
Exploring the In Vitro and In Vivo Therapeutic Potential of BRAF and MEK Inhibitor Combination in NRAS-Mutated Melanoma
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Culture of Human Cells
2.2. Signaling Pathway Inhibitors and Treatments
2.3. Viability Assay
2.4. Cell Cycle Analysis
2.5. RNA Isolation and cDNA Synthesis
2.6. Quantitative Reverse Transcription–Polymerase Chain Reaction
2.7. Western Blot
2.8. Tumor Slice Culture
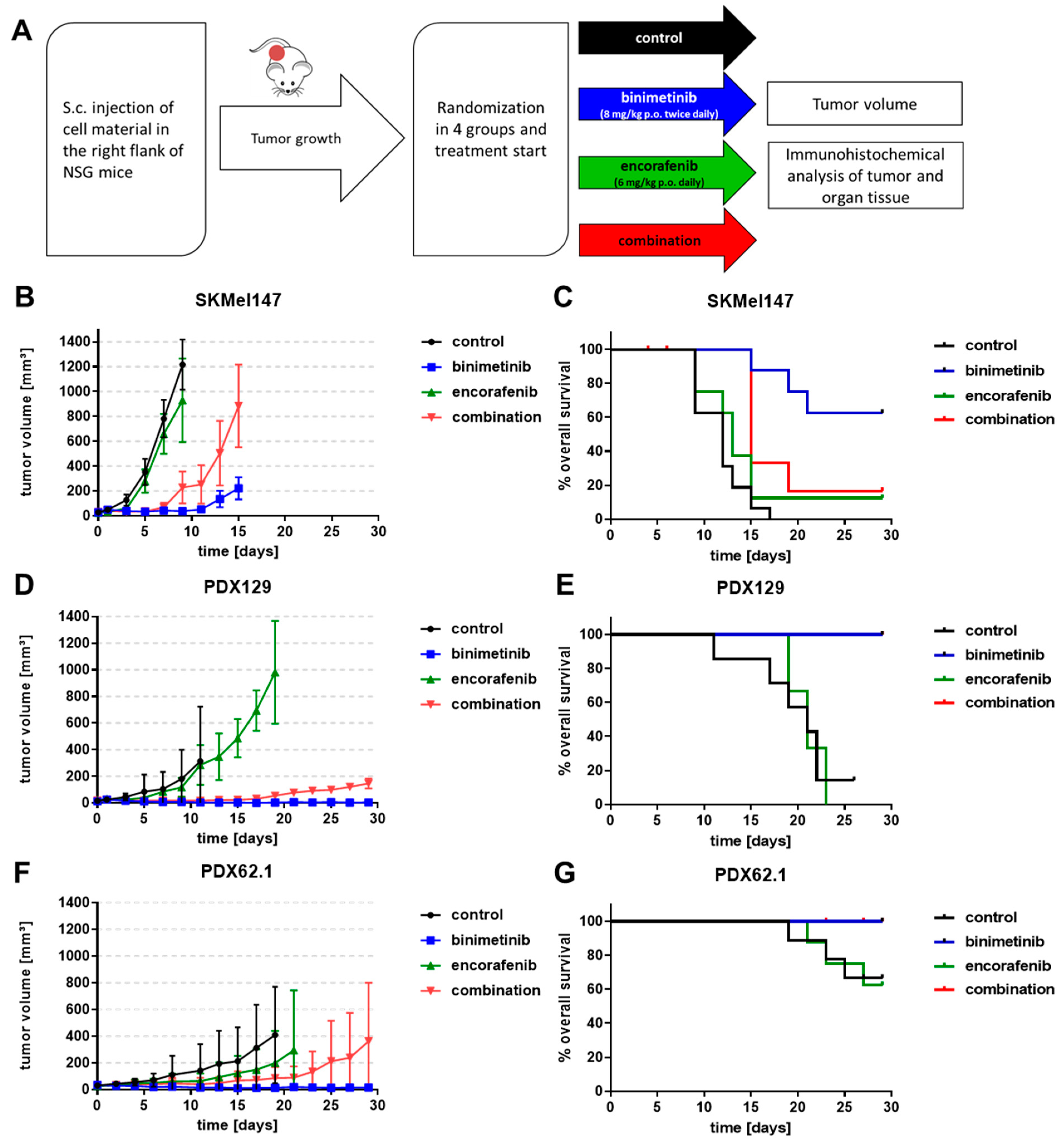
2.9. In Vivo Mouse Experiment
2.10. Immunohistochemistry of Mouse Tumors
2.11. Immunohistochemistry of Human Tumors
2.12. Individual Healing Experiment
2.13. Statistics
3. Results
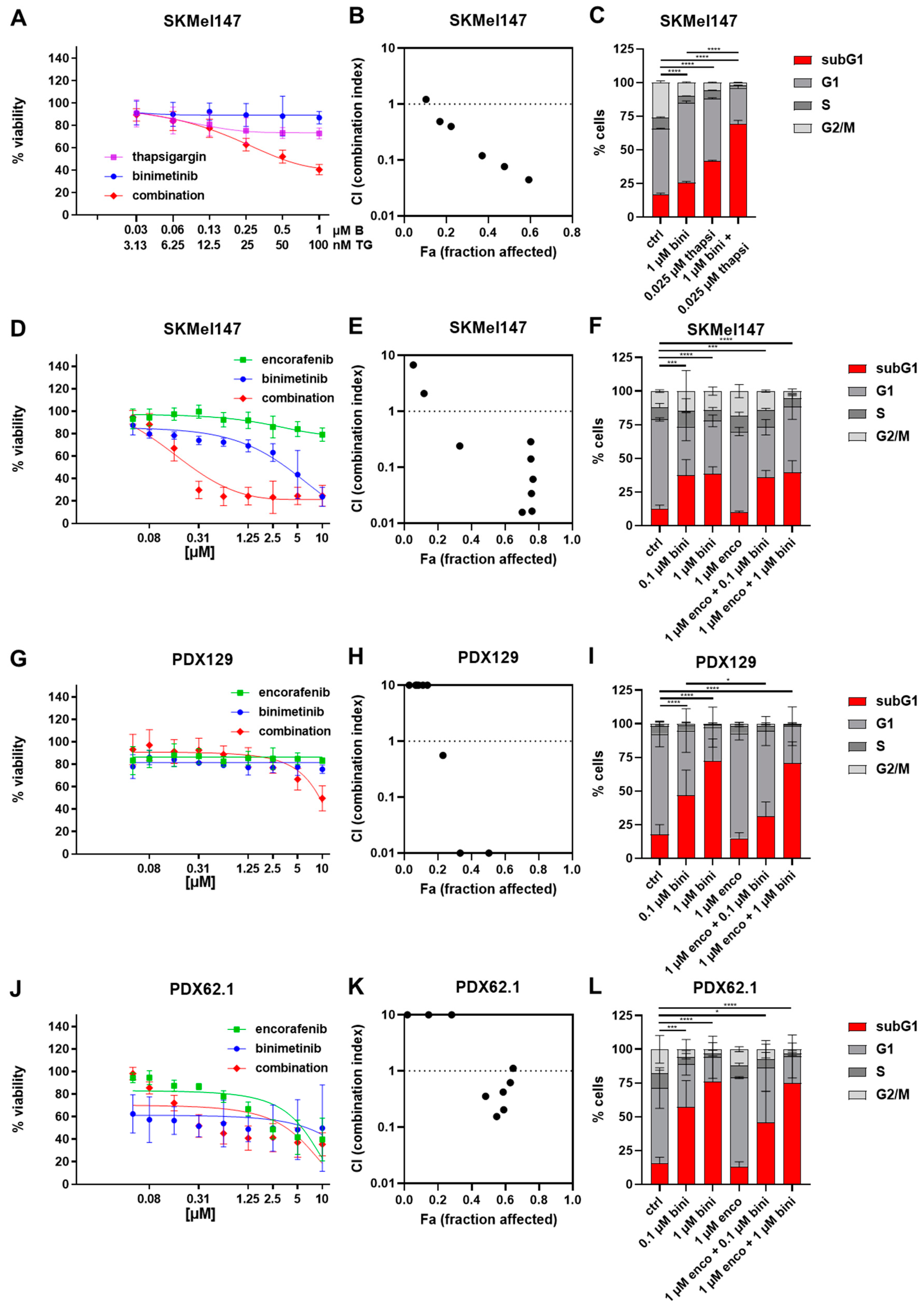
3.1. Effects of Thapsigargin/Encorafenib and/or Binimetinib on the Viability and Cell Cycle of Melanoma Cells
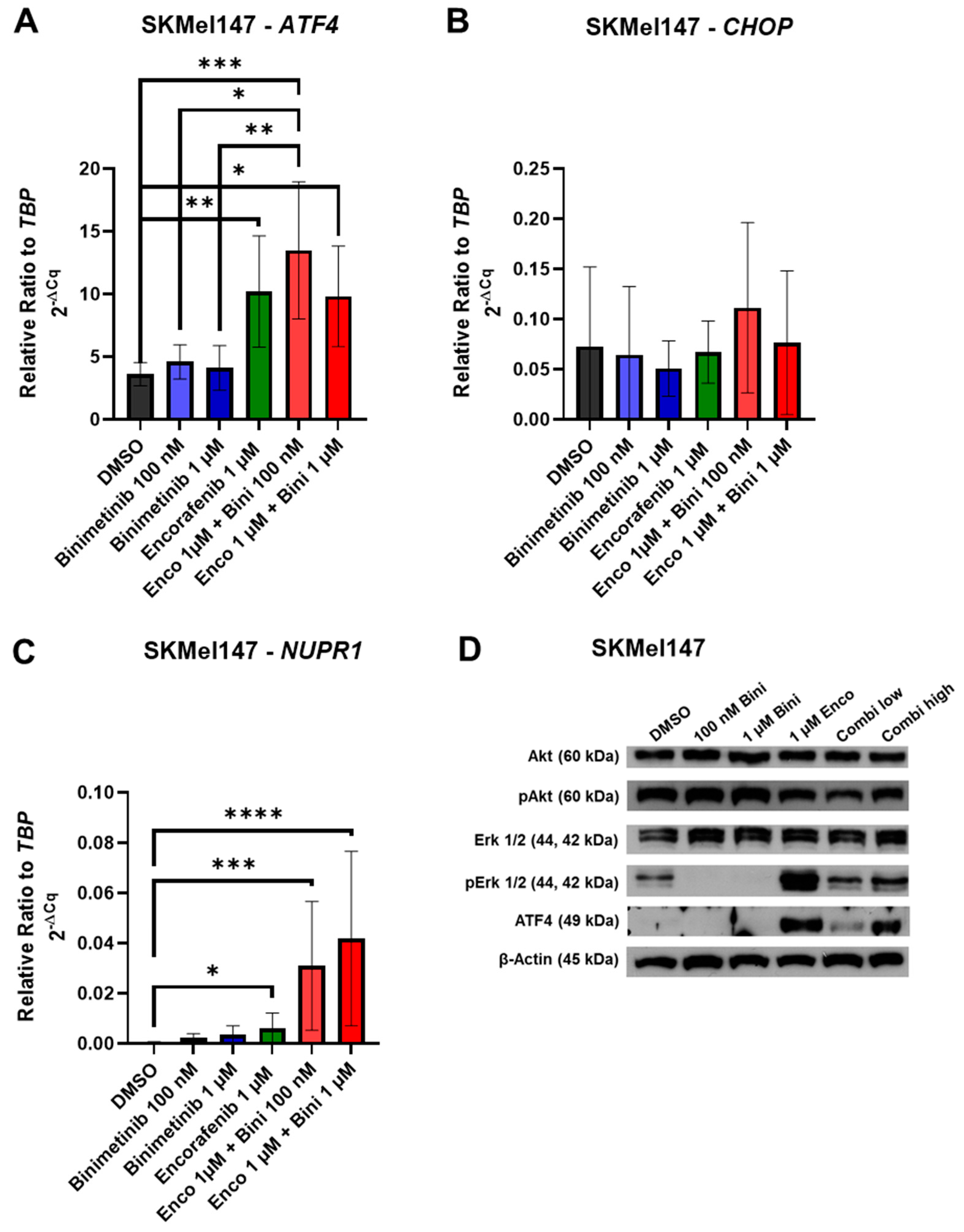
3.2. Effects of Binimetinib and Encorafenib on ER Stress Gene mRNA and Protein Expression
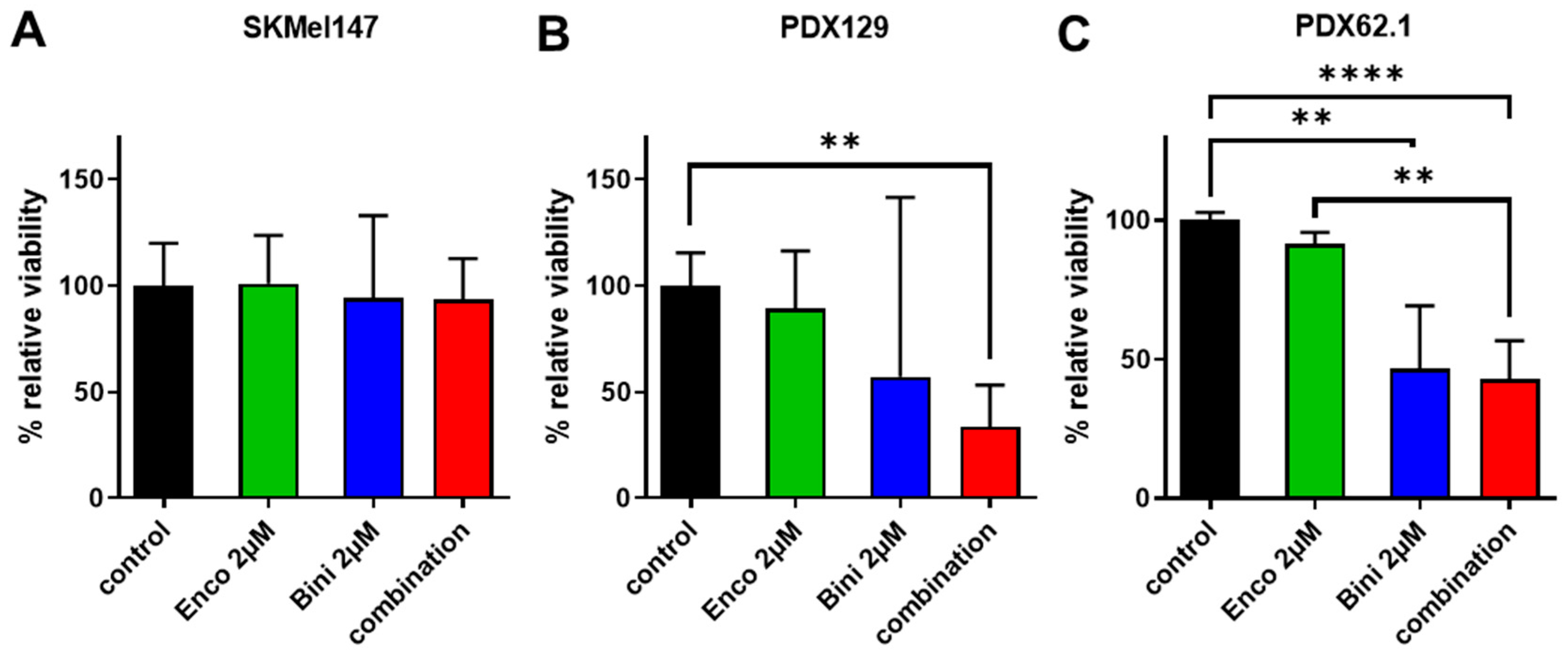
3.3. Effects of Binimetinib and Encorafenib on the Ex Vivo Tumor Viability of NRAS-Mutated Tumors
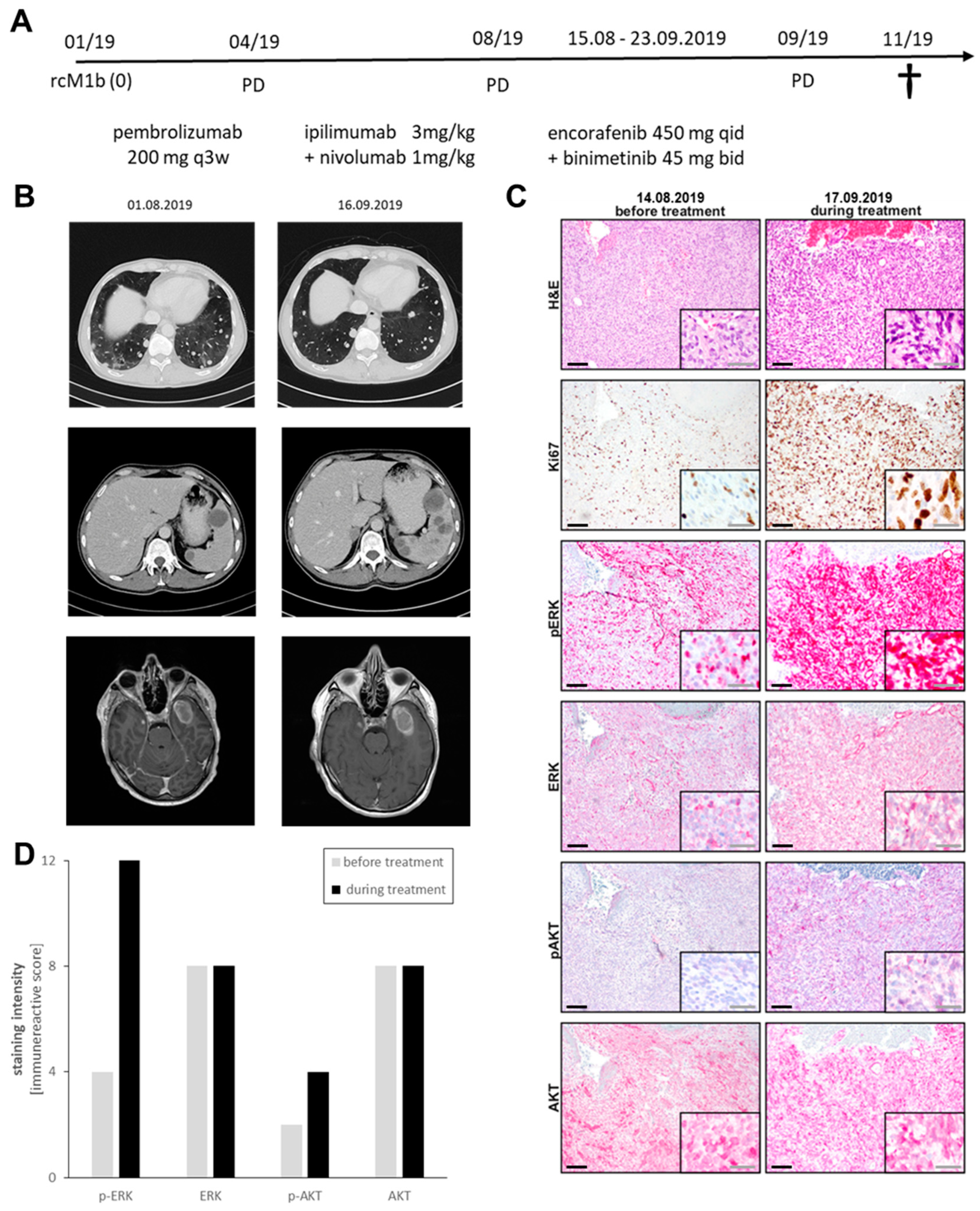
3.4. Case Report of Binimetinib and Encorafenib in a Patient with Advanced NRAS-Mutated Melanoma
3.5. Effects of Binimetinib and Encorafenib In Vivo
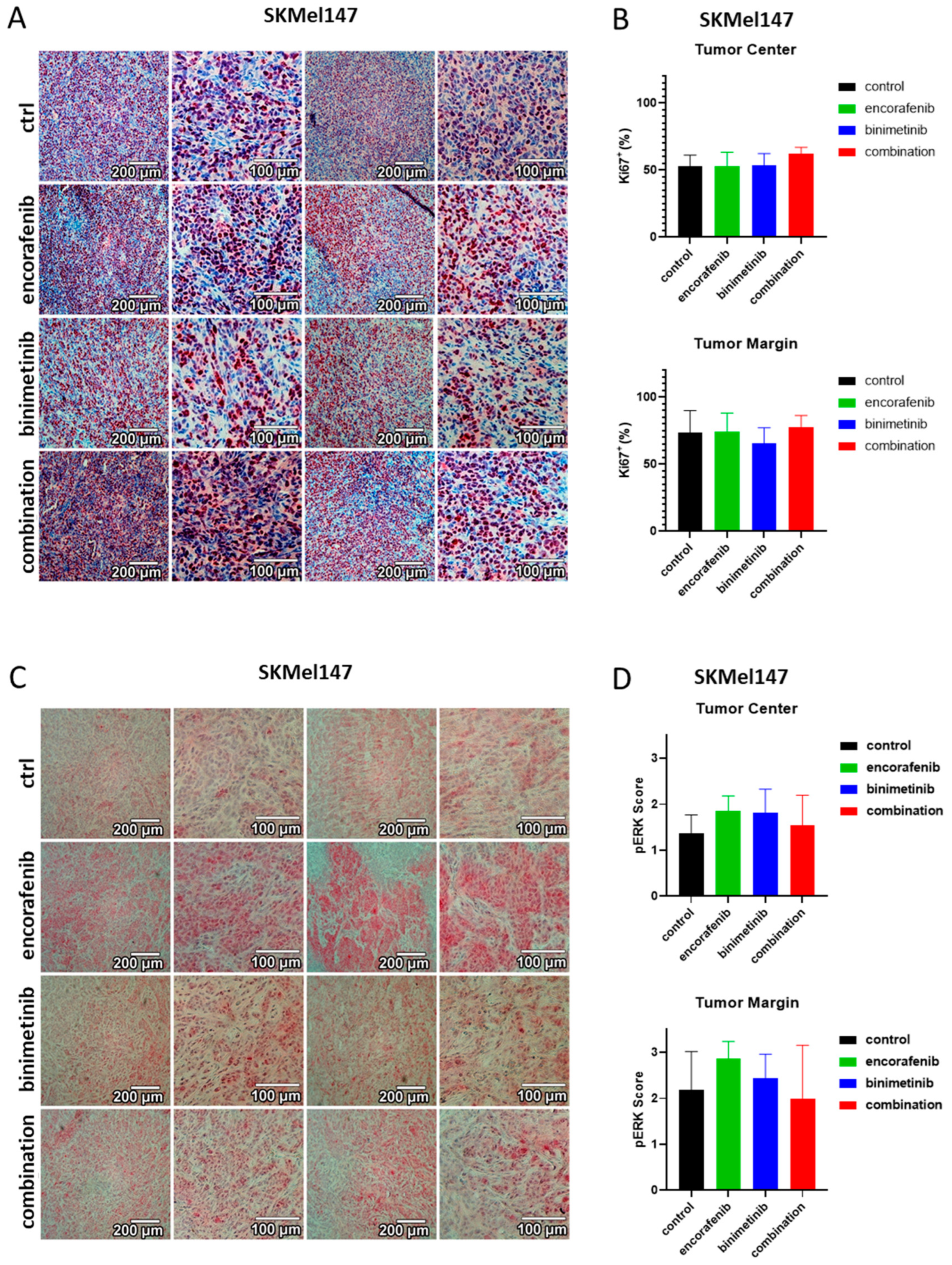
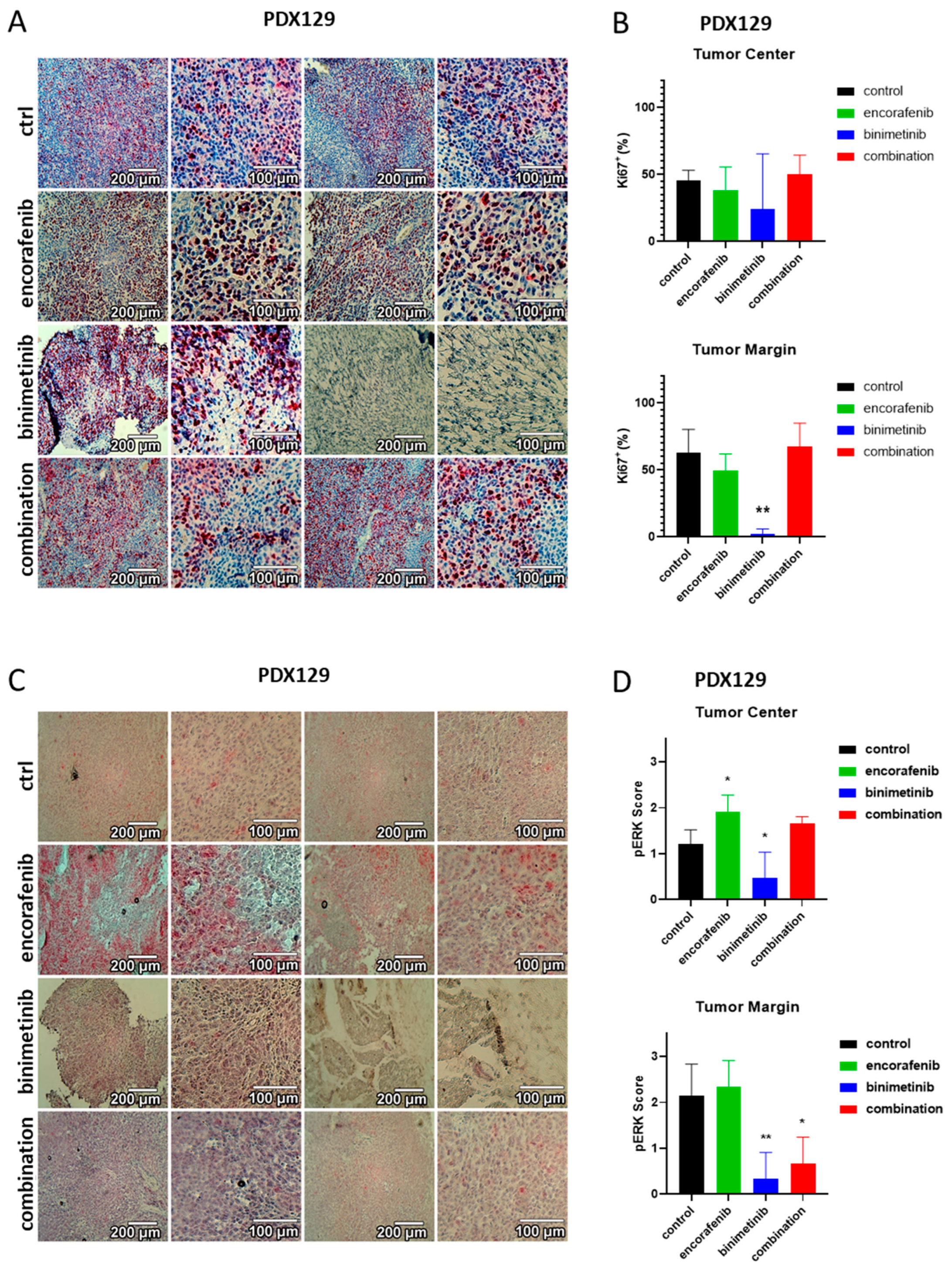
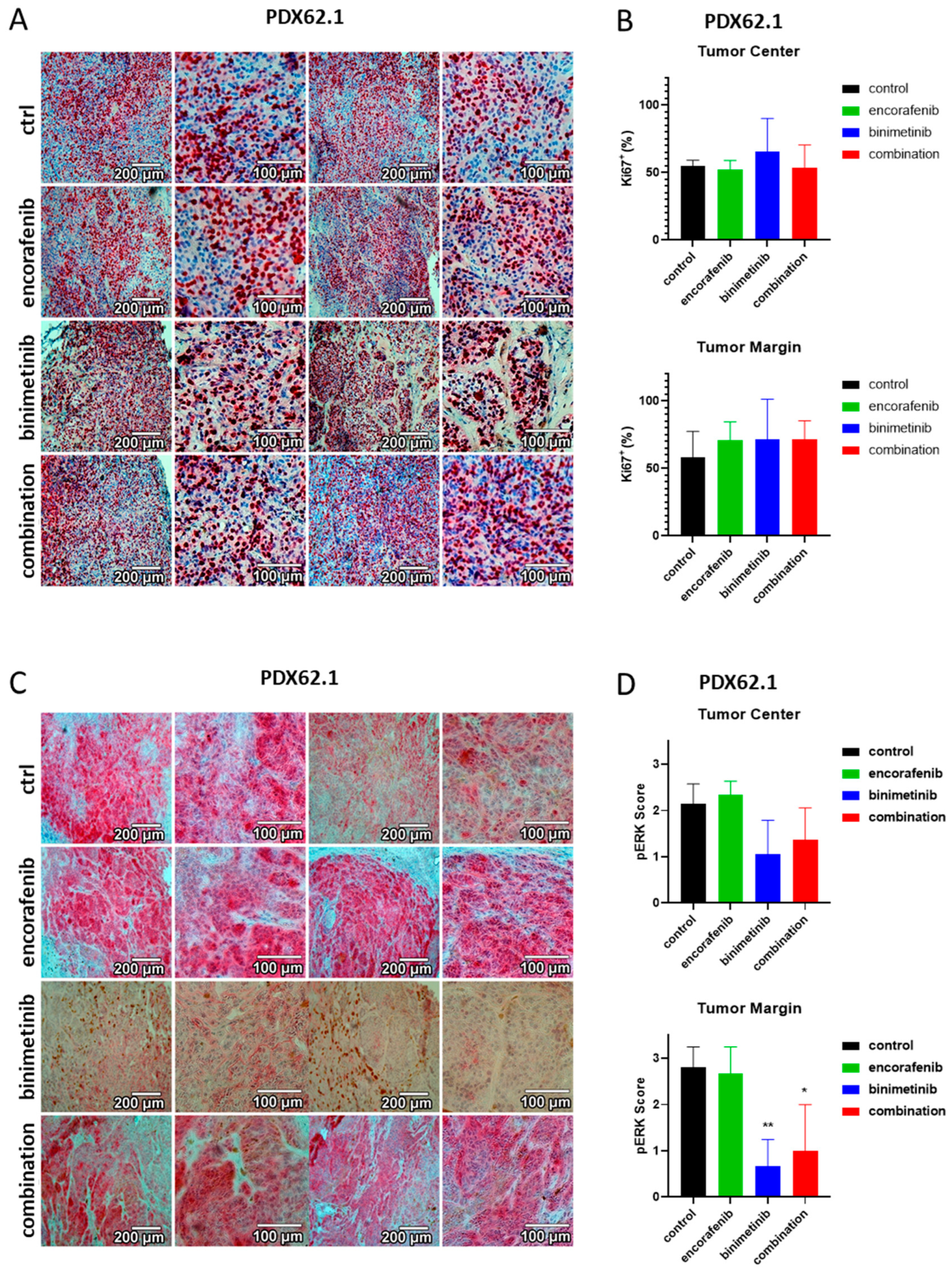
3.6. Immunohistochemical Evaluation of the PDX Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Atkins, M.B.; Hsu, J.; Lee, S.; Cohen, G.I.; Flaherty, L.E.; Sosman, J.A.; Sondak, V.K.; Kirkwood, J.M.; Eastern Cooperative Oncology Group. Phase III trial comparing concurrent biochemotherapy with cisplatin, vinblastine, dacarbazine, interleukin-2, and interferon alfa-2b with cisplatin, vinblastine, and dacarbazine alone in patients with metastatic malignant melanoma (E3695): A trial coordinated by the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2008, 26, 5748–5754. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Neyns, B.; Linette, G.; Negrier, S.; Lutzky, J.; Thomas, L.; Waterfield, W.; Schadendorf, D.; Smylie, M.; Guthrie, T., Jr.; et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: A randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010, 11, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.M.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Results of COLUMBUS Part 1: A Phase 3 Trial of Encorafenib (ENCO) Plus Binimeitnib (BINI) Versus Vemurafenib (VEM) or ENCO in BRAF-Mutant Melanoma. In Proceedings of the Eighteenth International Congress, Boston, MA, USA, 6–9 November 2016. [Google Scholar]
- Young, K.; Minchom, A.; Larkin, J. BRIM-1, -2 and -3 trials: Improved survival with vemurafenib in metastatic melanoma patients with a BRAF(V600E) mutation. Future Oncol. 2012, 8, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Rohaan, M.W.; Borch, T.H.; van den Berg, J.H.; Met, O.; Kessels, R.; Geukes Foppen, M.H.; Stoltenborg Granhoj, J.; Nuijen, B.; Nijenhuis, C.; Jedema, I.; et al. Tumor-Infiltrating Lymphocyte Therapy or Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2022, 387, 2113–2125. [Google Scholar] [CrossRef]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Heppt, M.V.; Siepmann, T.; Engel, J.; Schubert-Fritschle, G.; Eckel, R.; Mirlach, L.; Kirchner, T.; Jung, A.; Gesierich, A.; Ruzicka, T.; et al. Prognostic significance of BRAF and NRAS mutations in melanoma: A German study from routine care. BMC Cancer 2017, 17, 536. [Google Scholar] [CrossRef] [PubMed]
- Zablocka, T.; Kreismane, M.; Pjanova, D.; Isajevs, S. Effects of BRAF V600E and NRAS mutational status on the progression-free survival and clinicopathological characteristics of patients with melanoma. Oncol. Lett. 2023, 25, 27. [Google Scholar] [CrossRef]
- Niessner, H.; Sinnberg, T.; Kosnopfel, C.; Smalley, K.S.M.; Beck, D.; Praetorius, C.; Mai, M.; Beissert, S.; Kulms, D.; Schaller, M.; et al. BRAF Inhibitors Amplify the Proapoptotic Activity of MEK Inhibitors by Inducing ER Stress in NRAS-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 6203–6214. [Google Scholar] [CrossRef]
- Beck, D.; Niessner, H.; Smalley, K.S.; Flaherty, K.; Paraiso, K.H.; Busch, C.; Sinnberg, T.; Vasseur, S.; Iovanna, J.L.; Driessen, S.; et al. Vemurafenib potently induces endoplasmic reticulum stress-mediated apoptosis in BRAFV600E melanoma cells. Sci. Signal 2013, 6, ra7. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Leli, N.M.; Onorati, A.; Piao, S.; Verginadis, I.I.; Tameire, F.; Rebecca, V.W.; Chude, C.I.; Murugan, S.; Fennelly, C.; et al. ER Translocation of the MAPK Pathway Drives Therapy Resistance in BRAF-Mutant Melanoma. Cancer Discov. 2019, 9, 396–415. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- O’Bryan, J.P. Pharmacological targeting of RAS: Recent success with direct inhibitors. Pharmacol. Res. 2019, 139, 503–511. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Sullivan, R.J. NRAS mutant melanoma: An overview for the clinician for melanoma management. Melanoma Manag. 2016, 3, 47–59. [Google Scholar] [CrossRef]
- Boespflug, A.; Caramel, J.; Dalle, S.; Thomas, L. Treatment of NRAS-mutated advanced or metastatic melanoma: Rationale, current trials and evidence to date. Ther. Adv. Med. Oncol. 2017, 9, 481–492. [Google Scholar] [CrossRef]
- Jakob, J.A.; Bassett, R.L., Jr.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Rohlfs, M.L.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef]
- Sanchez-Laorden, B.; Viros, A.; Girotti, M.R.; Pedersen, M.; Saturno, G.; Zambon, A.; Niculescu-Duvaz, D.; Turajlic, S.; Hayes, A.; Gore, M.; et al. BRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci. Signal 2014, 7, ra30. [Google Scholar] [CrossRef]
- Krepler, C.; Xiao, M.; Sproesser, K.; Brafford, P.A.; Shannan, B.; Beqiri, M.; Liu, Q.; Xu, W.; Garman, B.; Nathanson, K.L.; et al. Personalized Preclinical Trials in BRAF Inhibitor-Resistant Patient-Derived Xenograft Models Identify Second-Line Combination Therapies. Clin. Cancer Res. 2016, 22, 1592–1602. [Google Scholar] [CrossRef]
- Remmele, W.; Stegner, H.E. Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue. Pathologe 1987, 8, 138–140. [Google Scholar] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; Garcia, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. Onco Targets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Moffat, J.; Schaefer, G.; Chan, J.; Wang, X.; Orr, C.; Cheng, J.; Hunsaker, T.; Shao, L.; Wang, S.J.; et al. Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS ONE 2017, 12, e0185862. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Kun, E.; Tsang, Y.T.M.; Ng, C.W.; Gershenson, D.M.; Wong, K.K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat. Rev. 2021, 92, 102137. [Google Scholar] [CrossRef]
- Adelmann, C.H.; Ching, G.; Du, L.; Saporito, R.C.; Bansal, V.; Pence, L.J.; Liang, R.; Lee, W.; Tsai, K.Y. Comparative profiles of BRAF inhibitors: The paradox index as a predictor of clinical toxicity. Oncotarget 2016, 7, 30453–30460. [Google Scholar] [CrossRef]
- Anforth, R.; Menzies, A.; Byth, K.; Carlos, G.; Chou, S.; Sharma, R.; Scolyer, R.A.; Kefford, R.; Long, G.V.; Fernandez-Penas, P. Factors influencing the development of cutaneous squamous cell carcinoma in patients on BRAF inhibitor therapy. J. Am. Acad. Dermatol. 2015, 72, 809–815.e801. [Google Scholar] [CrossRef]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Holderfield, M.; Nagel, T.E.; Stuart, D.D. Mechanism and consequences of RAF kinase activation by small-molecule inhibitors. Br. J. Cancer 2014, 111, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Stuart, D.D. Preclinical profile of LGX818: A potent and selective RAF kinase inhibitor. Cancer Res. 2012, 72, 3790. [Google Scholar] [CrossRef]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Duggan, M.C.; Stiff, A.R.; Bainazar, M.; Regan, K.; Olaverria Salavaggione, G.N.; Maharry, S.; Blachly, J.S.; Krischak, M.; Walker, C.J.; Latchana, N.; et al. Identification of NRAS isoform 2 overexpression as a mechanism facilitating BRAF inhibitor resistance in malignant melanoma. Proc. Natl. Acad. Sci. USA 2017, 114, 9629–9634. [Google Scholar] [CrossRef]
- Chatziioannou, E.; Rossner, J.; Aung, T.N.; Rimm, D.L.; Niessner, H.; Keim, U.; Serna-Higuita, L.M.; Bonzheim, I.; Kuhn Cuellar, L.; Westphal, D.; et al. Deep learning-based scoring of tumour-infiltrating lymphocytes is prognostic in primary melanoma and predictive to PD-1 checkpoint inhibition in melanoma metastases. EBioMedicine 2023, 93, 104644. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef]
- Gogas, H.; Dreno, B.; Larkin, J.; Demidov, L.; Stroyakovskiy, D.; Eroglu, Z.; Francesco Ferrucci, P.; Pigozzo, J.; Rutkowski, P.; Mackiewicz, J.; et al. Cobimetinib plus atezolizumab in BRAF(V600) wild-type melanoma: Primary results from the randomized phase III IMspire170 study. Ann. Oncol. 2021, 32, 384–394. [Google Scholar] [CrossRef]
- Eichhoff, O.M.; Stoffel, C.I.; Kasler, J.; Briker, L.; Turko, P.; Karsai, G.; Zila, N.; Paulitschke, V.; Cheng, P.F.; Leitner, A.; et al. ROS Induction Targets Persister Cancer Cells with Low Metabolic Activity in NRAS-Mutated Melanoma. Cancer Res. 2023, 83, 1128–1146. [Google Scholar] [CrossRef] [PubMed]
- Meraz-Torres, F. Augmenting MEK inhibitor Efficacy in BRAF Wild-Type Melanoma: Synergistic Effects of Disulfiram Combination Therapy. Res. Sq. Prepr. 2023. [Google Scholar] [CrossRef]
- Atefi, M.; Titz, B.; Avramis, E.; Ng, C.; Wong, D.J.; Lassen, A.; Cerniglia, M.; Escuin-Ordinas, H.; Foulad, D.; Comin-Anduix, B.; et al. Combination of pan-RAF and MEK inhibitors in NRAS mutant melanoma. Mol. Cancer 2015, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.J. A phase Ib trial of belvarafenib in combination with cobimetinib in patients with advanced solid tumors: Interim results of dose-escalation and patients with NRAS-mutant melanoma of dose-expansion. J. Clin. Oncol. 2021, 39, 3007. [Google Scholar] [CrossRef]
- Yen, I.; Shanahan, F.; Lee, J.; Hong, Y.S.; Shin, S.J.; Moore, A.R.; Sudhamsu, J.; Chang, M.T.; Bae, I.; Dela Cruz, D.; et al. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature 2021, 594, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Arafeh, R.; Flores, K.; Keren-Paz, A.; Maik-Rachline, G.; Gutkind, N.; Rosenberg, S.; Seger, R.; Samuels, Y. Combined inhibition of MEK and nuclear ERK translocation has synergistic antitumor activity in melanoma cells. Sci. Rep. 2017, 7, 16345. [Google Scholar] [CrossRef]
- Echevarria-Vargas, I.M.; Reyes-Uribe, P.I.; Guterres, A.N.; Yin, X.; Kossenkov, A.V.; Liu, Q.; Zhang, G.; Krepler, C.; Cheng, C.; Wei, Z.; et al. Co-targeting BET and MEK as salvage therapy for MAPK and checkpoint inhibitor-resistant melanoma. EMBO Mol. Med. 2018, 10, e8446. [Google Scholar] [CrossRef]
- Posch, C.; Moslehi, H.; Feeney, L.; Green, G.A.; Ebaee, A.; Feichtenschlager, V.; Chong, K.; Peng, L.; Dimon, M.T.; Phillips, T.; et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 4015–4020. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Oligonucleotide Primer 5′→3′ | |
|---|---|---|
| TBP | for | tgc aca gga gcc aag agt gaa |
| rev | cac atc aca gct ccc cac ca | |
| ATF4 | for | tgg gga aag ggg aag agg ttg taa |
| rev | agt cgg gtt tgg ggg ctg aag | |
| CHOP | for | aag gca ctg agc gta tca tgt |
| rev | tga aga tac act tcc ttc ttg aac ac | |
| p8/NUPR1 | for | cca ttc cta cct cgg gcc tct catc |
| rev | tct tgg tgc gac ctt tcc ggc | |
| Sample | Mutational Status | Tumor Stage | Metastasis Site | Prior Treatment |
|---|---|---|---|---|
| PDX129 | NRASQ61R | IV | small intestine | Pembrolizumab; ipilimumab + nivolumab |
| PDX62.1 | NRASQ61R | IV | brain | Ipilimumab + nivolumab; DTIC |
| Protein | Company | Retrieval | Dilution | Incubation | Detection |
|---|---|---|---|---|---|
| AKT | #4691 (Cell Signaling) | CC1 (#950-124 Ventana), 60 min | 1:300 | 32 min, 37 °C | AP red (#760-501, Ventana) |
| pAKT | #4060 (Cell Signaling) | CC1 (#950-124 Ventana), 60 min | 1:20 | 2 h, RT | AP red (#760-501, Ventana) |
| ERK | #9102 (Cell Signaling) | CC1 (#950-124 Ventana), 60 min | 1:50 | 32 min, 37 °C | AP red (#760-501, Ventana) |
| pERK | #4376 (Cell Signaling) | CC1 (#950-124 Ventana), 60 min | 1:200 | 2 h, RT | AP red (#760-501, Ventana) |
| Ki67 | M7240 (Dako) | CC1 (#950-124 Ventana), 60 min | 1:50 | 28 min, 37 °C | DAB brown uView (#760-700, Ventana) |
| % Positive Cells | Staining Intensity | IRS—% Positive Cells × Staining Intensity |
|---|---|---|
| 0 = no positive cells | 0 = no color reaction | 0–1 = negative |
| 1 = <10% positive cells | 1 = mild reaction | 2–3 = mild |
| 2 = 10–50% positive cells | 2 = moderate reaction | 4–8 = moderate |
| 3 = 51–80% positive cells | 3 = intense reaction | 9–12 = strongly positive |
| 4 = >80% positive cells |
| Marker | Before Treatment | During treatment | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Intensity | % Positive Cells | IRS | Comments | Intensity | % Positive Cells | IRS | Comments | |||
| Score | Score | Score | Score | Score | Score | |||||
| HE | Apoptosis, also necrosis | Little apoptosis, hardly any necrosis | ||||||||
| MIB/Ki67 | 15–35% * | n.a. | 30–60% * | n.a. | ||||||
| p-ERK | 2 | 50% | 2 | 4 | Heterogeneous | 3 | 95% | 4 | 12 | |
| ERK | 2 | 90% | 4 | 8 | 2 | 90% | 4 | 8 | ||
| p-AKT | 1 | 10% | 2 | 2 | 2 | 50% | 2 | 4 | ||
| AKT | 2 | 100% | 4 | 8 | 2 | 100% | 4 | 8 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niessner, H.; Hüsch, A.; Kosnopfel, C.; Meinhardt, M.; Westphal, D.; Meier, F.; Schilling, B.; Sinnberg, T. Exploring the In Vitro and In Vivo Therapeutic Potential of BRAF and MEK Inhibitor Combination in NRAS-Mutated Melanoma. Cancers 2023, 15, 5521. https://doi.org/10.3390/cancers15235521
Niessner H, Hüsch A, Kosnopfel C, Meinhardt M, Westphal D, Meier F, Schilling B, Sinnberg T. Exploring the In Vitro and In Vivo Therapeutic Potential of BRAF and MEK Inhibitor Combination in NRAS-Mutated Melanoma. Cancers. 2023; 15(23):5521. https://doi.org/10.3390/cancers15235521
Chicago/Turabian StyleNiessner, Heike, Anna Hüsch, Corinna Kosnopfel, Matthias Meinhardt, Dana Westphal, Friedegund Meier, Bastian Schilling, and Tobias Sinnberg. 2023. "Exploring the In Vitro and In Vivo Therapeutic Potential of BRAF and MEK Inhibitor Combination in NRAS-Mutated Melanoma" Cancers 15, no. 23: 5521. https://doi.org/10.3390/cancers15235521
APA StyleNiessner, H., Hüsch, A., Kosnopfel, C., Meinhardt, M., Westphal, D., Meier, F., Schilling, B., & Sinnberg, T. (2023). Exploring the In Vitro and In Vivo Therapeutic Potential of BRAF and MEK Inhibitor Combination in NRAS-Mutated Melanoma. Cancers, 15(23), 5521. https://doi.org/10.3390/cancers15235521