
TIL-Derived CAR T Cells Improve Immune Cell Infiltration and Survival in the Treatment of CD19-Humanized Mouse Colorectal Cancer

Abstract
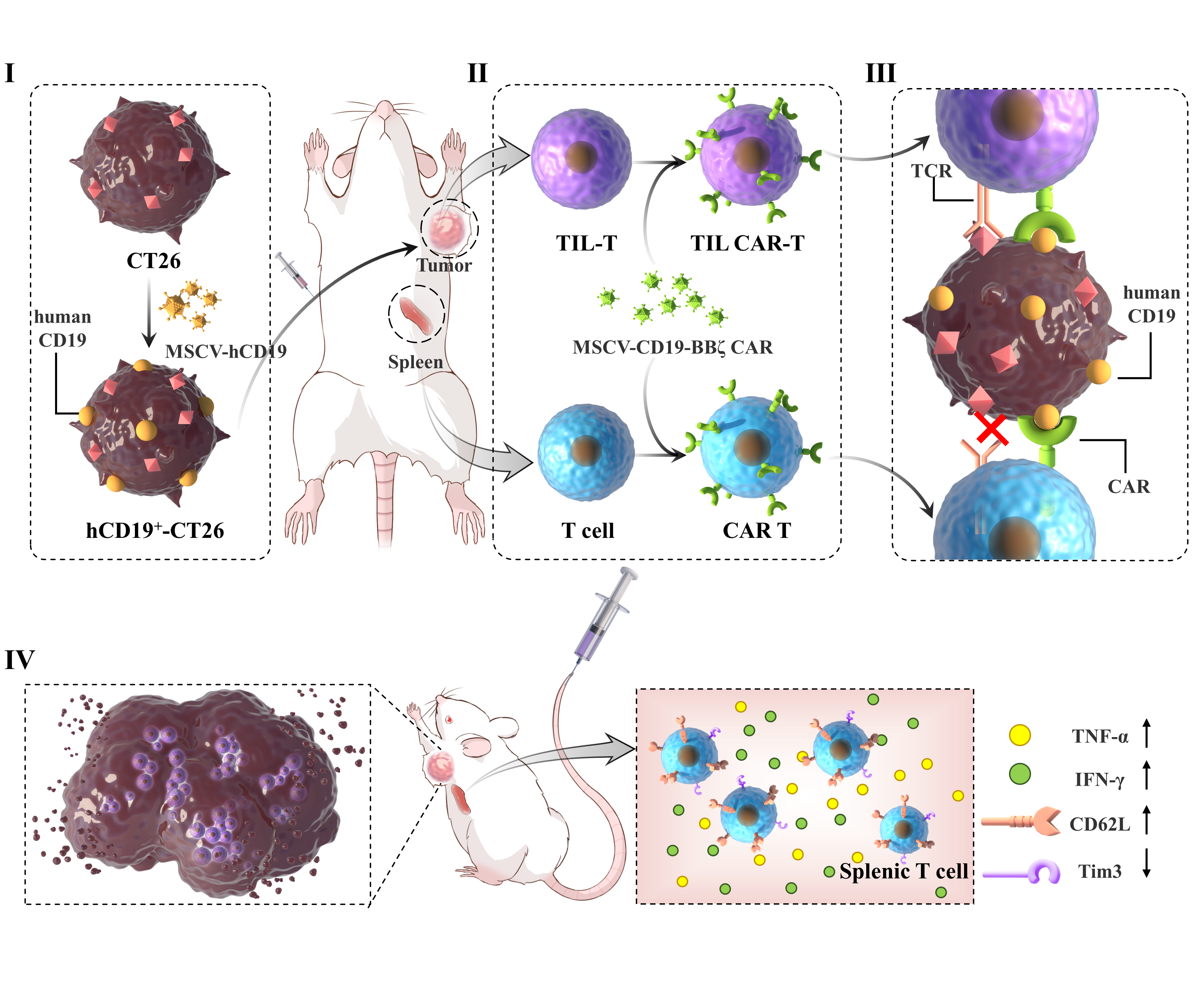
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Establishment of Human CD19-Expressed Tumor Cell Line
2.2. Construction of CD19-BBζ CAR Expression Plasmid and Retrovirus
2.3. Activation of Splenic T Cells from the BALB/c Mice
2.4. Construction of CD19-BBζ CAR T Cells (CAR T)
2.5. Establishment of Tumor-Bearing Mice
2.6. Preparation of TIL CD19-BBζ CAR-T Cells (TIL CAR-T)
2.7. Assessment of CD19-BBζ CAR T Cytotoxicity
2.8. Adoptive Cell Therapy (ACT)
2.9. Flow Cytometry Analysis
2.10. Histological Analysis
2.11. Statistical Analysis
3. Results
3.1. Infected Ratio of MSCV-CD19-BBζ CAR Retrovirus
3.2. Construction of hCD19-Expressed Tumor Cells
3.3. Effect of TIL CAR-T Cells on hCD19-Humanized CT26
3.4. TIL CAR-T Suppressed Tumor Growth In Vivo
3.5. TIL CAR-T Promoted Immune Cell Infiltration in the Tumor Tissues
3.6. TIL CAR-T Enhanced the Cytotoxicity of Splenic T Cells
3.7. TIL CAR-T Cells Increased the Frequency of Memory T Cells
3.8. TIL CAR-T Cells Downregulated the Expression of Tim3 in the Splenic T Cells
3.9. TIL CAR-T Did Not Harm Organs In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef]
- Han, E.Q.; Li, X.L.; Wang, C.R.; Li, T.F.; Han, S.Y. Chimeric antigen receptor-engineered T cells for cancer immunotherapy: Progress and challenges. J. Hematol. Oncol. 2013, 6, 47. [Google Scholar] [CrossRef]
- Abramson, J.S. Anti-CD19 CAR T-Cell Therapy for B-Cell Non-Hodgkin Lymphoma. Transfus. Med. Rev. 2020, 34, 29–33. [Google Scholar] [CrossRef]
- Annesley, C.E.; Summers, C.; Ceppi, F.; Gardner, R.A. The Evolution and Future of CAR T Cells for B-Cell Acute Lymphoblastic Leukemia. Clin. Pharmacol. Ther. 2018, 103, 591–598. [Google Scholar] [CrossRef]
- Young, R.M.; Engel, N.W.; Uslu, U.; Wellhausen, N.; June, C.H. Next-Generation CAR T-cell Therapies. Cancer Discov. 2022, 12, 1625–1633. [Google Scholar] [CrossRef]
- Sur, D.; Havasi, A.; Cainap, C.; Samasca, G.; Burz, C.; Balacescu, O.; Lupan, I.; Deleanu, D.; Irimie, A. Chimeric Antigen Receptor T-Cell Therapy for Colorectal Cancer. J. Clin. Med. 2020, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Razavi, A.; Keshavarz-Fathi, M.; Pawelek, J.; Rezaei, N. Chimeric antigen receptor T-cell therapy for melanoma. Expert Rev. Clin. Immunol. 2021, 17, 209–223. [Google Scholar] [CrossRef]
- Corti, C.; Venetis, K.; Sajjadi, E.; Zattoni, L.; Curigliano, G.; Fusco, N. CAR-T cell therapy for triple-negative breast cancer and other solid tumors: Preclinical and clinical progress. Expert Opin. Investig. Drugs 2022, 31, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front. Neurosci. 2021, 15, 662064. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, F.; Li, J.; Pu, Y.; Yang, C.; Wang, Y.; Lei, Y.; Huang, Y. CAR-T cell therapy for lung cancer: Potential and perspective. Thorac. Cancer 2022, 13, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, J.; Chen, K.; Ma, P.; Lei, Q.; Xing, S.; Cao, Z.; Sun, S.; Yu, Z.; Liu, Y.; et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021, 19, 140. [Google Scholar] [CrossRef] [PubMed]
- Granhøj, J.S.; Witness Præst Jensen, A.; Presti, M.; Met, Ö.; Svane, I.M.; Donia, M. Tumor-infiltrating lymphocytes for adoptive cell therapy: Recent advances, challenges, and future directions. Expert Opin. Biol. Ther. 2022, 22, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Zhao, Y.Y.; Shen, J.; Sun, X.; Liu, Y.; Liu, H.; Wang, Y.; Wang, J. Nanoenabled Modulation of Acidic Tumor Microenvironment Reverses Anergy of Infiltrating T Cells and Potentiates Anti-PD-1 Therapy. Nano Lett. 2019, 19, 2774–2783. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G. MHC class II-restricted tumor antigens recognized by CD4+ T cells: New strategies for cancer vaccine design. J. Immunother. 2001, 24, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wang, G.; Cheng, H.; Wei, C.; Qi, K.; Sang, W.; Zhenyu, L.; Shi, M.; Li, H.; Qiao, J.; et al. Potent anti-leukemia activities of humanized CD19-targeted Chimeric antigen receptor T (CAR-T) cells in patients with relapsed/refractory acute lymphoblastic leukemia. Am. J. Hematol. 2018, 93, 851–858. [Google Scholar] [CrossRef]
- Pacella, I.; Grimaldi, A.; Piconese, S. Assessment of lipid load in tumor-infiltrating Tregs by flow cytometry. Methods Enzymol. 2020, 632, 283–294. [Google Scholar] [CrossRef]
- Yamazumi, K.; Nakayama, T.; Kusaba, T.; Wen, C.Y.; Yoshizaki, A.; Yakata, Y.; Nagayasu, T.; Sekine, I. Expression of interleukin-11 and interleukin-11 receptor alpha in human colorectal adenocarcinoma; immunohistochemical analyses and correlation with clinicopathological factors. World J. Gastroenterol. 2006, 12, 317–321. [Google Scholar] [CrossRef]
- Yoshizaki, A.; Nakayama, T.; Yamazumi, K.; Yakata, Y.; Taba, M.; Sekine, I. Expression of interleukin (IL)-11 and IL-11 receptor in human colorectal adenocarcinoma: IL-11 up-regulation of the invasive and proliferative activity of human colorectal carcinoma cells. Int. J. Oncol. 2006, 29, 869–876. [Google Scholar] [CrossRef][Green Version]
- Cardó-Vila, M.; Marchiò, S.; Sato, M.; Staquicini, F.I.; Smith, T.L.; Bronk, J.K.; Yin, G.; Zurita, A.J.; Sun, M.; Behrens, C.; et al. Interleukin-11 Receptor Is a Candidate Target for Ligand-Directed Therapy in Lung Cancer: Analysis of Clinical Samples and BMTP-11 Preclinical Activity. Am. J. Pathol. 2016, 186, 2162–2170. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; He, Y.; Gu, C.; Jiang, J.; Zhou, H.; Zhou, S. Binding Specificity of Radiolabeled Cyclic Peptide 153Sm-DTPA-c(CGRRAGGSC) to MHCC97-H Human Liver Cancer Cells and its Antitumor Effects in vivo. Technol. Cancer Res. Treat. 2016, 15, NP1–NP9. [Google Scholar] [CrossRef]
- Jacobson, C.A. CD19 Chimeric Antigen Receptor Therapy for Refractory Aggressive B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Gong, J.; Li, J.; Liu, D.; Qin, Y.; Ge, S.; Zhang, M.; Peng, Z.; Zhou, J.; Cao, Y.; et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat. Med. 2022, 28, 1189–1198. [Google Scholar] [CrossRef]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.; Gileadi, T.; et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Guo, Y.; Dai, H.; Wang, Y.; Li, X.; Jia, H.; Han, W. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci. China Life Sci. 2016, 59, 468–479. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef]
- Castellarin, M.; Sands, C.; Da, T.; Scholler, J.; Graham, K.; Buza, E.; Fraietta, J.A.; Zhao, Y.; June, C.H. A rational mouse model to detect on-target, off-tumor CAR T cell toxicity. JCI Insight 2020, 5, e136012. [Google Scholar] [CrossRef]
- Guha, P.; Heatherton, K.R.; O’Connell, K.P.; Alexander, I.S.; Katz, S.C. Assessing the Future of Solid Tumor Immunotherapy. Biomedicines 2022, 10, 655. [Google Scholar] [CrossRef]
- Flugel, C.L.; Majzner, R.G.; Krenciute, G.; Dotti, G.; Riddell, S.R.; Wagner, D.L.; Abou-El-Enein, M. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat. Rev. Clin. Oncol. 2023, 20, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Du, L.; Li, H.; Zhu, X.; Cui, L.; Li, X. Tumor-infiltrating lymphocytes: Warriors fight against tumors powerfully. Biomed. Pharmacother. 2020, 132, 110873. [Google Scholar] [CrossRef] [PubMed]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309–5337. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, E.M.V.; Riise, R.; Saellström, S.; Karlsson, J.; Alsén, S.; Bucher, V.; Hemminki, A.E.; Olofsson Bagge, R.; Ny, L.; Nilsson, L.M.; et al. Treatment with Anti-HER2 Chimeric Antigen Receptor Tumor-Infiltrating Lymphocytes (CAR-TILs) Is Safe and Associated with Antitumor Efficacy in Mice and Companion Dogs. Cancers 2023, 15, 648. [Google Scholar] [CrossRef]
- Uche, I.K.; Stanfield, B.A.; Rudd, J.S.; Kousoulas, K.G.; Rider, P.J.F. Utility of a Recombinant HSV-1 Vaccine Vector for Personalized Cancer Vaccines. Front. Mol. Biosci. 2022, 9, 832393. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, C.; Zhao, Y.; He, J.; Zhao, H.; Ni, L.; Cheng, X.; Chen, Y.; Mu, L.; Zhou, X.; Shi, Q.; et al. TIL-Derived CAR T Cells Improve Immune Cell Infiltration and Survival in the Treatment of CD19-Humanized Mouse Colorectal Cancer. Cancers 2023, 15, 5567. https://doi.org/10.3390/cancers15235567
Zhu C, Zhao Y, He J, Zhao H, Ni L, Cheng X, Chen Y, Mu L, Zhou X, Shi Q, et al. TIL-Derived CAR T Cells Improve Immune Cell Infiltration and Survival in the Treatment of CD19-Humanized Mouse Colorectal Cancer. Cancers. 2023; 15(23):5567. https://doi.org/10.3390/cancers15235567
Chicago/Turabian StyleZhu, Can, Yuanyuan Zhao, Jiaheng He, Huan Zhao, Li Ni, Xinyi Cheng, Yida Chen, Liqian Mu, Xiaojun Zhou, Qin Shi, and et al. 2023. "TIL-Derived CAR T Cells Improve Immune Cell Infiltration and Survival in the Treatment of CD19-Humanized Mouse Colorectal Cancer" Cancers 15, no. 23: 5567. https://doi.org/10.3390/cancers15235567
APA StyleZhu, C., Zhao, Y., He, J., Zhao, H., Ni, L., Cheng, X., Chen, Y., Mu, L., Zhou, X., Shi, Q., & Sun, J. (2023). TIL-Derived CAR T Cells Improve Immune Cell Infiltration and Survival in the Treatment of CD19-Humanized Mouse Colorectal Cancer. Cancers, 15(23), 5567. https://doi.org/10.3390/cancers15235567