
STAT2 Controls Colorectal Tumorigenesis and Resistance to Anti-Cancer Drugs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Mouse Models of CRC
2.2. Generation and Assessment of ApcMin/+ Tumoroids
2.2.1. Tumoroid Generation
2.2.2. Assessment of the Effects of Anti-Cancer Drugs on Tumoroids
2.3. Immunofluorescence and TUNEL
2.4. Western Blot
2.5. Quantitative Real-Time Polymerase Chain Reaction
3. Results
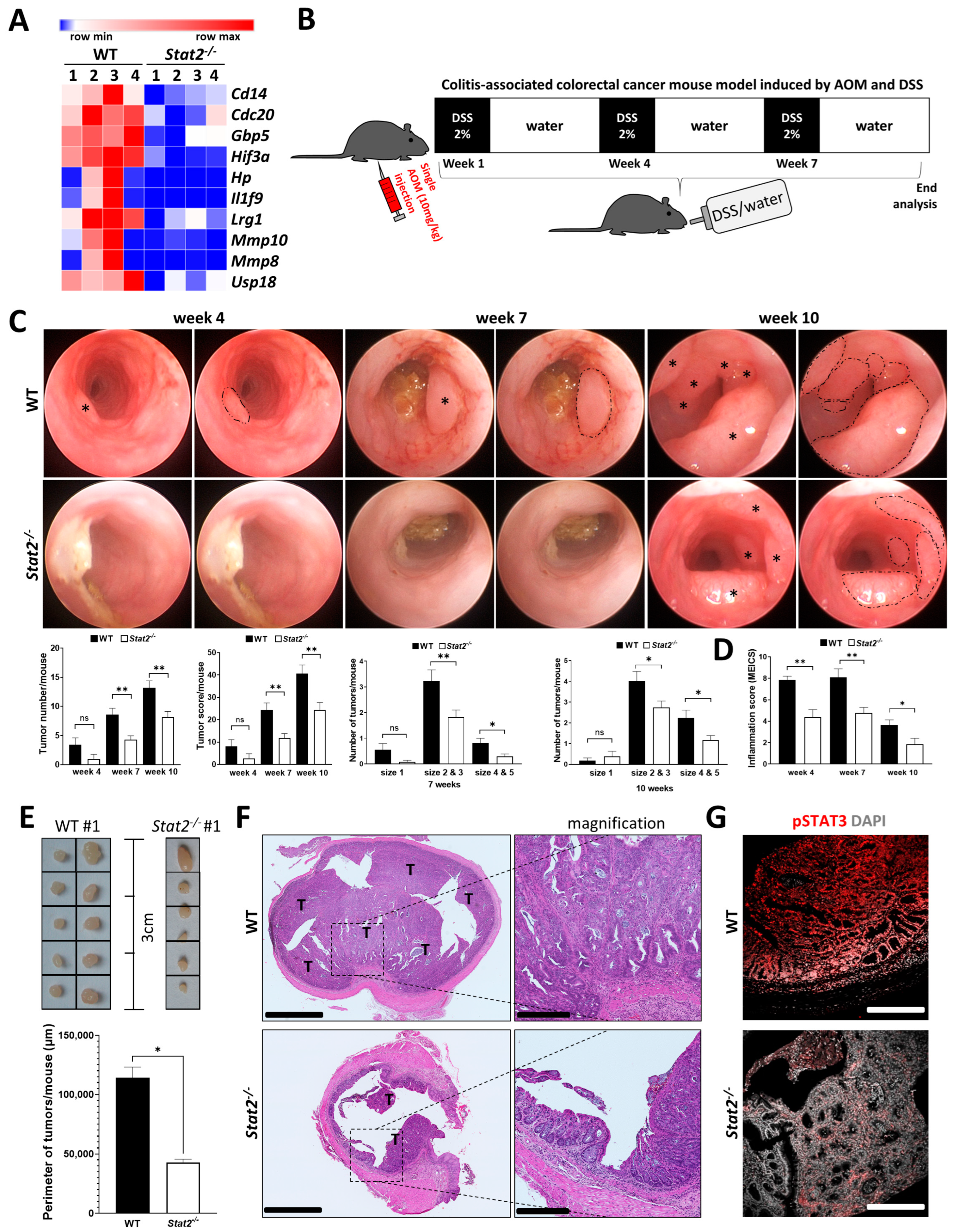
3.1. STAT2-Dependent Signaling Regulates Intestinal Inflammation and the Development of Inflammation-Driven Colorectal Tumors
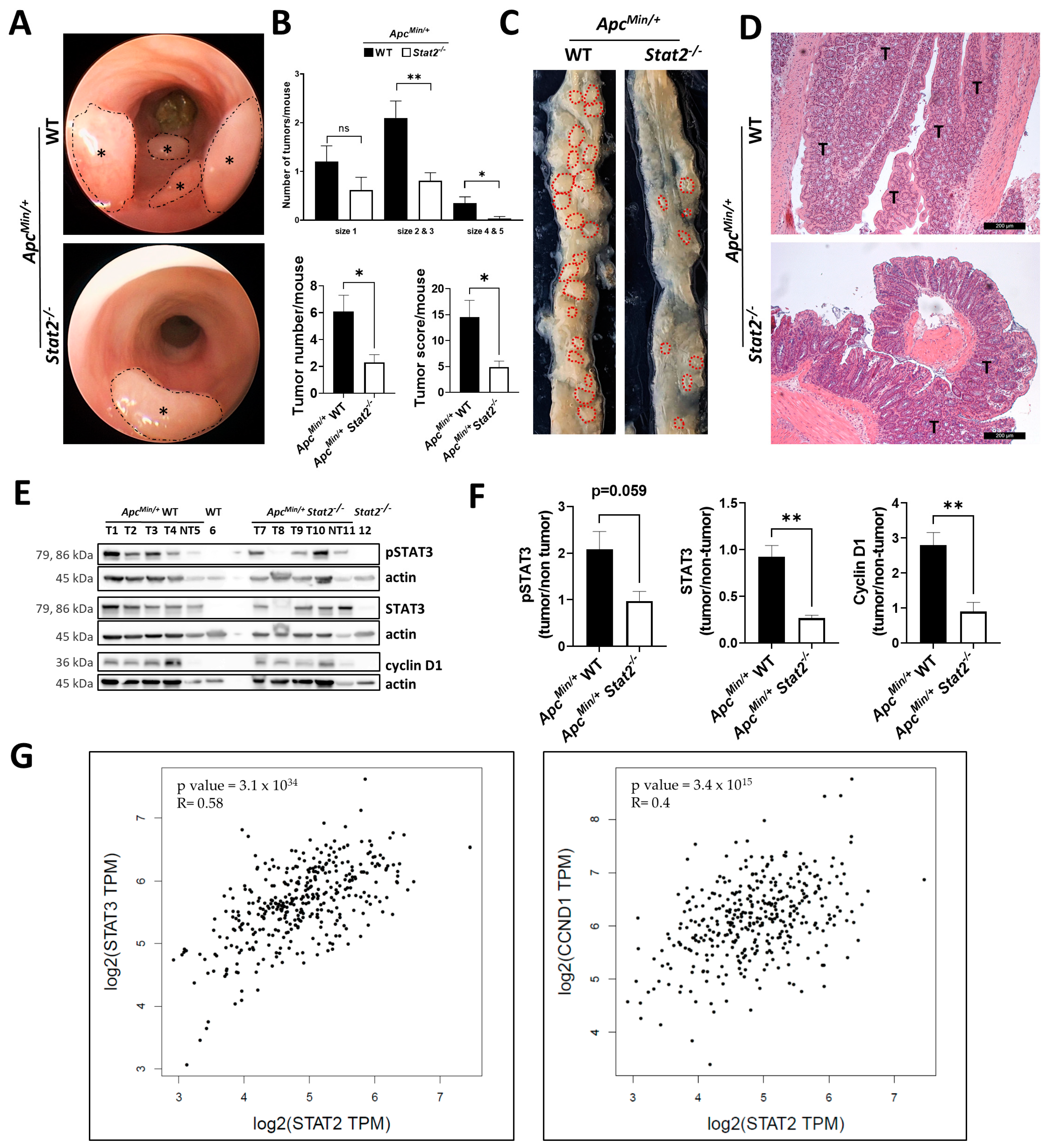
3.2. STAT2 Drives Tumorigenesis Independent of Intestinal Inflammation in ApcMin/+ Mice
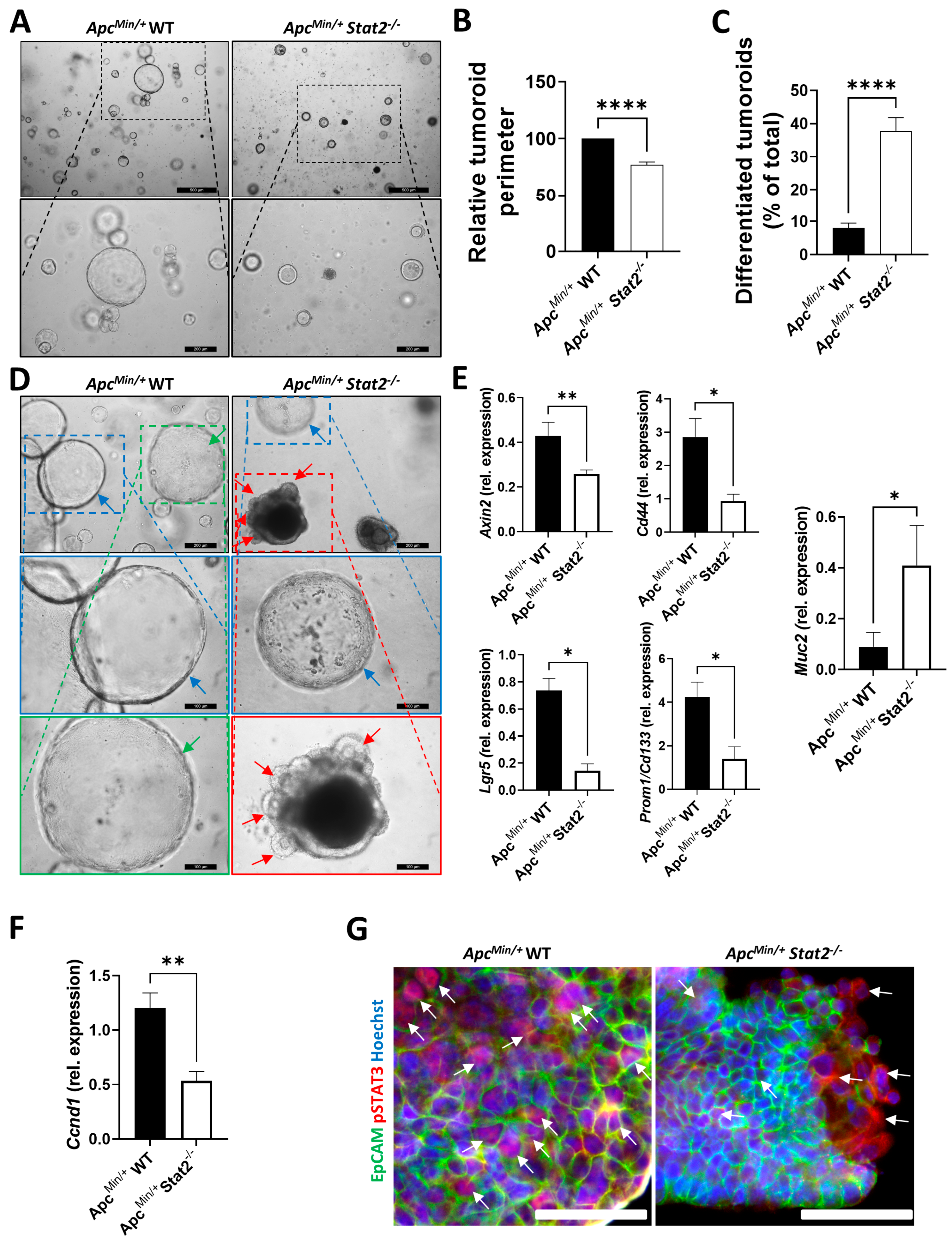
3.3. Tumoroids Derived from ApcMin/+ Stat2−/− Mice Proliferate Slower, Remain Smaller, and Become More Differentiated Compared with Tumoroids Derived from ApcMin/+ WT Mice
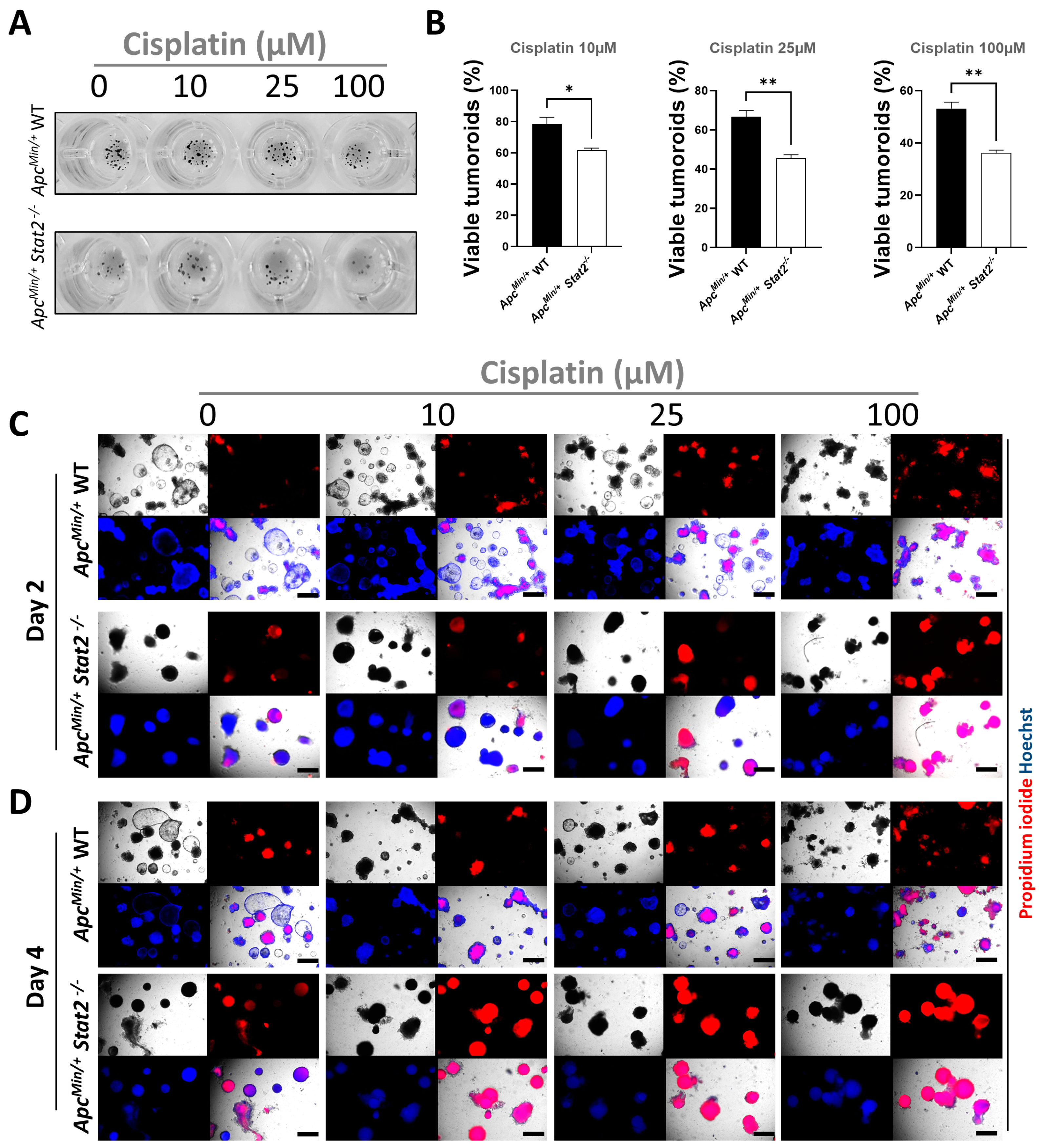
3.4. Tumoroids Derived from ApcMin/+ Stat2−/− Mice Are Highly Susceptible to Killing by Anti-Cancer Agents
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J Clin 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Beaugerie, L.; Itzkowitz, S.H. Cancers complicating inflammatory bowel disease. N. Engl. J. Med. 2015, 372, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.R.; Chang, D.K. Colorectal cancer in inflammatory bowel disease: The risk, pathogenesis, prevention and diagnosis. World J. Gastroenterol. WJG 2014, 20, 9872–9881. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Fuss, I.; Mannon, P. The fundamental basis of inflammatory bowel disease. J. Clin. Investig. 2007, 117, 514–521. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Neurath, M.F.; Travis, S.P. Mucosal healing in inflammatory bowel diseases: A systematic review. Gut 2012, 61, 1619–1635. [Google Scholar] [CrossRef]
- Travis, M.A.; Sheppard, D. TGF-beta activation and function in immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef]
- Canar, J.; Darling, K.; Dadey, R.; Gamero, A.M. The duality of STAT2 mediated type I interferon signaling in the tumor microenvironment and chemoresistance. Cytokine 2023, 161, 156081. [Google Scholar] [CrossRef]
- Lucafo, M.; Curci, D.; Franzin, M.; Decorti, G.; Stocco, G. Inflammatory Bowel Disease and Risk of Colorectal Cancer: An Overview From Pathophysiology to Pharmacological Prevention. Front. Pharmacol. 2021, 12, 772101. [Google Scholar] [CrossRef] [PubMed]
- Tschurtschenthaler, M.; Wang, J.; Fricke, C.; Fritz, T.M.; Niederreiter, L.; Adolph, T.E.; Sarcevic, E.; Kunzel, S.; Offner, F.A.; Kalinke, U.; et al. Type I interferon signalling in the intestinal epithelium affects Paneth cells, microbial ecology and epithelial regeneration. Gut 2014, 63, 1921–1931. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Li, P.; Shang, X.; Jiao, Q.; Mi, Q.; Zhu, M.; Ren, Y.; Li, J.; Li, L.; Liu, J.; Wang, C.; et al. Alteration of chromatin high-order conformation associated with oxaliplatin resistance acquisition in colorectal cancer cells. Exploration 2023, 3, 20220136. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Celi, N.; Zhang, D.; Cai, J. Magnetic Biohybrid Microrobot Multimers Based on Chlorella Cells for Enhanced Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2022, 14, 6320–6330. [Google Scholar] [CrossRef]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1957, 147, 258–267. [Google Scholar]
- Isaacs, A.; Lindenmann, J.; Valentine, R.C. Virus interference. II. Some properties of interferon. Proc. R. Soc. London. Ser. B Biol. Sci. 1957, 147, 268–273. [Google Scholar] [CrossRef]
- Schindler, C.; Fu, X.Y.; Improta, T.; Aebersold, R.; Darnell, J.E., Jr. Proteins of transcription factor ISGF-3: One gene encodes the 91- and 84-kDa ISGF-3 proteins that are activated by interferon alpha. Proc. Natl. Acad. Sci. USA 1992, 89, 7836–7839. [Google Scholar] [CrossRef]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef]
- Philips, R.L.; Wang, Y.; Cheon, H.; Kanno, Y.; Gadina, M.; Sartorelli, V.; Horvath, C.M.; Darnell, J.E., Jr.; Stark, G.R.; O’Shea, J.J. The JAK-STAT pathway at 30: Much learned, much more to do. Cell 2022, 185, 3857–3876. [Google Scholar] [CrossRef]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef]
- Chiriac, M.T.; Hracsko, Z.; Gunther, C.; Gonzalez-Acera, M.; Atreya, R.; Stolzer, I.; Wittner, L.; Dressel, A.; Schickedanz, L.; Gamez-Belmonte, R.; et al. IL-20 controls resolution of experimental colitis by regulating epithelial IFN/STAT2 signalling. Gut 2023. [Google Scholar] [CrossRef]
- Picaud, S.; Bardot, B.; De Maeyer, E.; Seif, I. Enhanced tumor development in mice lacking a functional type I interferon receptor. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2002, 22, 457–462. [Google Scholar] [CrossRef]
- Clifford, J.L.; Yang, X.; Walch, E.; Wang, M.; Lippman, S.M. Dominant negative signal transducer and activator of transcription 2 (STAT2) protein: Stable expression blocks interferon alpha action in skin squamous cell carcinoma cells. Mol. Cancer Ther. 2003, 2, 453–459. [Google Scholar] [PubMed]
- Du, Z.; Fan, M.; Kim, J.G.; Eckerle, D.; Lothstein, L.; Wei, L.; Pfeffer, L.M. Interferon-resistant Daudi cell line with a Stat2 defect is resistant to apoptosis induced by chemotherapeutic agents. J. Biol. Chem. 2009, 284, 27808–27815. [Google Scholar] [CrossRef] [PubMed]
- Gamero, A.M.; Larner, A.C. Vanadate facilitates interferon alpha-mediated apoptosis that is dependent on the Jak/Stat pathway. J. Biol. Chem. 2001, 276, 13547–13553. [Google Scholar] [CrossRef] [PubMed]
- Romero-Weaver, A.L.; Wang, H.W.; Steen, H.C.; Scarzello, A.J.; Hall, V.L.; Sheikh, F.; Donnelly, R.P.; Gamero, A.M. Resistance to IFN-alpha-induced apoptosis is linked to a loss of STAT2. Mol. Cancer Res. MCR 2010, 8, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pham-Mitchell, N.; Schindler, C.; Campbell, I.L. Dysregulated Sonic hedgehog signaling and medulloblastoma consequent to IFN-alpha-stimulated STAT2-independent production of IFN-gamma in the brain. J. Clin. Investig. 2003, 112, 535–543. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gamero, A.M.; Young, M.R.; Mentor-Marcel, R.; Bobe, G.; Scarzello, A.J.; Wise, J.; Colburn, N.H. STAT2 contributes to promotion of colorectal and skin carcinogenesis. Cancer Prev. Res. 2010, 3, 495–504. [Google Scholar] [CrossRef]
- Yue, C.; Xu, J.; Tan Estioko, M.D.; Kotredes, K.P.; Lopez-Otalora, Y.; Hilliard, B.A.; Baker, D.P.; Gallucci, S.; Gamero, A.M. Host STAT2/type I interferon axis controls tumor growth. Int. J. Cancer 2015, 136, 117–126. [Google Scholar] [CrossRef]
- Park, C.; Li, S.; Cha, E.; Schindler, C. Immune response in Stat2 knockout mice. Immunity 2000, 13, 795–804. [Google Scholar] [CrossRef]
- Becker, C.; Fantini, M.C.; Wirtz, S.; Nikolaev, A.; Kiesslich, R.; Lehr, H.A.; Galle, P.R.; Neurath, M.F. In vivo imaging of colitis and colon cancer development in mice using high resolution chromoendoscopy. Gut 2005, 54, 950–954. [Google Scholar] [CrossRef]
- Moser, A.R.; Pitot, H.C.; Dove, W.F. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990, 247, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Lindemans, C.A.; Calafiore, M.; Mertelsmann, A.M.; O’Connor, M.H.; Dudakov, J.A.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Lawrence, G.; et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 2015, 528, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Grabinger, T.; Luks, L.; Kostadinova, F.; Zimberlin, C.; Medema, J.P.; Leist, M.; Brunner, T. Ex vivo culture of intestinal crypt organoids as a model system for assessing cell death induction in intestinal epithelial cells and enteropathy. Cell Death Dis. 2014, 5, e1228. [Google Scholar] [CrossRef]
- Bode, K.J.; Mueller, S.; Schweinlin, M.; Metzger, M.; Brunner, T. A fast and simple fluorometric method to detect cell death in 3D intestinal organoids. Biotechniques 2019, 67, 23–28. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; van den Brink, S.; Geurts, V.; Beumer, J.; Clevers, H. Establishment and Culture of Human Intestinal Organoids Derived from Adult Stem Cells. Curr. Protoc. Immunol. 2020, 130, e106. [Google Scholar] [CrossRef]
- Sun, W.; Gao, J.; Yang, B.; Chen, X.; Kang, N.; Liu, W. Protocol for colitis-associated colorectal cancer murine model induced by AOM and DSS. STAR Protoc. 2023, 4, 102105. [Google Scholar] [CrossRef]
- Tanaka, T.; Kohno, H.; Suzuki, R.; Yamada, Y.; Sugie, S.; Mori, H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003, 94, 965–973. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Ahmad, S.I.; Kirk, S.H.; Eisenstark, A. Thymine metabolism and thymineless death in prokaryotes and eukaryotes. Annu. Rev. Microbiol. 1998, 52, 591–625. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ng, A.S.; Cai, S.; Li, Q.; Yang, L.; Kerr, D. Novel therapeutic strategies: Targeting epithelial-mesenchymal transition in colorectal cancer. Lancet Oncol. 2021, 22, e358–e368. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Huang, J.L.; Tseng, L.C.; Yu, P.H.; Chen, S.Y.; Lin, C.S. High Expression of Interferon Pathway Genes CXCL10 and STAT2 Is Associated with Activated T-Cell Signature and Better Outcome of Oral Cancer Patients. J. Pers. Med. 2022, 12, 140. [Google Scholar] [CrossRef]
- Friedman, K.; Brodsky, A.S.; Lu, S.; Wood, S.; Gill, A.J.; Lombardo, K.; Yang, D.; Resnick, M.B. Medullary carcinoma of the colon: A distinct morphology reveals a distinctive immunoregulatory microenvironment. Mod. Pathol. 2016, 29, 528–541. [Google Scholar] [CrossRef]
- Xue, X.; Jungles, K.; Onder, G.; Samhoun, J.; Gyorffy, B.; Hardiman, K.M. HIF-3alpha1 promotes colorectal tumor cell growth by activation of JAK-STAT3 signaling. Oncotarget 2016, 7, 11567–11579. [Google Scholar] [CrossRef]
- Marino-Crespo, O.; Cuevas-Alvarez, E.; Harding, A.L.; Murdoch, C.; Fernandez-Briera, A.; Gil-Martin, E. Haptoglobin expression in human colorectal cancer. Histol. Histopathol. 2019, 34, 953–963. [Google Scholar] [CrossRef]
- Yang, W.; Dong, H.P.; Wang, P.; Xu, Z.G.; Xian, J.; Chen, J.; Wu, H.; Lou, Y.; Lin, D.; Zhong, B. IL-36gamma and IL-36Ra Reciprocally Regulate Colon Inflammation and Tumorigenesis by Modulating the Cell-Matrix Adhesion Network and Wnt Signaling. Adv. Sci. 2022, 9, e2103035. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, X.; Zhang, J.; Fang, J.; Ge, Z.; Li, X. LRG1 promotes proliferation and inhibits apoptosis in colorectal cancer cells via RUNX1 activation. PLoS ONE 2017, 12, e0175122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, N.; Li, X.; Wu, W.; Zhang, Y.; Wang, J. High expression of USP18 is associated with the growth of colorectal carcinoma. Histol. Histopathol. 2021, 36, 697–704. [Google Scholar] [CrossRef]
- Chen, D.; Wang, H. The clinical and immune features of CD14 in colorectal cancer identified via large-scale analysis. Int. Immunopharmacol. 2020, 88, 106966. [Google Scholar] [CrossRef]
- Wu, W.J.; Hu, K.S.; Wang, D.S.; Zeng, Z.L.; Zhang, D.S.; Chen, D.L.; Bai, L.; Xu, R.H. CDC20 overexpression predicts a poor prognosis for patients with colorectal cancer. J. Transl. Med. 2013, 11, 142. [Google Scholar] [CrossRef]
- Sirnio, P.; Tuomisto, A.; Tervahartiala, T.; Sorsa, T.; Klintrup, K.; Karhu, T.; Herzig, K.H.; Makela, J.; Karttunen, T.J.; Salo, T.; et al. High-serum MMP-8 levels are associated with decreased survival and systemic inflammation in colorectal cancer. Br. J. Cancer 2018, 119, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Klupp, F.; Neumann, L.; Kahlert, C.; Diers, J.; Halama, N.; Franz, C.; Schmidt, T.; Koch, M.; Weitz, J.; Schneider, M.; et al. Serum MMP7, MMP10 and MMP12 level as negative prognostic markers in colon cancer patients. BMC Cancer 2016, 16, 494. [Google Scholar] [CrossRef]
- Arber, N.; Hibshoosh, H.; Moss, S.F.; Sutter, T.; Zhang, Y.; Begg, M.; Wang, S.; Weinstein, I.B.; Holt, P.R. Increased expression of cyclin D1 is an early event in multistage colorectal carcinogenesis. Gastroenterology 1996, 110, 669–674. [Google Scholar] [CrossRef]
- Horst, D.; Kriegl, L.; Engel, J.; Kirchner, T.; Jung, A. CD133 expression is an independent prognostic marker for low survival in colorectal cancer. Br. J. Cancer 2008, 99, 1285–1289. [Google Scholar] [CrossRef]
- Li, C.; Zuo, D.; Yin, L.; Lin, Y.; Li, C.; Liu, T.; Wang, L. Prognostic Value of MUC2 Expression in Colorectal Cancer: A Systematic Review and Meta-Analysis. Gastroenterol. Res. Pract. 2018, 2018, 6986870. [Google Scholar] [CrossRef]
- Hsu, H.C.; Liu, Y.S.; Tseng, K.C.; Hsu, C.L.; Liang, Y.; Yang, T.S.; Chen, J.S.; Tang, R.P.; Chen, S.J.; Chen, H.C. Overexpression of Lgr5 correlates with resistance to 5-FU-based chemotherapy in colorectal cancer. Int. J. Colorectal Dis. 2013, 28, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.P.; Pirnie, R.; Mesaros, C.; Blair, I.A. Cisplatin Dependent Secretion of Immunomodulatory High Mobility Group Box 1 (HMGB1) Protein from Lung Cancer Cells. Biomolecules 2023, 13, 1335. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Dominguez, R.L.; Perez-Medina, M.A.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Matias-Florentino, M.; Avila-Rios, S.; Rumbo-Nava, U.; Salgado-Aguayo, A.; Gonzalez-Gonzalez, C.; Aguilar-Cazares, D. Role of HMGB1 in Cisplatin-Persistent Lung Adenocarcinoma Cell Lines. Front. Oncol. 2021, 11, 750677. [Google Scholar] [CrossRef]
- Aalinkeel, R.; Hu, Z.; Nair, B.B.; Sykes, D.E.; Reynolds, J.L.; Mahajan, S.D.; Schwartz, S.A. Genomic Analysis Highlights the Role of the JAK-STAT Signaling in the Anti-proliferative Effects of Dietary Flavonoid-’Ashwagandha’ in Prostate Cancer Cells. Evid. Based Complement. Altern. Med. 2010, 7, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Nan, J.; Wang, Y.; Yang, J.; Stark, G.R. IRF9 and unphosphorylated STAT2 cooperate with NF-kappaB to drive IL6 expression. Proc. Natl. Acad. Sci. USA 2018, 115, 3906–3911. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Fantini, M.C.; Schramm, C.; Lehr, H.A.; Wirtz, S.; Nikolaev, A.; Burg, J.; Strand, S.; Kiesslich, R.; Huber, S.; et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004, 21, 491–501. [Google Scholar] [CrossRef]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef]
- Kolosenko, I.; Fryknas, M.; Forsberg, S.; Johnsson, P.; Cheon, H.; Holvey-Bates, E.G.; Edsbacker, E.; Pellegrini, P.; Rassoolzadeh, H.; Brnjic, S.; et al. Cell crowding induces interferon regulatory factor 9, which confers resistance to chemotherapeutic drugs. Int. J. Cancer 2015, 136, E51–E61. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef]
- Cheon, H.; Wang, Y.; Wightman, S.M.; Jackson, M.W.; Stark, G.R. How cancer cells make and respond to interferon-I. Trends Cancer 2023, 9, 83–92. [Google Scholar] [CrossRef]
- Wang, C.; Nan, J.; Holvey-Bates, E.; Chen, X.; Wightman, S.; Latif, M.B.; Zhao, J.; Li, X.; Sen, G.C.; Stark, G.R.; et al. STAT2 hinders STING intracellular trafficking and reshapes its activation in response to DNA damage. Proc. Natl. Acad. Sci. USA 2023, 120, e2216953120. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiriac, M.T.; Hracsko, Z.; Becker, C.; Neurath, M.F. STAT2 Controls Colorectal Tumorigenesis and Resistance to Anti-Cancer Drugs. Cancers 2023, 15, 5423. https://doi.org/10.3390/cancers15225423
Chiriac MT, Hracsko Z, Becker C, Neurath MF. STAT2 Controls Colorectal Tumorigenesis and Resistance to Anti-Cancer Drugs. Cancers. 2023; 15(22):5423. https://doi.org/10.3390/cancers15225423
Chicago/Turabian StyleChiriac, Mircea T., Zsuzsanna Hracsko, Christoph Becker, and Markus F. Neurath. 2023. "STAT2 Controls Colorectal Tumorigenesis and Resistance to Anti-Cancer Drugs" Cancers 15, no. 22: 5423. https://doi.org/10.3390/cancers15225423
APA StyleChiriac, M. T., Hracsko, Z., Becker, C., & Neurath, M. F. (2023). STAT2 Controls Colorectal Tumorigenesis and Resistance to Anti-Cancer Drugs. Cancers, 15(22), 5423. https://doi.org/10.3390/cancers15225423