

New Insights into YAP/TAZ-TEAD-Mediated Gene Regulation and Biological Processes in Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
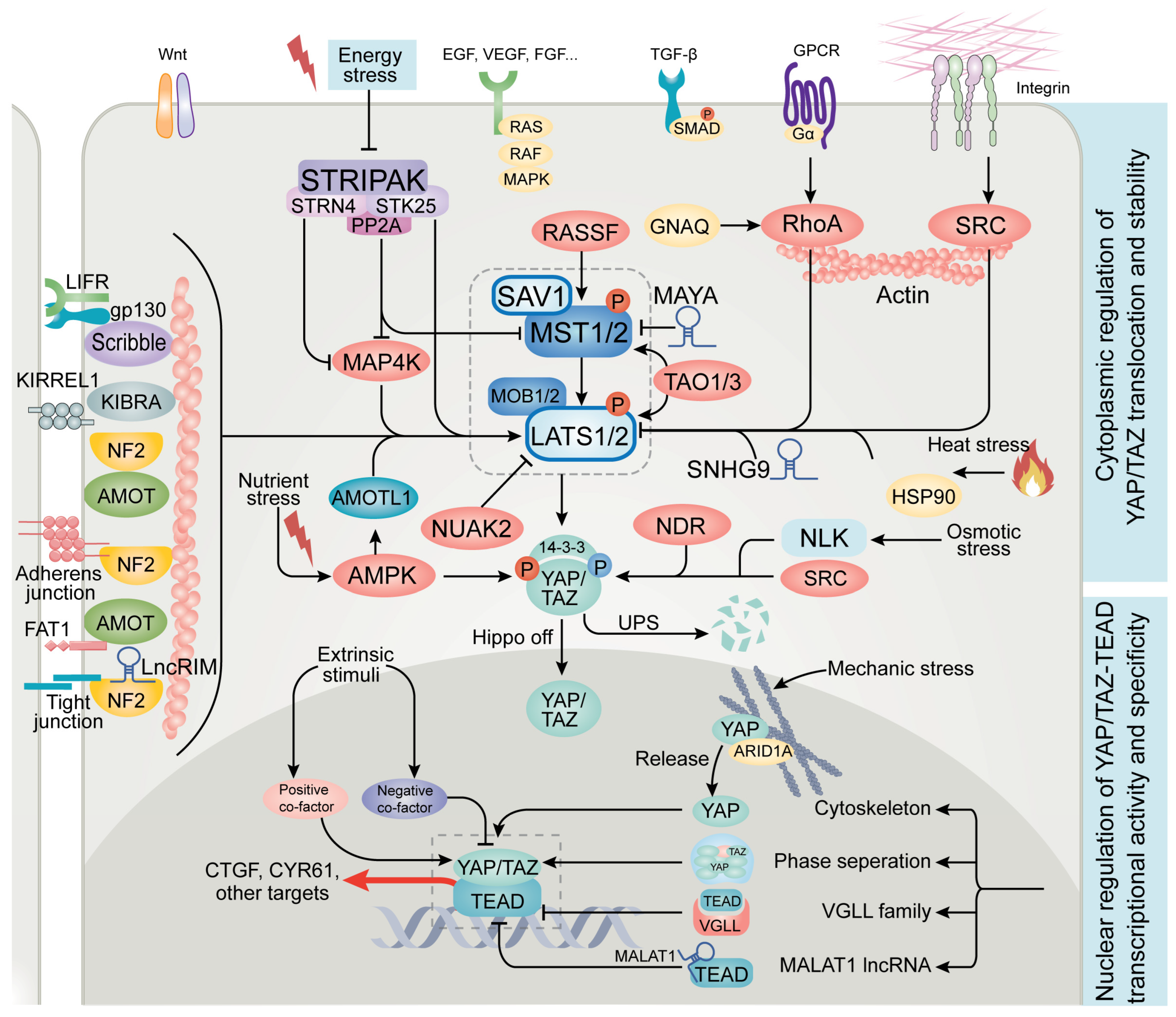
2. Advances in Upstream Regulators of Hippo Signaling
3. Emerging Regulators of YAP/TAZ and TEAD
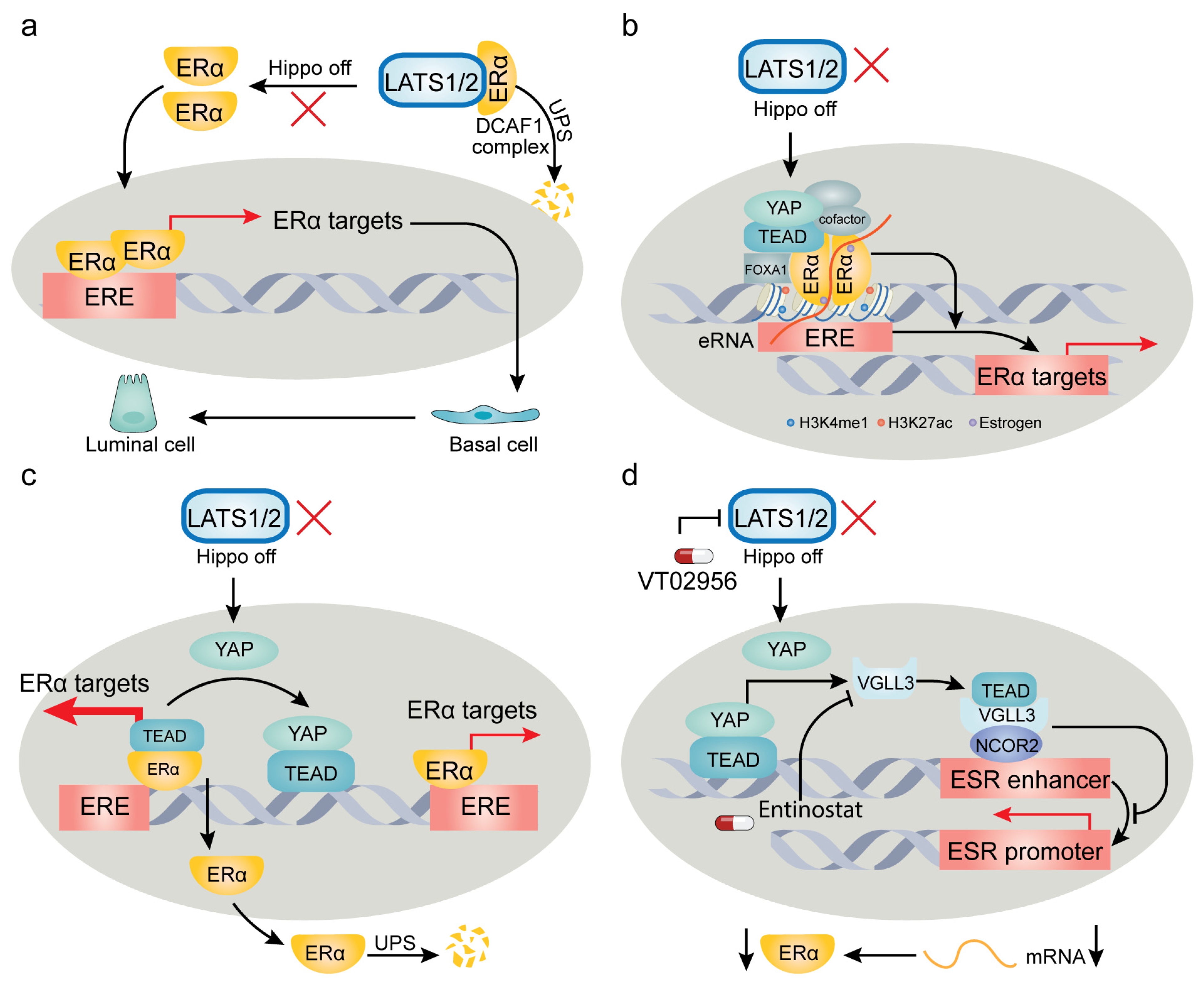
4. YAP/TAZ-TEAD Target Genes and Context-Dependent Regulation of Their Expression
5. Cancer-Type-Specific Alterations in the Hippo Pathway
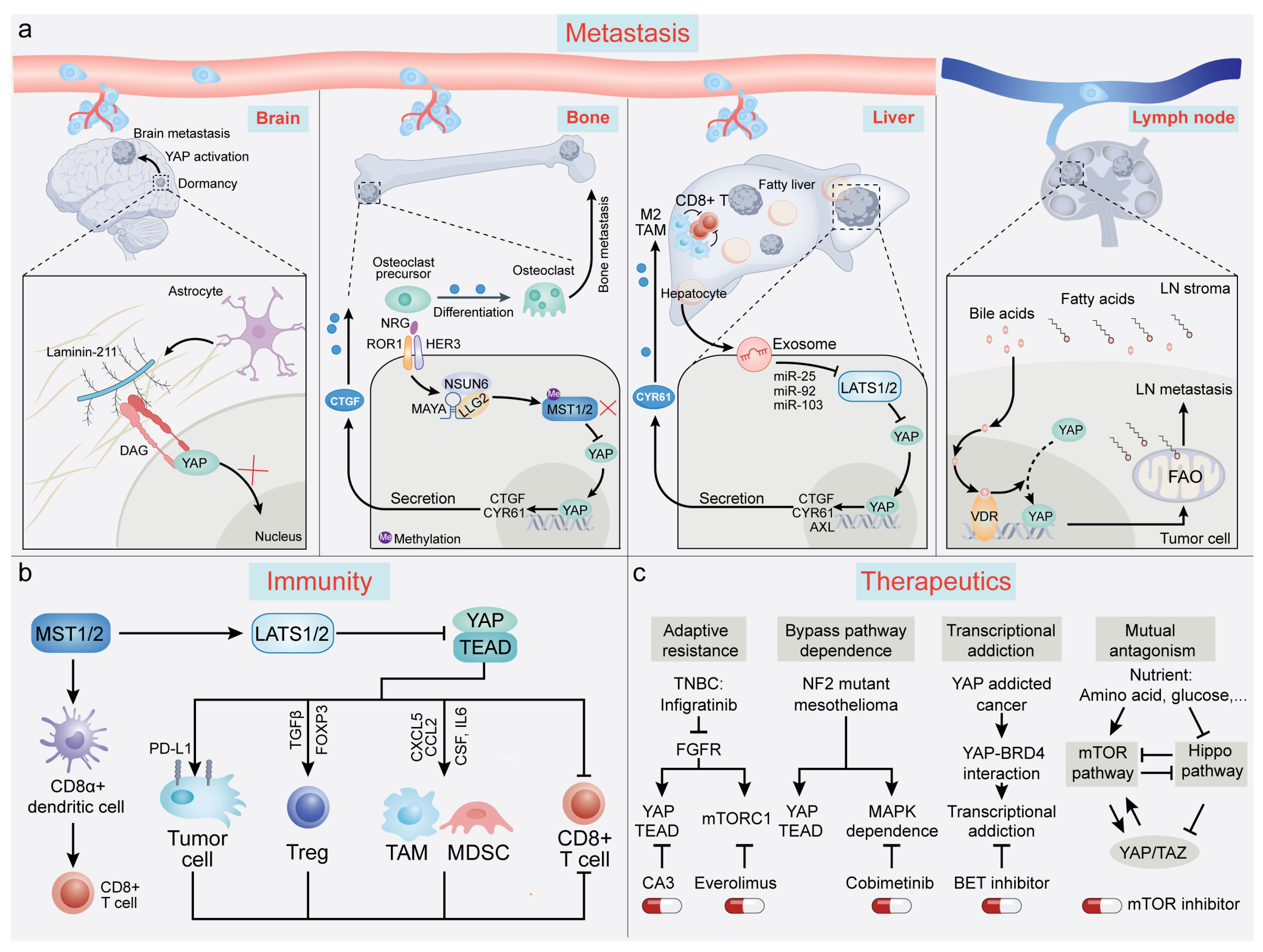
6. Advances in YAP/TAZ-TEAD-Mediated Regulation of Tumor Dormancy, Metastatic Relapse, and Organ Tropism
7. Advances in YAP/TAZ-TEAD-Mediated Regulation of Tumor Immunity
8. Advances in YAP/TAZ-TEAD-Mediated Regulation of Therapeutic Response
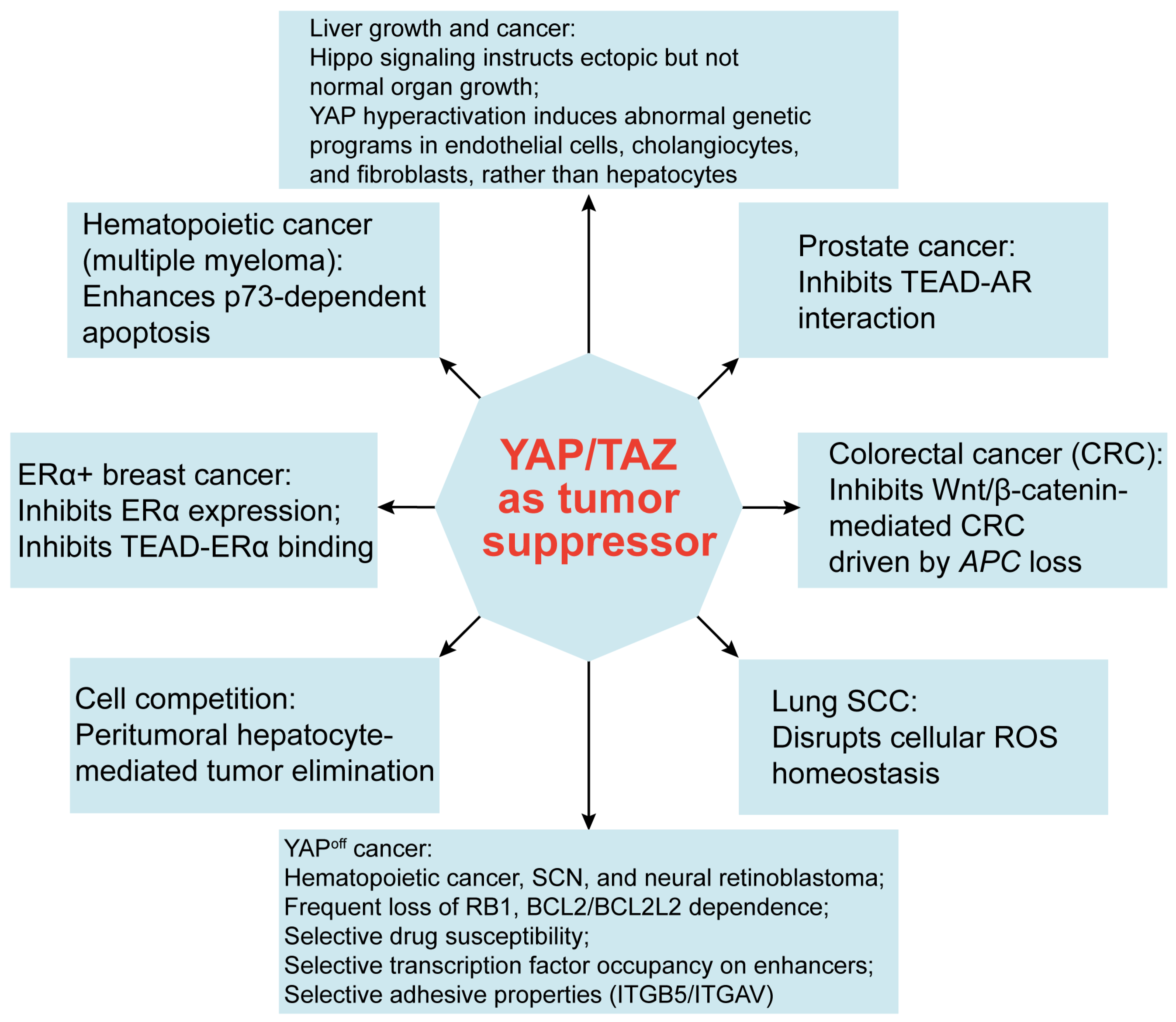
9. Emerging Roles of YAP/TAZ in Tumor Suppression
10. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, D.C.; Schiripo, T.A.; Haber, D.; Hariharan, I.K. Salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002, 110, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wang, W.; Zhang, S.; Stewart, R.A.; Yu, W. Identifying tumor suppressors in genetic mosaics: The Drosophila lats gene encodes a putative protein kinase. Development 1995, 121, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Huang, J.; Dong, J.; Pan, D. Hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 2003, 114, 445–456. [Google Scholar] [CrossRef]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Cunningham, R.; Hansen, C.G. The Hippo pathway in cancer: YAP/TAZ and TEAD as therapeutic targets in cancer. Clin. Sci. 2022, 136, 197–222. [Google Scholar] [CrossRef]
- Franklin, J.M.; Wu, Z.; Guan, K.L. Insights into recent findings and clinical application of YAP and TAZ in cancer. Nat. Rev. Cancer 2023, 23, 512–525. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Ashayeri, N.; Baghaie, L.; Sambi, M.; Satari, K.; Baluch, N.; Bosykh, D.A.; Szewczuk, M.R.; Chakraborty, S. The Hippo Pathway Effectors YAP/TAZ-TEAD Oncoproteins as Emerging Therapeutic Targets in the Tumor Microenvironment. Cancers 2023, 15, 3468. [Google Scholar] [CrossRef]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Reggiani, F.; Gobbi, G.; Ciarrocchi, A.; Ambrosetti, D.C.; Sancisi, V. Multiple roles and context-specific mechanisms underlying YAP and TAZ-mediated resistance to anti-cancer therapy. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188341. [Google Scholar] [CrossRef] [PubMed]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 2020, 133, jcs230425. [Google Scholar] [CrossRef]
- Manning, S.A.; Kroeger, B.; Harvey, K.F. The regulation of Yorkie, YAP and TAZ: New insights into the Hippo pathway. Development 2020, 147, dev179069. [Google Scholar] [CrossRef]
- Kwon, H.; Kim, J.; Jho, E.H. Role of the Hippo pathway and mechanisms for controlling cellular localization of YAP/TAZ. FEBS J. 2022, 289, 5798–5818. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, X.; Maglic, D.; Dill, M.T.; Mojumdar, K.; Ng, P.K.; Jeong, K.J.; Tsang, Y.H.; Moreno, D.; Bhavana, V.H.; et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018, 25, 1304–1317.e1305. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Ibar, C.; Irvine, K.D. Integration of Hippo-YAP Signaling with Metabolism. Dev. Cell 2020, 54, 256–267. [Google Scholar] [CrossRef]
- Garner, H.; de Visser, K.E. Immune crosstalk in cancer progression and metastatic spread: A complex conversation. Nat. Rev. Immunol. 2020, 20, 483–497. [Google Scholar] [CrossRef] [PubMed]
- van Rensburg, H.J.J.; Yang, X. The roles of the Hippo pathway in cancer metastasis. Cell. Signal. 2016, 28, 1761–1772. [Google Scholar] [CrossRef]
- Pearson, J.D.; Huang, K.; Pacal, M.; McCurdy, S.R.; Lu, S.; Aubry, A.; Yu, T.; Wadosky, K.M.; Zhang, L.; Wang, T.; et al. Binary pan-cancer classes with distinct vulnerabilities defined by pro- or anti-cancer YAP/TEAD activity. Cancer Cell 2021, 39, 1115–1134.e1112. [Google Scholar] [CrossRef] [PubMed]
- Rausch, V.; Hansen, C.G. The Hippo Pathway, YAP/TAZ, and the Plasma Membrane. Trends Cell Biol. 2020, 30, 32–48. [Google Scholar] [CrossRef]
- Genevet, A.; Wehr, M.C.; Brain, R.; Thompson, B.J.; Tapon, N. Kibra is a regulator of the Salvador/Warts/Hippo signaling network. Dev. Cell 2010, 18, 300–308. [Google Scholar] [CrossRef]
- Yin, F.; Yu, J.; Zheng, Y.; Chen, Q.; Zhang, N.; Pan, D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 2013, 154, 1342–1355. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Choi, K.; Su, T.; Li, B.; Wu, X.; Zhang, R.; Driskill, J.H.; Li, H.; Lei, H.; Guo, P.; et al. Multiphase coalescence mediates Hippo pathway activation. Cell 2022, 185, 4376–4393.e4318. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.; Guan, K.L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef]
- Paul, A.; Annunziato, S.; Lu, B.; Sun, T.; Evrova, O.; Planas-Paz, L.; Orsini, V.; Terracciano, L.M.; Charlat, O.; Loureiro, Z.Y.; et al. Cell adhesion molecule KIRREL1 is a feedback regulator of Hippo signaling recruiting SAV1 to cell-cell contact sites. Nat. Commun. 2022, 13, 930. [Google Scholar] [CrossRef]
- Wang, C.; Feng, X.; Su, D.; Chen, Z.; Wang, S.; Tang, M.; Huang, M.; Nie, L.; Zhang, H.; Li, S.; et al. Integrated screens uncover a cell surface tumor suppressor gene KIRREL involved in Hippo pathway. Proc. Natl. Acad. Sci. USA 2022, 119, e2121779119. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, Y.; Sha, Z.; He, C.; Zhu, Y.; Li, J.; Yu, A.; Zhong, Z.; Wang, X.; Sun, Y.; et al. Transmembrane protein KIRREL1 regulates Hippo signaling via a feedback loop and represents a therapeutic target in YAP/TAZ-active cancers. Cell Rep. 2022, 40, 111296. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Degese, M.S.; Vitale-Cross, L.; Iglesias-Bartolome, R.; Valera, J.L.C.; Wang, Z.; Feng, X.; Yeerna, H.; Vadmal, V.; Moroishi, T.; et al. Assembly and activation of the Hippo signalome by FAT1 tumor suppressor. Nat. Commun. 2018, 9, 2372. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Sun, Y.; Wei, Y.; Zhang, P.; Rezaeian, A.H.; Teruya-Feldstein, J.; Gupta, S.; Liang, H.; Lin, H.K.; Hung, M.C.; et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat. Med. 2012, 18, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef]
- Pastushenko, I.; Mauri, F.; Song, Y.; de Cock, F.; Meeusen, B.; Swedlund, B.; Impens, F.; Van Haver, D.; Opitz, M.; Thery, M.; et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 2021, 589, 448–455. [Google Scholar] [CrossRef]
- Avruch, J.; Zhou, D.; Fitamant, J.; Bardeesy, N.; Mou, F.; Barrufet, L.R. Protein kinases of the Hippo pathway: Regulation and substrates. Semin. Cell Dev. Biol. 2012, 23, 770–784. [Google Scholar] [CrossRef]
- Guo, C.; Zhang, X.; Pfeifer, G.P. The tumor suppressor RASSF1A prevents dephosphorylation of the mammalian STE20-like kinases MST1 and MST2. J. Biol. Chem. 2011, 286, 6253–6261. [Google Scholar] [CrossRef]
- Yee, K.S.; Grochola, L.; Hamilton, G.; Grawenda, A.; Bond, E.E.; Taubert, H.; Wurl, P.; Bond, G.L.; O’Neill, E. A RASSF1A polymorphism restricts p53/p73 activation and associates with poor survival and accelerated age of onset of soft tissue sarcoma. Cancer Res. 2012, 72, 2206–2217. [Google Scholar] [CrossRef]
- Grawenda, A.M.; O’Neill, E. Clinical utility of RASSF1A methylation in human malignancies. Br. J. Cancer 2015, 113, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, S.; Xing, Z.; Lin, A.; Liang, K.; Song, J.; Hu, Q.; Yao, J.; Chen, Z.; Park, P.K.; et al. A ROR1-HER3-lncRNA signalling axis modulates the Hippo-YAP pathway to regulate bone metastasis. Nat. Cell Biol. 2017, 19, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Li, R.H.; Tian, T.; Ge, Q.W.; He, X.Y.; Shi, C.Y.; Li, J.H.; Zhang, Z.; Liu, F.Z.; Sang, L.J.; Yang, Z.Z.; et al. A phosphatidic acid-binding lncRNA SNHG9 facilitates LATS1 liquid-liquid phase separation to promote oncogenic YAP signaling. Cell Res. 2021, 31, 1088–1105. [Google Scholar] [CrossRef] [PubMed]
- He, X.Y.; Fan, X.; Qu, L.; Wang, X.; Jiang, L.; Sang, L.J.; Shi, C.Y.; Lin, S.; Yang, J.C.; Yang, Z.Z.; et al. LncRNA modulates Hippo-YAP signaling to reprogram iron metabolism. Nat. Commun. 2023, 14, 2253. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Teng, H.; Yao, F.; Yap, S.; Sun, Y.; Ma, L. Challenges and Strategies in Ascribing Functions to Long Noncoding RNAs. Cancers 2020, 12, 1458. [Google Scholar] [CrossRef]
- Plouffe, S.W.; Meng, Z.; Lin, K.C.; Lin, B.; Hong, A.W.; Chun, J.V.; Guan, K.L. Characterization of Hippo Pathway Components by Gene Inactivation. Mol. Cell 2016, 64, 993–1008. [Google Scholar] [CrossRef]
- Boggiano, J.C.; Vanderzalm, P.J.; Fehon, R.G. Tao-1 phosphorylates Hippo/MST kinases to regulate the Hippo-Salvador-Warts tumor suppressor pathway. Dev. Cell 2011, 21, 888–895. [Google Scholar] [CrossRef]
- Poon, C.L.; Lin, J.I.; Zhang, X.; Harvey, K.F. The sterile 20-like kinase Tao-1 controls tissue growth by regulating the Salvador-Warts-Hippo pathway. Dev. Cell 2011, 21, 896–906. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Mottier-Pavie, V.; Plouffe, S.W.; Hansen, C.G.; Hong, A.W.; Park, H.W.; Mo, J.S.; Lu, W.; Lu, S.; et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 2015, 6, 8357. [Google Scholar] [CrossRef]
- Seo, G.; Han, H.; Vargas, R.E.; Yang, B.; Li, X.; Wang, W. MAP4K Interactome Reveals STRN4 as a Key STRIPAK Complex Component in Hippo Pathway Regulation. Cell Rep. 2020, 32, 107860. [Google Scholar] [CrossRef]
- Kim, J.W.; Berrios, C.; Kim, M.; Schade, A.E.; Adelmant, G.; Yeerna, H.; Damato, E.; Iniguez, A.B.; Florens, L.; Washburn, M.P.; et al. STRIPAK directs PP2A activity toward MAP4K4 to promote oncogenic transformation of human cells. Elife 2020, 9, e53003. [Google Scholar] [CrossRef]
- Couzens, A.L.; Knight, J.D.; Kean, M.J.; Teo, G.; Weiss, A.; Dunham, W.H.; Lin, Z.Y.; Bagshaw, R.D.; Sicheri, F.; Pawson, T.; et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal. 2013, 6, rs15. [Google Scholar] [CrossRef]
- Ribeiro, P.S.; Josué, F.; Wepf, A.; Wehr, M.C.; Rinner, O.; Kelly, G.; Tapon, N.; Gstaiger, M. Combined functional genomic and proteomic approaches identify a PP2A complex as a negative regulator of Hippo signaling. Mol. Cell 2010, 39, 521–534. [Google Scholar] [CrossRef]
- Chen, R.; Xie, R.; Meng, Z.; Ma, S.; Guan, K.L. STRIPAK integrates upstream signals to initiate the Hippo kinase cascade. Nat. Cell Biol. 2019, 21, 1565–1577. [Google Scholar] [CrossRef]
- Tang, Y.; Fang, G.; Guo, F.; Zhang, H.; Chen, X.; An, L.; Chen, M.; Zhou, L.; Wang, W.; Ye, T.; et al. Selective Inhibition of STRN3-Containing PP2A Phosphatase Restores Hippo Tumor-Suppressor Activity in Gastric Cancer. Cancer Cell 2020, 38, 115–128.e119. [Google Scholar] [CrossRef]
- Lim, S.; Hermance, N.; Mudianto, T.; Mustaly, H.M.; Mauricio, I.P.M.; Vittoria, M.A.; Quinton, R.J.; Howell, B.W.; Cornils, H.; Manning, A.L.; et al. Identification of the kinase STK25 as an upstream activator of LATS signaling. Nat. Commun. 2019, 10, 1547. [Google Scholar] [CrossRef]
- Bae, S.J.; Ni, L.; Luo, X. STK25 suppresses Hippo signaling by regulating SAV1-STRIPAK antagonism. Elife 2020, 9, e54863. [Google Scholar] [CrossRef]
- Bae, S.J.; Ni, L.; Osinski, A.; Tomchick, D.R.; Brautigam, C.A.; Luo, X. SAV1 promotes Hippo kinase activation through antagonizing the PP2A phosphatase STRIPAK. Elife 2017, 6, e30278. [Google Scholar] [CrossRef]
- Seo, G.; Yu, C.; Han, H.; Xing, L.; Kattan, R.E.; An, J.; Kizhedathu, A.; Yang, B.; Luo, A.; Buckle, A.L.; et al. The Hippo pathway noncanonically drives autophagy and cell survival in response to energy stress. Mol. Cell 2023, 83, 3155–3170.e8. [Google Scholar] [CrossRef]
- Mo, J.S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.S.; Guan, K.L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef]
- DeRan, M.; Yang, J.; Shen, C.H.; Peters, E.C.; Fitamant, J.; Chan, P.; Hsieh, M.; Zhu, S.; Asara, J.M.; Zheng, B.; et al. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 2014, 9, 495–503. [Google Scholar] [CrossRef]
- Gill, M.K.; Christova, T.; Zhang, Y.Y.; Gregorieff, A.; Zhang, L.; Narimatsu, M.; Song, S.; Xiong, S.; Couzens, A.L.; Tong, J.; et al. A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat. Commun. 2018, 9, 3510. [Google Scholar] [CrossRef]
- Yuan, W.C.; Pepe-Mooney, B.; Galli, G.G.; Dill, M.T.; Huang, H.T.; Hao, M.; Wang, Y.; Liang, H.; Calogero, R.A.; Camargo, F.D. NUAK2 is a critical YAP target in liver cancer. Nat. Commun. 2018, 9, 4834. [Google Scholar] [CrossRef]
- Huang, H.L.; Wang, S.; Yin, M.X.; Dong, L.; Wang, C.; Wu, W.; Lu, Y.; Feng, M.; Dai, C.; Guo, X.; et al. Par-1 regulates tissue growth by influencing hippo phosphorylation status and hippo-salvador association. PLoS Biol. 2013, 11, e1001620. [Google Scholar] [CrossRef]
- Mohseni, M.; Sun, J.; Lau, A.; Curtis, S.; Goldsmith, J.; Fox, V.L.; Wei, C.; Frazier, M.; Samson, O.; Wong, K.K.; et al. A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat. Cell Biol. 2014, 16, 108–117. [Google Scholar] [CrossRef]
- Heidary Arash, E.; Shiban, A.; Song, S.; Attisano, L. MARK4 inhibits Hippo signaling to promote proliferation and migration of breast cancer cells. EMBO Rep. 2017, 18, 420–436. [Google Scholar] [CrossRef]
- Kwan, J.; Sczaniecka, A.; Heidary Arash, E.; Nguyen, L.; Chen, C.C.; Ratkovic, S.; Klezovitch, O.; Attisano, L.; McNeill, H.; Emili, A.; et al. DLG5 connects cell polarity and Hippo signaling protein networks by linking PAR-1 with MST1/2. Genes Dev. 2016, 30, 2696–2709. [Google Scholar] [CrossRef]
- Wehr, M.C.; Holder, M.V.; Gailite, I.; Saunders, R.E.; Maile, T.M.; Ciirdaeva, E.; Instrell, R.; Jiang, M.; Howell, M.; Rossner, M.J.; et al. Salt-inducible kinases regulate growth through the Hippo signalling pathway in Drosophila. Nat. Cell Biol. 2013, 15, 61–71. [Google Scholar] [CrossRef]
- Zhang, L.; Tang, F.; Terracciano, L.; Hynx, D.; Kohler, R.; Bichet, S.; Hess, D.; Cron, P.; Hemmings, B.A.; Hergovich, A.; et al. NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr. Biol. 2015, 25, 296–305. [Google Scholar] [CrossRef]
- Hergovich, A.; Stegert, M.R.; Schmitz, D.; Hemmings, B.A. NDR kinases regulate essential cell processes from yeast to humans. Nat. Rev. Mol. Cell Biol. 2006, 7, 253–264. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef]
- Wada, K.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhang, Y.; Park, H.W.; Jewell, J.L.; Chen, Q.; Deng, Y.; Pan, D.; Taylor, S.S.; Lai, Z.C.; Guan, K.L. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 2013, 27, 1223–1232. [Google Scholar] [CrossRef]
- Meng, Z.; Qiu, Y.; Lin, K.C.; Kumar, A.; Placone, J.K.; Fang, C.; Wang, K.C.; Lu, S.; Pan, M.; Hong, A.W.; et al. RAP2 mediates mechanoresponses of the Hippo pathway. Nature 2018, 560, 655–660. [Google Scholar] [CrossRef]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef]
- Esposito, D.; Pant, I.; Shen, Y.; Qiao, R.F.; Yang, X.; Bai, Y.; Jin, J.; Poulikakos, P.I.; Aaronson, S.A. ROCK1 mechano-signaling dependency of human malignancies driven by TEAD/YAP activation. Nat. Commun. 2022, 13, 703. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Li, P.; Silvis, M.R.; Honaker, Y.; Lien, W.H.; Arron, S.T.; Vasioukhin, V. αE-catenin inhibits a Src-YAP1 oncogenic module that couples tyrosine kinases and the effector of Hippo signaling pathway. Genes Dev. 2016, 30, 798–811. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Vlahov, N.; Scrace, S.; Soto, M.S.; Grawenda, A.M.; Bradley, L.; Pankova, D.; Papaspyropoulos, A.; Yee, K.S.; Buffa, F.; Goding, C.R.; et al. Alternate RASSF1 Transcripts Control SRC Activity, E-Cadherin Contacts, and YAP-Mediated Invasion. Curr. Biol. 2015, 25, 3019–3034. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Wu, L.W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.G.; Gumbiner, B.M. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol. 2015, 210, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Lamar, J.M.; Xiao, Y.; Norton, E.; Jiang, Z.G.; Gerhard, G.M.; Kooner, S.; Warren, J.S.A.; Hynes, R.O. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J. Biol. Chem. 2019, 294, 2302–2317. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Zuidema, A.; Wang, W.; Sonnenberg, A. Crosstalk between Cell Adhesion Complexes in Regulation of Mechanotransduction. Bioessays 2020, 42, e2000119. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.Y.; Yu, J.; Guan, K.L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Elbediwy, A.; Vincent-Mistiaen, Z.I.; Spencer-Dene, B.; Stone, R.K.; Boeing, S.; Wculek, S.K.; Cordero, J.; Tan, E.H.; Ridgway, R.; Brunton, V.G.; et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 2016, 143, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Meng, Z.; Moroishi, T.; Lin, K.C.; Shen, G.; Mo, F.; Shao, B.; Wei, X.; Zhang, P.; Wei, Y.; et al. Heat stress activates YAP/TAZ to induce the heat shock transcriptome. Nat. Cell Biol. 2020, 22, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.W.; Meng, Z.; Yuan, H.X.; Plouffe, S.W.; Moon, S.; Kim, W.; Jho, E.H.; Guan, K.L. Osmotic stress-induced phosphorylation by NLK at Ser128 activates YAP. EMBO Rep. 2017, 18, 72–86. [Google Scholar] [CrossRef]
- Lin, K.C.; Moroishi, T.; Meng, Z.; Jeong, H.S.; Plouffe, S.W.; Sekido, Y.; Han, J.; Park, H.W.; Guan, K.L. Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat. Cell Biol. 2017, 19, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Chen, Y.; Chen, L.; Cheng, H.; Mu, C.; Li, J.; Gao, R.; Zhou, C.; Cao, L.; Liu, J.; et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat. Cell Biol. 2015, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Cheng, H.; Gao, R.; Mu, C.; Chen, L.; Wu, S.; Chen, Q.; Zhu, Y. Zyxin-Siah2-Lats2 axis mediates cooperation between Hippo and TGF-β signalling pathways. Nat. Commun. 2016, 7, 11123. [Google Scholar] [CrossRef]
- Guo, Y.; Zhu, Z.; Huang, Z.; Cui, L.; Yu, W.; Hong, W.; Zhou, Z.; Du, P.; Liu, C.Y. CK2-induced cooperation of HHEX with the YAP-TEAD4 complex promotes colorectal tumorigenesis. Nat. Commun. 2022, 13, 4995. [Google Scholar] [CrossRef]
- Peng, C.; Zhu, Y.; Zhang, W.; Liao, Q.; Chen, Y.; Zhao, X.; Guo, Q.; Shen, P.; Zhen, B.; Qian, X.; et al. Regulation of the Hippo-YAP Pathway by Glucose Sensor O-GlcNAcylation. Mol. Cell 2017, 68, 591–604.e595. [Google Scholar] [CrossRef]
- Liu, Q.; Li, J.; Zhang, W.; Xiao, C.; Zhang, S.; Nian, C.; Li, J.; Su, D.; Chen, L.; Zhao, Q.; et al. Glycogen accumulation and phase separation drives liver tumor initiation. Cell 2021, 184, 5559–5576.e5519. [Google Scholar] [CrossRef]
- Han, H.; Qi, R.; Zhou, J.J.; Ta, A.P.; Yang, B.; Nakaoka, H.J.; Seo, G.; Guan, K.L.; Luo, R.; Wang, W. Regulation of the Hippo Pathway by Phosphatidic Acid-Mediated Lipid-Protein Interaction. Mol. Cell 2018, 72, 328–340.e328. [Google Scholar] [CrossRef]
- Lee, C.K.; Jeong, S.H.; Jang, C.; Bae, H.; Kim, Y.H.; Park, I.; Kim, S.K.; Koh, G.Y. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 2019, 363, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Gailite, I.; Aerne, B.L.; Tapon, N. Differential control of Yorkie activity by LKB1/AMPK and the Hippo/Warts cascade in the central nervous system. Proc. Natl. Acad. Sci. USA 2015, 112, E5169–E5178. [Google Scholar] [CrossRef]
- O’Neill, E.; Rushworth, L.; Baccarini, M.; Kolch, W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science 2004, 306, 2267–2270. [Google Scholar] [CrossRef] [PubMed]
- Romano, D.; Nguyen, L.K.; Matallanas, D.; Halasz, M.; Doherty, C.; Kholodenko, B.N.; Kolch, W. Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nat. Cell Biol. 2014, 16, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Slemmons, K.K.; Crose, L.E.S.; Riedel, S.; Sushnitha, M.; Belyea, B.; Linardic, C.M. A Novel Notch-YAP Circuit Drives Stemness and Tumorigenesis in Embryonal Rhabdomyosarcoma. Mol. Cancer Res. 2017, 15, 1777–1791. [Google Scholar] [CrossRef]
- Pefani, D.E.; Pankova, D.; Abraham, A.G.; Grawenda, A.M.; Vlahov, N.; Scrace, S.; O’Neill, E. TGF-β Targets the Hippo Pathway Scaffold RASSF1A to Facilitate YAP/SMAD2 Nuclear Translocation. Mol. Cell 2016, 63, 156–166. [Google Scholar] [CrossRef]
- Smoot, R.L.; Werneburg, N.W.; Sugihara, T.; Hernandez, M.C.; Yang, L.; Mehner, C.; Graham, R.P.; Bronk, S.F.; Truty, M.J.; Gores, G.J. Platelet-derived growth factor regulates YAP transcriptional activity via Src family kinase dependent tyrosine phosphorylation. J. Cell. Biochem. 2018, 119, 824–836. [Google Scholar] [CrossRef]
- Cai, D.; Feliciano, D.; Dong, P.; Flores, E.; Gruebele, M.; Porat-Shliom, N.; Sukenik, S.; Liu, Z.; Lippincott-Schwartz, J. Phase separation of YAP reorganizes genome topology for long-term YAP target gene expression. Nat. Cell Biol. 2019, 21, 1578–1589. [Google Scholar] [CrossRef]
- Tang, Y.; Rowe, R.G.; Botvinick, E.L.; Kurup, A.; Putnam, A.J.; Seiki, M.; Weaver, V.M.; Keller, E.T.; Goldstein, S.; Dai, J.; et al. MT1-MMP-dependent control of skeletal stem cell commitment via a β1-integrin/YAP/TAZ signaling axis. Dev. Cell 2013, 25, 402–416. [Google Scholar] [CrossRef]
- Brielle, S.; Bavli, D.; Motzik, A.; Kan-Tor, Y.; Sun, X.; Kozulin, C.; Avni, B.; Ram, O.; Buxboim, A. Delineating the heterogeneity of matrix-directed differentiation toward soft and stiff tissue lineages via single-cell profiling. Proc. Natl. Acad. Sci. USA 2021, 118, e2016322118. [Google Scholar] [CrossRef]
- Bertero, T.; Cottrill, K.A.; Lu, Y.; Haeger, C.M.; Dieffenbach, P.; Annis, S.; Hale, A.; Bhat, B.; Kaimal, V.; Zhang, Y.Y.; et al. Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit. Cell Rep. 2015, 13, 1016–1032. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Wang, K.C.; Meng, Z. Mechanoregulation of YAP and TAZ in Cellular Homeostasis and Disease Progression. Front. Cell Dev. Biol. 2021, 9, 673599. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.; Hoorens, A.; Reynaert, H.; Halder, G.; van Grunsven, L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, H.; Wang, J.; Liu, Y.; Luo, T.; Hua, H. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J. Hematol. Oncol. 2022, 15, 34. [Google Scholar] [CrossRef]
- Deng, B.; Zhao, Z.; Kong, W.; Han, C.; Shen, X.; Zhou, C. Biological role of matrix stiffness in tumor growth and treatment. J. Transl. Med. 2022, 20, 540. [Google Scholar] [CrossRef]
- Yang, N.; Chen, T.; Wang, L.; Liu, R.; Niu, Y.; Sun, L.; Yao, B.; Wang, Y.; Yang, W.; Liu, Q.; et al. CXCR4 mediates matrix stiffness-induced downregulation of UBTD1 driving hepatocellular carcinoma progression via YAP signaling pathway. Theranostics 2020, 10, 5790–5801. [Google Scholar] [CrossRef]
- Fan, Y.; Sun, Q.; Li, X.; Feng, J.; Ao, Z.; Li, X.; Wang, J. Substrate Stiffness Modulates the Growth, Phenotype, and Chemoresistance of Ovarian Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 718834. [Google Scholar] [CrossRef]
- Huang, Q.; Hu, X.; He, W.; Zhao, Y.; Hao, S.; Wu, Q.; Li, S.; Zhang, S.; Shi, M. Fluid shear stress and tumor metastasis. Am. J. Cancer Res. 2018, 8, 763–777. [Google Scholar] [PubMed]
- Lee, H.J.; Diaz, M.F.; Price, K.M.; Ozuna, J.A.; Zhang, S.; Sevick-Muraca, E.M.; Hagan, J.P.; Wenzel, P.L. Fluid shear stress activates YAP1 to promote cancer cell motility. Nat. Commun. 2017, 8, 14122. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Ewere, A.; Diaz, M.F.; Wenzel, P.L. TAZ responds to fluid shear stress to regulate the cell cycle. Cell Cycle 2018, 17, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Li, J.; Sun, J.; Liu, L.; Chen, D.; Liu, Y. Low shear stress induces ERK nuclear localization and YAP activation to control the proliferation of breast cancer cells. Biochem. Biophys. Res. Commun. 2019, 510, 219–223. [Google Scholar] [CrossRef]
- Scutigliani, E.M.; Liang, Y.; Crezee, H.; Kanaar, R.; Krawczyk, P.M. Modulating the Heat Stress Response to Improve Hyperthermia-Based Anticancer Treatments. Cancers 2021, 13, 1243. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, W.; Cheng, J.; Deng, Z.; Zhang, P.; Zhang, A.; Xu, Z.; Pan, S.; Liao, X.; Cui, D. Heat-induced manganese-doped magnetic nanocarriers combined with Yap-siRNA for MRI/NIR-guided mild photothermal and gene therapy of hepatocellular carcinoma. Chem. Eng. J. 2021, 426, 130746. [Google Scholar] [CrossRef]
- Liu, M.; Yan, M.; Lv, H.; Wang, B.; Lv, X.; Zhang, H.; Xiang, S.; Du, J.; Liu, T.; Tian, Y.; et al. Macrophage K63-Linked Ubiquitination of YAP Promotes Its Nuclear Localization and Exacerbates Atherosclerosis. Cell Rep. 2020, 32, 107990. [Google Scholar] [CrossRef]
- Mittal, P.; Roberts, C.W.M. The SWI/SNF complex in cancer-biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, L.; Dai, T.; Qin, Z.; Lu, H.; Zhang, L.; Zhou, F. Liquid-liquid phase separation in human health and diseases. Signal Transduct. Target. Ther. 2021, 6, 290. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wu, T.; Gutman, O.; Lu, H.; Zhou, Q.; Henis, Y.I.; Luo, K. Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat. Cell Biol. 2020, 22, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Peng, Z.; Qin, M.; Liu, Y.; Wang, J.; Zhang, C.; Lin, J.; Dong, T.; Wang, L.; Li, S.; et al. Interferon-γ induces tumor resistance to anti-PD-1 immunotherapy by promoting YAP phase separation. Mol. Cell 2021, 81, 1216–1230.e1219. [Google Scholar] [CrossRef]
- Hu, X.; Wu, X.; Berry, K.; Zhao, C.; Xin, D.; Ogurek, S.; Liu, X.; Zhang, L.; Luo, Z.; Sakabe, M.; et al. Nuclear condensates of YAP fusion proteins alter transcription to drive ependymoma tumourigenesis. Nat. Cell Biol. 2023, 25, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, J.; Liu, Y.; Xie, S.A.; Zhang, J.; Zhao, C.; Zhou, Y.; Pang, W.; Yao, W.; Peng, Q.; et al. Liquid-Liquid Phase Separation of DDR1 Counteracts the Hippo Pathway to Orchestrate Arterial Stiffening. Circ. Res. 2023, 132, 87–105. [Google Scholar] [CrossRef]
- Jia, Z.; Yang, S.; Li, M.; Lei, Z.; Ding, X.; Fan, M.; Wang, D.; Xie, D.; Zhou, H.; Qiu, Y.; et al. A novel NF2 splicing mutant causes neurofibromatosis type 2 via liquid-liquid phase separation with large tumor suppressor and Hippo pathway. iScience 2022, 25, 105275. [Google Scholar] [CrossRef]
- Huang, X.; Zheng, Z.; Wu, Y.; Gao, M.; Su, Z.; Huang, Y. 14-3-3 Proteins are Potential Regulators of Liquid-Liquid Phase Separation. Cell Biochem. Biophys. 2022, 80, 277–293. [Google Scholar] [CrossRef]
- Zhang, W.; Gao, Y.; Li, P.; Shi, Z.; Guo, T.; Li, F.; Han, X.; Feng, Y.; Zheng, C.; Wang, Z.; et al. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell Res. 2014, 24, 331–343. [Google Scholar] [CrossRef]
- Cai, J.; Choi, K.; Li, H.; Pulgar Prieto, K.D.; Zheng, Y.; Pan, D. YAP-VGLL4 antagonism defines the major physiological function of the Hippo signaling effector YAP. Genes Dev. 2022, 36, 1119–1128. [Google Scholar] [CrossRef]
- Kim, J.; Piao, H.L.; Kim, B.J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef]
- Liu, C.Y.; Zha, Z.Y.; Zhou, X.; Zhang, H.; Huang, W.; Zhao, D.; Li, T.; Chan, S.W.; Lim, C.J.; Hong, W.; et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFβ-TrCP E3 ligase. J. Biol. Chem. 2010, 285, 37159–37169. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(β-TRCP). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Zhou, Z.; Kim, J.; Hang, Q.; Xiao, Z.; Ton, B.N.; Chang, L.; Liu, N.; Zeng, L.; Wang, W.; et al. SKP2- and OTUD1-regulated non-proteolytic ubiquitination of YAP promotes YAP nuclear localization and activity. Nat. Commun. 2018, 9, 2269. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xie, F.; Chu, F.; Zhang, Z.; Yang, B.; Dai, T.; Gao, L.; Wang, L.; Ling, L.; Jia, J.; et al. YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKɛ-mediated phosphorylation. Nat. Immunol. 2017, 18, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Hu, P.; Zhang, Y.; Ji, Z.; Shan, X.; Ni, L.; Ning, N.; Wang, J.; Tian, H.; Shui, G.; et al. Serine metabolism antagonizes antiviral innate immunity by preventing ATP6V0d2-mediated YAP lysosomal degradation. Cell Metab. 2021, 33, 971–987.e976. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.O.; Camargo, F.D. Hippo signalling in the liver: Role in development, regeneration and disease. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 297–312. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, J.Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell Biol. 2009, 11, 1444–1450. [Google Scholar] [CrossRef]
- Hong, X.; Nguyen, H.T.; Chen, Q.; Zhang, R.; Hagman, Z.; Voorhoeve, P.M.; Cohen, S.M. Opposing activities of the Ras and Hippo pathways converge on regulation of YAP protein turnover. EMBO J. 2014, 33, 2447–2457. [Google Scholar] [CrossRef]
- Wu, B.K.; Mei, S.C.; Chen, E.H.; Zheng, Y.; Pan, D. YAP induces an oncogenic transcriptional program through TET1-mediated epigenetic remodeling in liver growth and tumorigenesis. Nat. Genet. 2022, 54, 1202–1213. [Google Scholar] [CrossRef]
- Kim, M.; Hwang, S.; Kim, B.; Shin, S.; Yang, S.; Gwak, J.; Jeong, S.M. YAP governs cellular adaptation to perturbation of glutamine metabolism by regulating ATF4-mediated stress response. Oncogene 2023, 42, 2828–2840. [Google Scholar] [CrossRef]
- Li, X.; Zhuo, S.; Zhuang, T.; Cho, Y.S.; Wu, G.; Liu, Y.; Mu, K.; Zhang, K.; Su, P.; Yang, Y.; et al. YAP inhibits ERα and ER(+) breast cancer growth by disrupting a TEAD-ERα signaling axis. Nat. Commun. 2022, 13, 3075. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Tang, T.; Probst, G.; Konradi, A.; Jin, C.; Li, F.; Gutkind, J.S.; Fu, X.D.; Guan, K.L. Transcriptional repression of estrogen receptor alpha by YAP reveals the Hippo pathway as therapeutic target for ER(+) breast cancer. Nat. Commun. 2022, 13, 1061. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Teng, H.; Wang, Y.; Liao, G.; Weng, L.; Li, Y.; Wang, X.; Jin, J.; Jiao, C.; Chen, L.; et al. SET1A-Mediated Mono-Methylation at K342 Regulates YAP Activation by Blocking Its Nuclear Export and Promotes Tumorigenesis. Cancer Cell 2018, 34, 103–118.e109. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Kimura, R.; Hirade, K.; Yanase, S.; Nishioka, Y.; Kasuga, N.; Yamaguchi, R.; Ebi, H. Scribble mis-localization induces adaptive resistance to KRAS G12C inhibitors through feedback activation of MAPK signaling mediated by YAP-induced MRAS. Nat. Cancer 2023, 4, 829–843. [Google Scholar] [CrossRef]
- Zheng, X.; Han, H.; Liu, G.P.; Ma, Y.X.; Pan, R.L.; Sang, L.J.; Li, R.H.; Yang, L.J.; Marks, J.R.; Wang, W.; et al. LncRNA wires up Hippo and Hedgehog signaling to reprogramme glucose metabolism. EMBO J. 2017, 36, 3325–3335. [Google Scholar] [CrossRef]
- Dai, X.; Liu, H.; Shen, S.; Guo, X.; Yan, H.; Ji, X.; Li, L.; Huang, J.; Feng, X.H.; Zhao, B. YAP activates the Hippo pathway in a negative feedback loop. Cell Res. 2015, 25, 1175–1178. [Google Scholar] [CrossRef]
- He, C.; Lv, X.; Huang, C.; Hua, G.; Ma, B.; Chen, X.; Angeletti, P.C.; Dong, J.; Zhou, J.; Wang, Z.; et al. YAP1-LATS2 feedback loop dictates senescent or malignant cell fate to maintain tissue homeostasis. EMBO Rep. 2019, 20, e44948. [Google Scholar] [CrossRef]
- Moroishi, T.; Park, H.W.; Qin, B.; Chen, Q.; Meng, Z.; Plouffe, S.W.; Taniguchi, K.; Yu, F.X.; Karin, M.; Pan, D.; et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015, 29, 1271–1284. [Google Scholar] [CrossRef]
- Galli, G.G.; Carrara, M.; Yuan, W.C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; de Laat, W.; et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol. Cell 2015, 60, 328–337. [Google Scholar] [CrossRef]
- Stein, C.; Bardet, A.F.; Roma, G.; Bergling, S.; Clay, I.; Ruchti, A.; Agarinis, C.; Schmelzle, T.; Bouwmeester, T.; Schübeler, D.; et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015, 11, e1005465. [Google Scholar] [CrossRef]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, K.; Fukaya, T. Molecular architecture of enhancer-promoter interaction. Curr. Opin. Cell Biol. 2022, 74, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Battilana, G.; Zanconato, F.; Piccolo, S. Mechanisms of YAP/TAZ transcriptional control. Cell Stress 2021, 5, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, H.; Rajurkar, M.; Li, Q.; Cotton, J.L.; Ou, J.; Zhu, L.J.; Goel, H.L.; Mercurio, A.M.; Park, J.S.; et al. Tead and AP1 Coordinate Transcription and Motility. Cell Rep. 2016, 14, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Lapi, E.; Di Agostino, S.; Donzelli, S.; Gal, H.; Domany, E.; Rechavi, G.; Pandolfi, P.P.; Givol, D.; Strano, S.; Lu, X.; et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol. Cell 2008, 32, 803–814. [Google Scholar] [CrossRef]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; O’Neill, E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 2007, 27, 962–975. [Google Scholar] [CrossRef]
- Speight, P.; Kofler, M.; Szászi, K.; Kapus, A. Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and TGFβ-regulated Smad3. Nat. Commun. 2016, 7, 11642. [Google Scholar] [CrossRef]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef]
- Hong, W. Angiomotin’g YAP into the nucleus for cell proliferation and cancer development. Sci. Signal. 2013, 6, pe27. [Google Scholar] [CrossRef] [PubMed]
- Elster, D.; Tollot, M.; Schlegelmilch, K.; Ori, A.; Rosenwald, A.; Sahai, E.; von Eyss, B. TRPS1 shapes YAP/TEAD-dependent transcription in breast cancer cells. Nat. Commun. 2018, 9, 3115. [Google Scholar] [CrossRef]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.S. Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Haymond, A.; Davis, J.B.; Williams, A.; Espina, V. Protein biomarkers for subtyping breast cancer and implications for future research. Expert Rev. Proteom. 2018, 15, 131–152. [Google Scholar] [CrossRef] [PubMed]
- Hergueta-Redondo, M.; Palacios, J.; Cano, A.; Moreno-Bueno, G. “New” molecular taxonomy in breast cancer. Clin. Transl. Oncol. 2008, 10, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.G.; Tilston-Lunel, A.M.; Federico, A.; Ning, B.; Mueller, A.; Peppler, G.B.; Stampouloglou, E.; Cheng, N.; Johnson, R.L.; Lenburg, M.E.; et al. Inactivation of LATS1/2 drives luminal-basal plasticity to initiate basal-like mammary carcinomas. Nat. Commun. 2022, 13, 7198. [Google Scholar] [CrossRef]
- Furth, N.; Pateras, I.S.; Rotkopf, R.; Vlachou, V.; Rivkin, I.; Schmitt, I.; Bakaev, D.; Gershoni, A.; Ainbinder, E.; Leshkowitz, D.; et al. LATS1 and LATS2 suppress breast cancer progression by maintaining cell identity and metabolic state. Life Sci. Alliance 2018, 1, e201800171. [Google Scholar] [CrossRef]
- Britschgi, A.; Duss, S.; Kim, S.; Couto, J.P.; Brinkhaus, H.; Koren, S.; De Silva, D.; Mertz, K.D.; Kaup, D.; Varga, Z.; et al. The Hippo kinases LATS1 and 2 control human breast cell fate via crosstalk with ERα. Nature 2017, 541, 541–545. [Google Scholar] [CrossRef]
- Ma, S.; Wu, Z.; Yang, F.; Zhang, J.; Johnson, R.L.; Rosenfeld, M.G.; Guan, K.L. Hippo signalling maintains ER expression and ER(+) breast cancer growth. Nature 2021, 591, E1–E10. [Google Scholar] [CrossRef]
- Zhu, C.; Li, L.; Zhang, Z.; Bi, M.; Wang, H.; Su, W.; Hernandez, K.; Liu, P.; Chen, J.; Chen, M.; et al. A Non-canonical Role of YAP/TEAD Is Required for Activation of Estrogen-Regulated Enhancers in Breast Cancer. Mol. Cell 2019, 75, 791–806.e798. [Google Scholar] [CrossRef]
- Qu, Y.; Han, B.; Yu, Y.; Yao, W.; Bose, S.; Karlan, B.Y.; Giuliano, A.E.; Cui, X. Evaluation of MCF10A as a Reliable Model for Normal Human Mammary Epithelial Cells. PLoS ONE 2015, 10, e0131285. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.K.; Guan, J.L. Breast Cancer: Multiple Subtypes within a Tumor? Trends Cancer 2017, 3, 753–760. [Google Scholar] [CrossRef] [PubMed]
- White, S.M.; Murakami, S.; Yi, C. The complex entanglement of Hippo-Yap/Taz signaling in tumor immunity. Oncogene 2019, 38, 2899–2909. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef]
- Suo, J.; Feng, X.; Li, J.; Wang, J.; Wang, Z.; Zhang, L.; Zou, W. VGLL4 promotes osteoblast differentiation by antagonizing TEADs-inhibited Runx2 transcription. Sci. Adv. 2020, 6, eaba4147. [Google Scholar] [CrossRef]
- Yap, T.A.; Kwiatkowski, D.J.; Desai, J.; Dagogo-Jack, I.; Millward, M.; Kindler, H.L.; Tolcher, A.W.; Frentzas, S.; Thurston, A.W.; Post, L. Abstract CT006: First-in-class, first-in-human phase 1 trial of VT3989, an inhibitor of yes-associated protein (YAP)/transcriptional enhancer activator domain (TEAD), in patients (pts) with advanced solid tumors enriched for malignant mesothelioma and other tumors with neurofibromatosis 2 (NF2) mutations. Cancer Res. 2023, 83, CT006. [Google Scholar]
- Pham, T.H.; Hagenbeek, T.J.; Lee, H.J.; Li, J.; Rose, C.M.; Lin, E.; Yu, M.; Martin, S.E.; Piskol, R.; Lacap, J.A.; et al. Machine-Learning and Chemicogenomics Approach Defines and Predicts Cross-Talk of Hippo and MAPK Pathways. Cancer Discov. 2021, 11, 778–793. [Google Scholar] [CrossRef]
- Kowalczyk, W.; Romanelli, L.; Atkins, M.; Hillen, H.; Bravo González-Blas, C.; Jacobs, J.; Xie, J.; Soheily, S.; Verboven, E.; Moya, I.M.; et al. Hippo signaling instructs ectopic but not normal organ growth. Science 2022, 378, eabg3679. [Google Scholar] [CrossRef]
- Silva-Rodríguez, P.; Fernández-Díaz, D.; Bande, M.; Pardo, M.; Loidi, L.; Blanco-Teijeiro, M.J. GNAQ and GNA11 Genes: A Comprehensive Review on Oncogenesis, Prognosis and Therapeutic Opportunities in Uveal Melanoma. Cancers 2022, 14, 3066. [Google Scholar] [CrossRef]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef]
- Barbosa, I.A.M.; Gopalakrishnan, R.; Mercan, S.; Mourikis, T.P.; Martin, T.; Wengert, S.; Sheng, C.; Ji, F.; Lopes, R.; Knehr, J.; et al. Cancer lineage-specific regulation of YAP responsive elements revealed through large-scale functional epigenomic screens. Nat. Commun. 2023, 14, 3907. [Google Scholar] [CrossRef] [PubMed]
- Garcia, K.; Gingras, A.C.; Harvey, K.F.; Tanas, M.R. TAZ/YAP fusion proteins: Mechanistic insights and therapeutic opportunities. Trends Cancer 2022, 8, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Pajtler, K.W.; Wei, Y.; Okonechnikov, K.; Silva, P.B.G.; Vouri, M.; Zhang, L.; Brabetz, S.; Sieber, L.; Gulley, M.; Mauermann, M.; et al. YAP1 subgroup supratentorial ependymoma requires TEAD and nuclear factor I-mediated transcriptional programmes for tumorigenesis. Nat. Commun. 2019, 10, 3914. [Google Scholar] [CrossRef]
- Merritt, N.; Garcia, K.; Rajendran, D.; Lin, Z.Y.; Zhang, X.; Mitchell, K.A.; Borcherding, N.; Fullenkamp, C.; Chimenti, M.S.; Gingras, A.C.; et al. TAZ-CAMTA1 and YAP-TFE3 alter the TAZ/YAP transcriptome by recruiting the ATAC histone acetyltransferase complex. Elife 2021, 10, e62857. [Google Scholar] [CrossRef]
- Szulzewsky, F.; Arora, S.; Hoellerbauer, P.; King, C.; Nathan, E.; Chan, M.; Cimino, P.J.; Ozawa, T.; Kawauchi, D.; Pajtler, K.W.; et al. Comparison of tumor-associated YAP1 fusions identifies a recurrent set of functions critical for oncogenesis. Genes Dev. 2020, 34, 1051–1064. [Google Scholar] [CrossRef]
- Kupp, R.; Ruff, L.; Terranova, S.; Nathan, E.; Ballereau, S.; Stark, R.; Sekhar Reddy Chilamakuri, C.; Hoffmann, N.; Wickham-Rahrmann, K.; Widdess, M.; et al. ZFTA Translocations Constitute Ependymoma Chromatin Remodeling and Transcription Factors. Cancer Discov. 2021, 11, 2216–2229. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef]
- Warren, J.S.A.; Xiao, Y.; Lamar, J.M. YAP/TAZ Activation as a Target for Treating Metastatic Cancer. Cancers 2018, 10, 115. [Google Scholar] [CrossRef]
- Piccolo, S.; Panciera, T.; Contessotto, P.; Cordenonsi, M. YAP/TAZ as master regulators in cancer: Modulation, function and therapeutic approaches. Nat. Cancer 2023, 4, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Risson, E.; Nobre, A.R.; Maguer-Satta, V.; Aguirre-Ghiso, J.A. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat. Cancer 2020, 1, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Jahanban-Esfahlan, R.; Seidi, K.; Manjili, M.H.; Jahanban-Esfahlan, A.; Javaheri, T.; Zare, P. Tumor Cell Dormancy: Threat or Opportunity in the Fight against Cancer. Cancers 2019, 11, 1207. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, J.S.; Nobre, A.R.; Mondal, C.; Taha, I.; Farias, E.F.; Fertig, E.J.; Naba, A.; Aguirre-Ghiso, J.A.; Bravo-Cordero, J.J. A tumor-derived type III collagen-rich ECM niche regulates tumor cell dormancy. Nat. Cancer 2022, 3, 90–107. [Google Scholar] [CrossRef] [PubMed]
- Barney, L.E.; Hall, C.L.; Schwartz, A.D.; Parks, A.N.; Sparages, C.; Galarza, S.; Platt, M.O.; Mercurio, A.M.; Peyton, S.R. Tumor cell-organized fibronectin maintenance of a dormant breast cancer population. Sci. Adv. 2020, 6, eaaz4157. [Google Scholar] [CrossRef]
- Mukherjee, A.; Bravo-Cordero, J.J. Regulation of dormancy during tumor dissemination: The role of the ECM. Cancer Metastasis Rev. 2023, 42, 99–112. [Google Scholar] [CrossRef]
- Dai, J.; Cimino, P.J.; Gouin, K.H., 3rd; Grzelak, C.A.; Barrett, A.; Lim, A.R.; Long, A.; Weaver, S.; Saldin, L.T.; Uzamere, A.; et al. Astrocytic laminin-211 drives disseminated breast tumor cell dormancy in brain. Nat. Cancer 2022, 3, 25–42. [Google Scholar] [CrossRef]
- Er, E.E.; Valiente, M.; Ganesh, K.; Zou, Y.; Agrawal, S.; Hu, J.; Griscom, B.; Rosenblum, M.; Boire, A.; Brogi, E.; et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol. 2018, 20, 966–978. [Google Scholar] [CrossRef]
- Ohta, Y.; Fujii, M.; Takahashi, S.; Takano, A.; Nanki, K.; Matano, M.; Hanyu, H.; Saito, M.; Shimokawa, M.; Nishikori, S.; et al. Cell-matrix interface regulates dormancy in human colon cancer stem cells. Nature 2022, 608, 784–794. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e112. [Google Scholar] [CrossRef]
- Montagner, M.; Dupont, S. Mechanical Forces as Determinants of Disseminated Metastatic Cell Fate. Cells 2020, 9, 250. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kim, S.Y.; Tu, W.; Kim, J.; Xu, A.; Yang, Y.M.; Matsuda, M.; Reolizo, L.; Tsuchiya, T.; Billet, S.; et al. Extracellular vesicles in fatty liver promote a metastatic tumor microenvironment. Cell Metab. 2023, 35, 1209–1226.e1213. [Google Scholar] [CrossRef]
- Sun, H.; Meng, Q.; Shi, C.; Yang, H.; Li, X.; Wu, S.; Familiari, G.; Relucenti, M.; Aschner, M.; Wang, X.; et al. Hypoxia-Inducible Exosomes Facilitate Liver-Tropic Premetastatic Niche in Colorectal Cancer. Hepatology 2021, 74, 2633–2651. [Google Scholar] [CrossRef] [PubMed]
- Heinz, M.C.; Peters, N.A.; Oost, K.C.; Lindeboom, R.G.H.; van Voorthuijsen, L.; Fumagalli, A.; van der Net, M.C.; de Medeiros, G.; Hageman, J.H.; Verlaan-Klink, I.; et al. Liver Colonization by Colorectal Cancer Metastases Requires YAP-Controlled Plasticity at the Micrometastatic Stage. Cancer Res. 2022, 82, 1953–1968. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Xu, Y.; Huo, L.; Wang, R.; Li, Y.; Wang, Y.; Pizzi, M.P.; Scott, A.; Harada, K.; Ma, L.; et al. YAP1 mediates gastric adenocarcinoma peritoneal metastases that are attenuated by YAP1 inhibition. Gut 2021, 70, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Lengel, H.B.; Mastrogiacomo, B.; Connolly, J.G.; Tan, K.S.; Liu, Y.; Fick, C.N.; Dunne, E.G.; He, D.; Lankadasari, M.B.; Satravada, B.A.; et al. Genomic mapping of metastatic organotropism in lung adenocarcinoma. Cancer Cell 2023, 41, 970–985.e973. [Google Scholar] [CrossRef]
- Shih, D.J.H.; Nayyar, N.; Bihun, I.; Dagogo-Jack, I.; Gill, C.M.; Aquilanti, E.; Bertalan, M.; Kaplan, A.; D’Andrea, M.R.; Chukwueke, U.; et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat. Genet. 2020, 52, 371–377. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Moroishi, T.; Hayashi, T.; Pan, W.W.; Fujita, Y.; Holt, M.V.; Qin, J.; Carson, D.A.; Guan, K.L. The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell 2016, 167, 1525–1539.e1517. [Google Scholar] [CrossRef]
- Liu, C.; Song, Y.; Li, D.; Wang, B. Regulation of the tumor immune microenvironment by the Hippo Pathway: Implications for cancer immunotherapy. Int. Immunopharmacol. 2023, 122, 110586. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, C.G.; Kim, S.K.; Shin, S.J.; Choe, E.A.; Park, S.H.; Shin, E.C.; Kim, J. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol. Res. 2018, 6, 255–266. [Google Scholar] [CrossRef]
- Lee, B.S.; Park, D.I.; Lee, D.H.; Lee, J.E.; Yeo, M.K.; Park, Y.H.; Lim, D.S.; Choi, W.; Lee, D.H.; Yoo, G.; et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2017, 491, 493–499. [Google Scholar] [CrossRef]
- Janse van Rensburg, H.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef]
- Miao, J.; Hsu, P.C.; Yang, Y.L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef]
- Guo, X.; Zhao, Y.; Yan, H.; Yang, Y.; Shen, S.; Dai, X.; Ji, X.; Ji, F.; Gong, X.G.; Li, L.; et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev. 2017, 31, 247–259. [Google Scholar] [CrossRef]
- Ni, X.; Tao, J.; Barbi, J.; Chen, Q.; Park, B.V.; Li, Z.; Zhang, N.; Lebid, A.; Ramaswamy, A.; Wei, P.; et al. YAP Is Essential for Treg-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2018, 8, 1026–1043. [Google Scholar] [CrossRef]
- Stampouloglou, E.; Cheng, N.; Federico, A.; Slaby, E.; Monti, S.; Szeto, G.L.; Varelas, X. Yap suppresses T-cell function and infiltration in the tumor microenvironment. PLoS Biol. 2020, 18, e3000591. [Google Scholar] [CrossRef]
- Du, X.; Wen, J.; Wang, Y.; Karmaus, P.W.F.; Khatamian, A.; Tan, H.; Li, Y.; Guy, C.; Nguyen, T.M.; Dhungana, Y.; et al. Hippo/Mst signalling couples metabolic state and immune function of CD8α(+) dendritic cells. Nature 2018, 558, 141–145. [Google Scholar] [CrossRef]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Meng, F.; Chen, S.; Plouffe, S.W.; Wu, S.; Liu, S.; Li, X.; Zhou, R.; Wang, J.; Zhao, B.; et al. Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat. Cell Biol. 2017, 19, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Yu, Z.; Zhang, D.; Chen, S.; Guan, H.; Zhou, R.; Wu, Q.; Zhang, Q.; Liu, S.; Venkat Ramani, M.K.; et al. Induced phase separation of mutant NF2 imprisons the cGAS-STING machinery to abrogate antitumor immunity. Mol. Cell 2021, 81, 4147–4164.e4147. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Guan, J.; Chen, M.; Wang, W.; Li, C.; Wang, Y.; Cheng, Y.; Zhou, Z. Targeting IRF3 as a YAP agonist therapy against gastric cancer. J. Exp. Med. 2018, 215, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rendueles, M.E.R.; Krishnamoorthy, G.; Saqcena, M.; Acuña-Ruiz, A.; Revilla, G.; de Stanchina, E.; Knauf, J.A.; Lester, R.; Xu, B.; Ghossein, R.A.; et al. Yap governs a lineage-specific neuregulin1 pathway-driven adaptive resistance to RAF kinase inhibitors. Mol. Cancer 2022, 21, 213. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Sun, H.; Robichaux, J.; Pfeifer, M.; McDermott, U.; Travers, J.; Diao, L.; Xi, Y.; Tong, P.; Shen, L.; et al. A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Sci. Transl. Med. 2020, 12, eaaz4589. [Google Scholar] [CrossRef]
- Okamoto, K.; Ando, T.; Izumi, H.; Kobayashi, S.S.; Shintani, T.; Gutkind, J.S.; Yanamoto, S.; Miyauchi, M.; Kajiya, M. AXL activates YAP through the EGFR-LATS1/2 axis and confers resistance to EGFR-targeted drugs in head and neck squamous cell carcinoma. Oncogene 2023, 42, 2869–2877. [Google Scholar] [CrossRef]
- Bauzone, M.; Souidi, M.; Dessein, A.F.; Wisztorski, M.; Vincent, A.; Gimeno, J.P.; Monté, D.; Van Seuningen, I.; Gespach, C.; Huet, G. Cross-talk between YAP and RAR-RXR Drives Expression of Stemness Genes to Promote 5-FU Resistance and Self-Renewal in Colorectal Cancer Cells. Mol. Cancer Res. 2021, 19, 612–622. [Google Scholar] [CrossRef]
- Yun, M.R.; Choi, H.M.; Lee, Y.W.; Joo, H.S.; Park, C.W.; Choi, J.W.; Kim, D.H.; Kang, H.N.; Pyo, K.H.; Shin, E.J.; et al. Targeting YAP to overcome acquired resistance to ALK inhibitors in ALK-rearranged lung cancer. EMBO Mol. Med. 2019, 11, e10581. [Google Scholar] [CrossRef]
- Chaib, I.; Karachaliou, N.; Pilotto, S.; Codony Servat, J.; Cai, X.; Li, X.; Drozdowskyj, A.; Servat, C.C.; Yang, J.; Hu, C.; et al. Co-activation of STAT3 and YES-Associated Protein 1 (YAP1) Pathway in EGFR-Mutant NSCLC. J. Natl. Cancer Inst. 2017, 109, djx014. [Google Scholar] [CrossRef]
- Li, Y.; Qiu, X.; Wang, X.; Liu, H.; Geck, R.C.; Tewari, A.K.; Xiao, T.; Font-Tello, A.; Lim, K.; Jones, K.L.; et al. FGFR-inhibitor-mediated dismissal of SWI/SNF complexes from YAP-dependent enhancers induces adaptive therapeutic resistance. Nat. Cell Biol. 2021, 23, 1187–1198. [Google Scholar] [CrossRef]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A.K. Mechanisms of disease: Oncogene addiction--a rationale for molecular targeting in cancer therapy. Nat. Clin. Pr. Oncol. 2006, 3, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Honda, D.; Okumura, M.; Chihara, T. Crosstalk between the mTOR and Hippo pathways. Dev. Growth Differ. 2023, 65, 337–347. [Google Scholar] [CrossRef]
- Gan, W.; Dai, X.; Dai, X.; Xie, J.; Yin, S.; Zhu, J.; Wang, C.; Liu, Y.; Guo, J.; Wang, M.; et al. LATS suppresses mTORC1 activity to directly coordinate Hippo and mTORC1 pathways in growth control. Nat. Cell Biol. 2020, 22, 246–256. [Google Scholar] [CrossRef]
- Liang, N.; Zhang, C.; Dill, P.; Panasyuk, G.; Pion, D.; Koka, V.; Gallazzini, M.; Olson, E.N.; Lam, H.; Henske, E.P.; et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J. Exp. Med. 2014, 211, 2249–2263. [Google Scholar] [CrossRef]
- Dai, M.O.; Yan, G.; Wang, N.; Daliah, G.; Edick, A.M.; Poulet, S.; Boudreault, J.; Ali, S.; Burgos, S.A.; Lebrun, J.J. In vivo genome-wide CRISPR screen reveals breast cancer vulnerabilities and synergistic mTOR/Hippo targeted combination therapy. Nat. Commun. 2021, 12, 3055. [Google Scholar] [CrossRef]
- Li, F.L.; Guan, K.L. The two sides of Hippo pathway in cancer. Semin. Cancer Biol. 2022, 85, 33–42. [Google Scholar] [CrossRef]
- Li, X.; Zhuo, S.; Cho, Y.S.; Liu, Y.; Yang, Y.; Zhu, J.; Jiang, J. YAP antagonizes TEAD-mediated AR signaling and prostate cancer growth. EMBO J. 2023, 42, e112184. [Google Scholar] [CrossRef]
- Lee, D.H.; Park, J.O.; Kim, T.S.; Kim, S.K.; Kim, T.H.; Kim, M.C.; Park, G.S.; Kim, J.H.; Kuninaka, S.; Olson, E.N.; et al. LATS-YAP/TAZ controls lineage specification by regulating TGFβ signaling and Hnf4α expression during liver development. Nat. Commun. 2016, 7, 11961. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, W.; Pan, Y.; Gao, Y.; Deng, L.; Li, F.; Li, F.; Ma, X.; Hou, S.; Xu, J.; et al. YAP Suppresses Lung Squamous Cell Carcinoma Progression via Deregulation of the DNp63-GPX2 Axis and ROS Accumulation. Cancer Res. 2017, 77, 5769–5781. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Xiol, J.; Dill, M.T.; Yuan, W.C.; Panero, R.; Roper, J.; Osorio, F.G.; Maglic, D.; Li, Q.; Gurung, B.; et al. Regenerative Reprogramming of the Intestinal Stem Cell State via Hippo Signaling Suppresses Metastatic Colorectal Cancer. Cell Stem Cell 2020, 27, 590–604.e599. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef]
- Barry, E.R.; Morikawa, T.; Butler, B.L.; Shrestha, K.; de la Rosa, R.; Yan, K.S.; Fuchs, C.S.; Magness, S.T.; Smits, R.; Ogino, S.; et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 2013, 493, 106–110. [Google Scholar] [CrossRef]
- Li, Q.; Sun, Y.; Jarugumilli, G.K.; Liu, S.; Dang, K.; Cotton, J.L.; Xiol, J.; Chan, P.Y.; DeRan, M.; Ma, L.; et al. Lats1/2 Sustain Intestinal Stem Cells and Wnt Activation through TEAD-Dependent and Independent Transcription. Cell Stem Cell 2020, 26, 675–692.e678. [Google Scholar] [CrossRef]
- Wu, Q.; Guo, J.; Liu, Y.; Zheng, Q.; Li, X.; Wu, C.; Fang, D.; Chen, X.; Ma, L.; Xu, P.; et al. YAP drives fate conversion and chemoresistance of small cell lung cancer. Sci. Adv. 2021, 7, eabg1850. [Google Scholar] [CrossRef]
- Moya, I.M.; Castaldo, S.A.; Van den Mooter, L.; Soheily, S.; Sansores-Garcia, L.; Jacobs, J.; Mannaerts, I.; Xie, J.; Verboven, E.; Hillen, H.; et al. Peritumoral activation of the Hippo pathway effectors YAP and TAZ suppresses liver cancer in mice. Science 2019, 366, 1029–1034. [Google Scholar] [CrossRef]
- Nagata, R.; Nakamura, M.; Sanaki, Y.; Igaki, T. Cell Competition Is Driven by Autophagy. Dev. Cell 2019, 51, 99–112.e114. [Google Scholar] [CrossRef]
- Maruyama, T.; Fujita, Y. Cell competition in mammals-novel homeostatic machinery for embryonic development and cancer prevention. Curr. Opin. Cell Biol. 2017, 48, 106–112. [Google Scholar] [CrossRef]
- Liu, Z.; Yee, P.P.; Wei, Y.; Liu, Z.; Kawasawa, Y.I.; Li, W. Differential YAP expression in glioma cells induces cell competition and promotes tumorigenesis. J. Cell Sci. 2019, 132, jcs225714. [Google Scholar] [CrossRef]
- Vishwakarma, M.; Piddini, E. Outcompeting cancer. Nat. Rev. Cancer 2020, 20, 187–198. [Google Scholar] [CrossRef]
- Parker, T.M.; Gupta, K.; Palma, A.M.; Yekelchyk, M.; Fisher, P.B.; Grossman, S.R.; Won, K.J.; Madan, E.; Moreno, E.; Gogna, R. Cell competition in intratumoral and tumor microenvironment interactions. EMBO J. 2021, 40, e107271. [Google Scholar] [CrossRef]
- Luo, M.; Xu, Y.; Chen, H.; Wu, Y.; Pang, A.; Hu, J.; Dong, X.; Che, J.; Yang, H. Advances of targeting the YAP/TAZ-TEAD complex in the hippo pathway for the treatment of cancers. Eur. J. Med. Chem. 2022, 244, 114847. [Google Scholar] [CrossRef]
- Lou, J.; Lu, Y.; Cheng, J.; Zhou, F.; Yan, Z.; Zhang, D.; Meng, X.; Zhao, Y. A chemical perspective on the modulation of TEAD transcriptional activities: Recent progress, challenges, and opportunities. Eur. J. Med. Chem. 2022, 243, 114684. [Google Scholar] [CrossRef]
- Sun, Y.; Hu, L.; Tao, Z.; Jarugumilli, G.K.; Erb, H.; Singh, A.; Li, Q.; Cotton, J.L.; Greninger, P.; Egan, R.K.; et al. Pharmacological blockade of TEAD-YAP reveals its therapeutic limitation in cancer cells. Nat. Commun. 2022, 13, 6744. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Sheldon, M.; Sun, Y.; Ma, L. New Insights into YAP/TAZ-TEAD-Mediated Gene Regulation and Biological Processes in Cancer. Cancers 2023, 15, 5497. https://doi.org/10.3390/cancers15235497
Zhao Y, Sheldon M, Sun Y, Ma L. New Insights into YAP/TAZ-TEAD-Mediated Gene Regulation and Biological Processes in Cancer. Cancers. 2023; 15(23):5497. https://doi.org/10.3390/cancers15235497
Chicago/Turabian StyleZhao, Yang, Marisela Sheldon, Yutong Sun, and Li Ma. 2023. "New Insights into YAP/TAZ-TEAD-Mediated Gene Regulation and Biological Processes in Cancer" Cancers 15, no. 23: 5497. https://doi.org/10.3390/cancers15235497
APA StyleZhao, Y., Sheldon, M., Sun, Y., & Ma, L. (2023). New Insights into YAP/TAZ-TEAD-Mediated Gene Regulation and Biological Processes in Cancer. Cancers, 15(23), 5497. https://doi.org/10.3390/cancers15235497