf1r as a therapeutic target in a mouse model of basal-like breast cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 2359–2364. [Google Scholar] [CrossRef]
- Sakamoto, K.; Radler, P.D.; Wehde, B.L.; Triplett, A.A.; Shrestha, H.; Ferraiuolo, R.M.; Amari, F.; Coppola, V.; Klinakis, A.; Efstratiadis, A.; et al. Efficient tissue-type specific expression of target genes in a tetracycline-controlled manner from the ubiquitously active Eef1a1 locus. Sci. Rep. 2020, 10, 207. [Google Scholar] [CrossRef]
- Ewald, D.; Li, M.; Efrat, S.; Auer, G.; Wall, R.J.; Furth, P.A.; Hennighausen, L. Time-sensitive reversal of hyperplasia in transgenic mice expressing SV40 T antigen. Science 1996, 273, 1384–1386. [Google Scholar] [CrossRef]
- Kisseberth, W.C.; Sandgren, E.P. Polyclonal development of mouse mammary preneoplastic nodules. Cancer Res. 2004, 64, 857–863. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wicker, M.N.; Wagner, K.-U. Cellular Plasticity in Mammary Gland Development and Breast Cancer. Cancers 2023, 15, 5605. https://doi.org/10.3390/cancers15235605
Wicker MN, Wagner K-U. Cellular Plasticity in Mammary Gland Development and Breast Cancer. Cancers. 2023; 15(23):5605. https://doi.org/10.3390/cancers15235605
Chicago/Turabian StyleWicker, Madison N., and Kay-Uwe Wagner. 2023. "Cellular Plasticity in Mammary Gland Development and Breast Cancer" Cancers 15, no. 23: 5605. https://doi.org/10.3390/cancers15235605
APA StyleWicker, M. N., & Wagner, K.-U. (2023). Cellular Plasticity in Mammary Gland Development and Breast Cancer. Cancers, 15(23), 5605. https://doi.org/10.3390/cancers15235605