
Self-Renewal Inhibition in Breast Cancer Stem Cells: Moonlight Role of PEDF in Breast Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Results
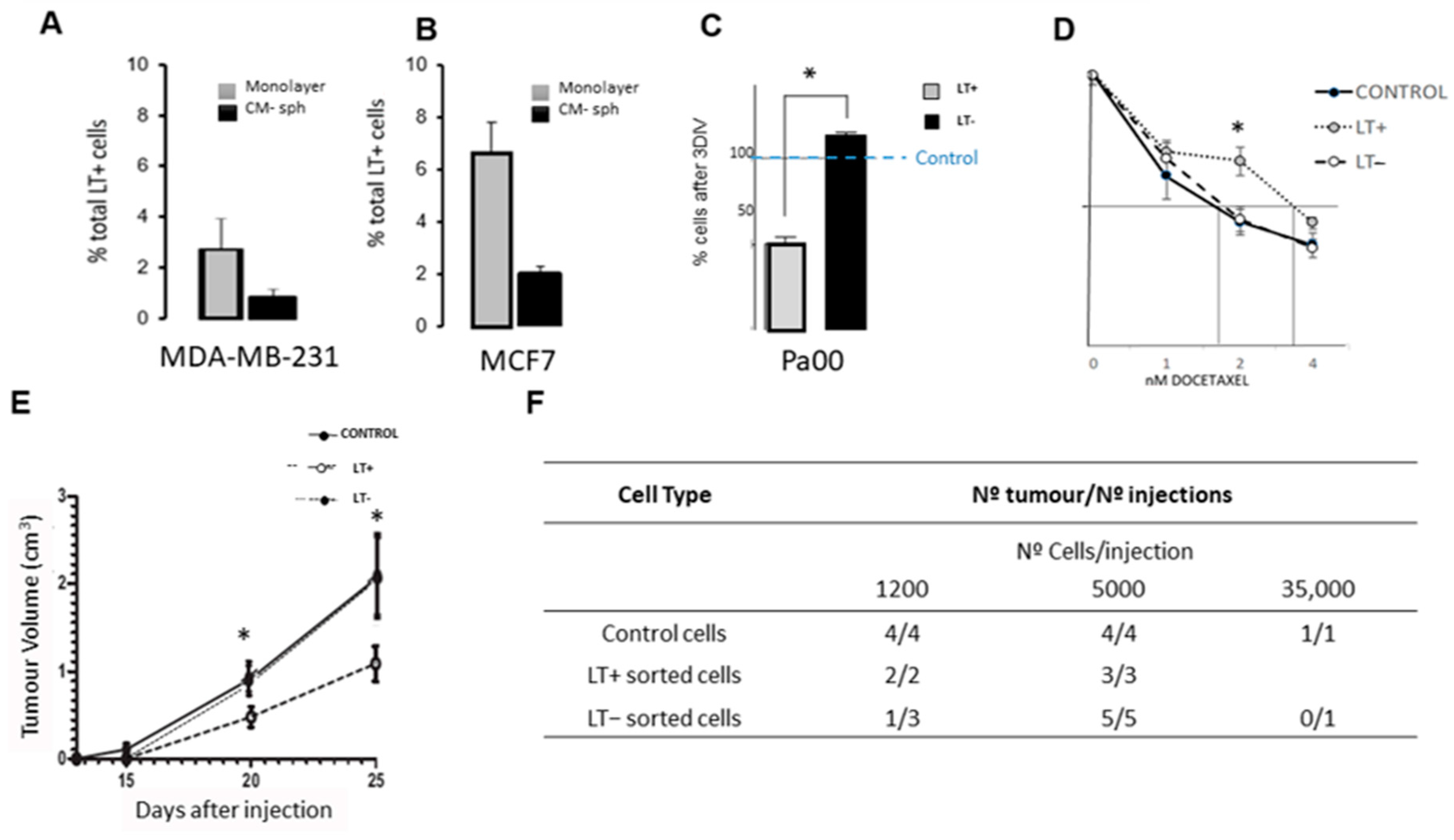
2.1. Long-Term Label-Retaining Cells Exhibit Characteristics of Cancer Stem Cells in Cancer Cell Lines and in Patient Cells
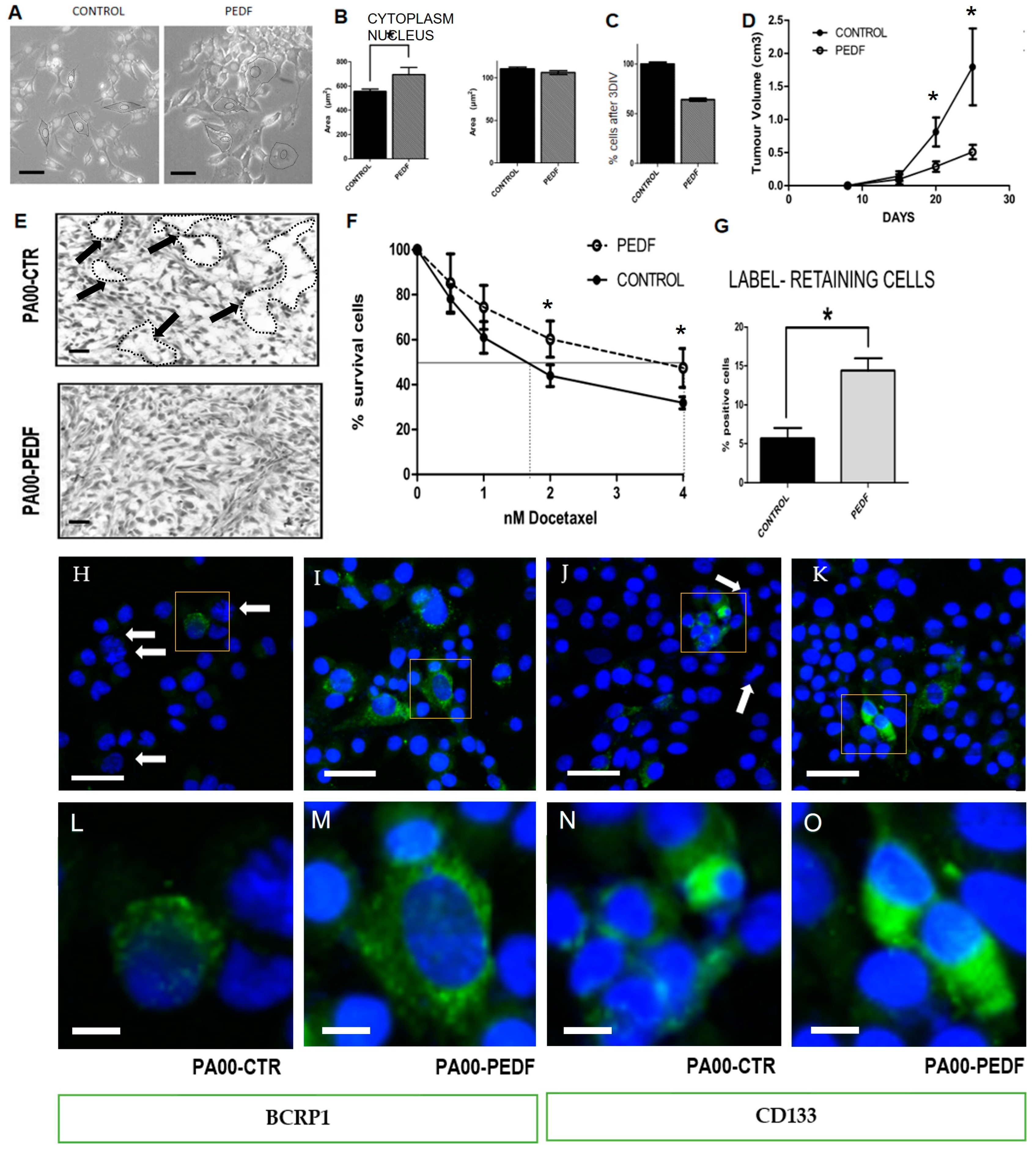
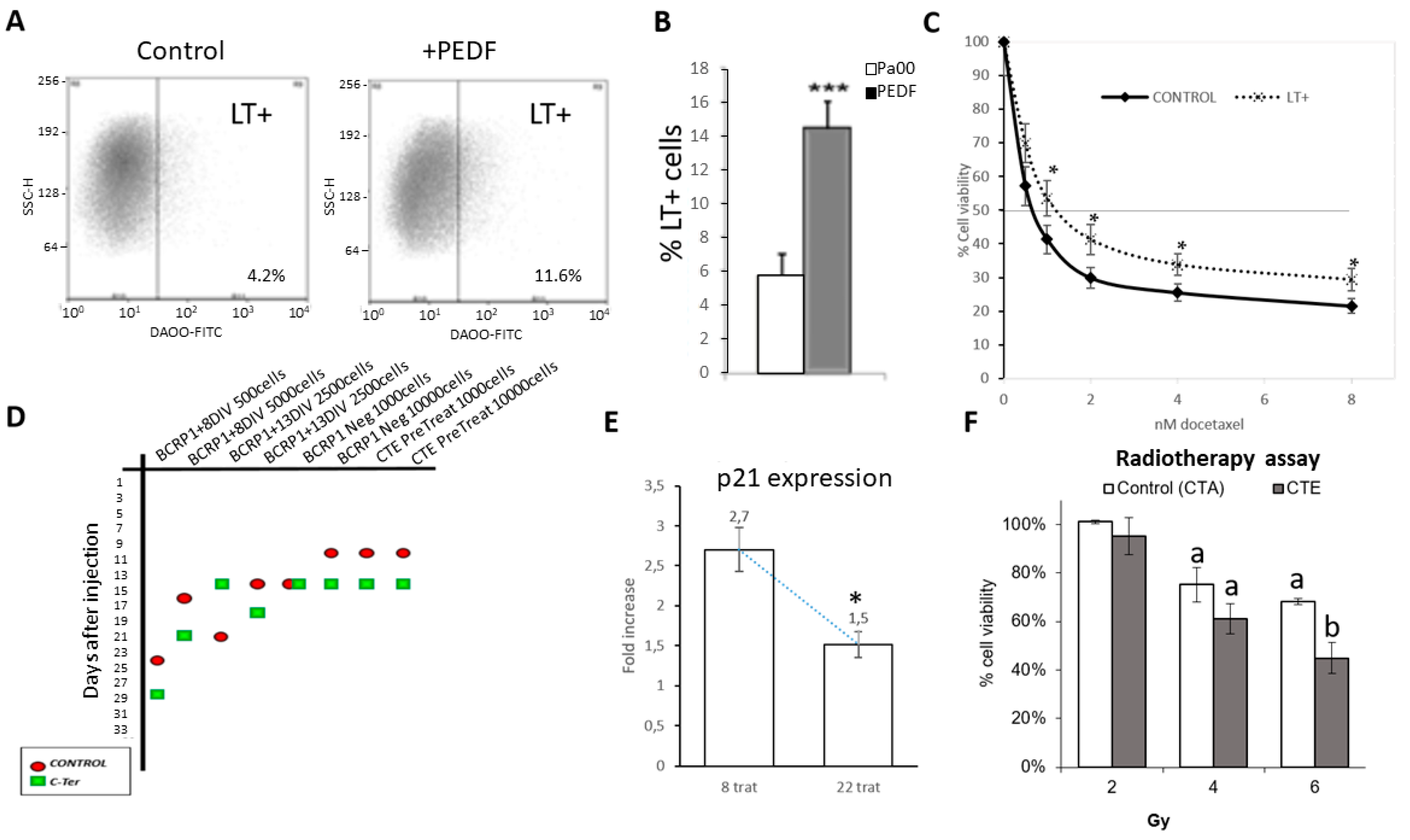
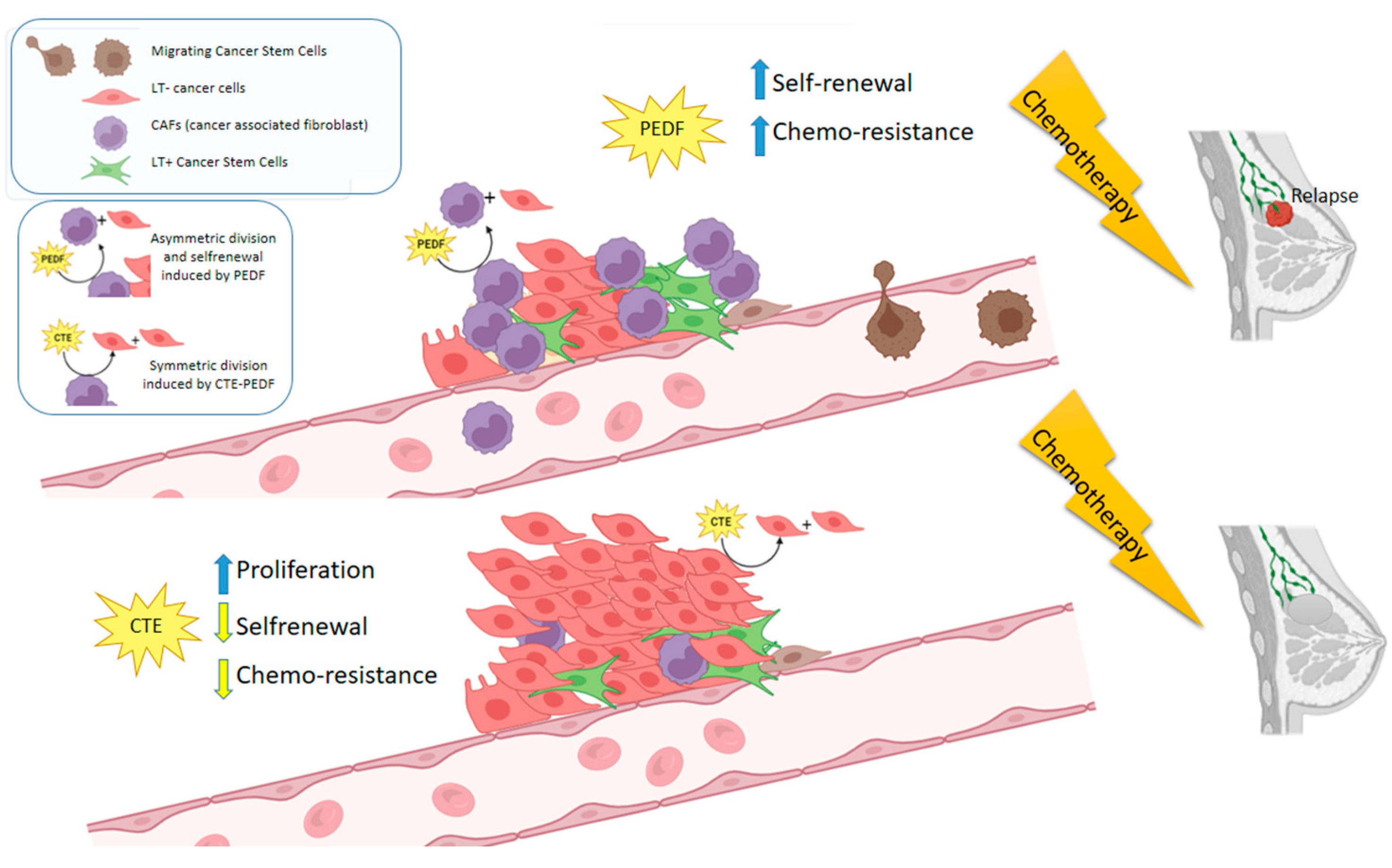
2.2. PEDF Modulate CSC Properties, Producing an Increase in Drug Resistance and Proportion of LT+ Cells
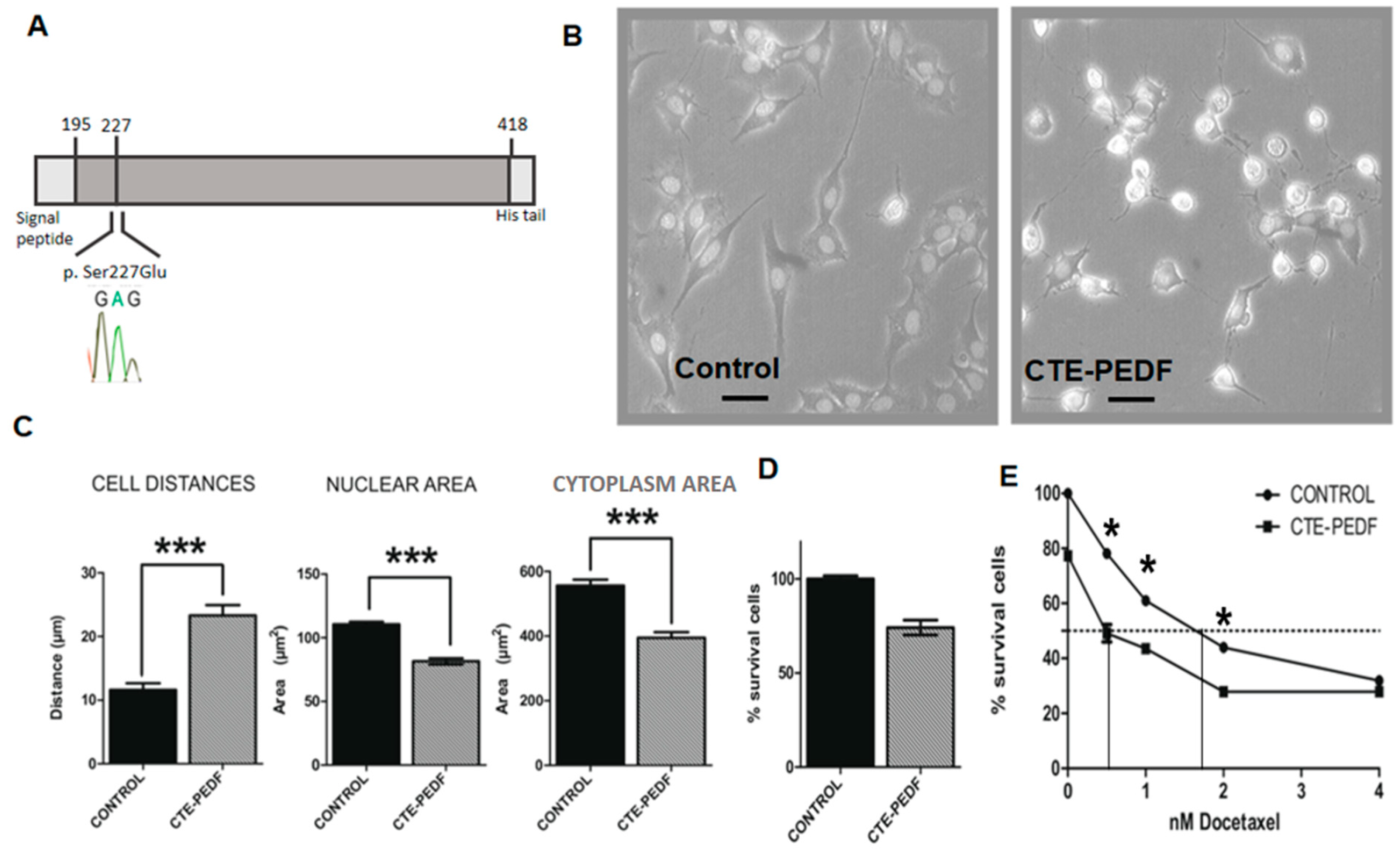
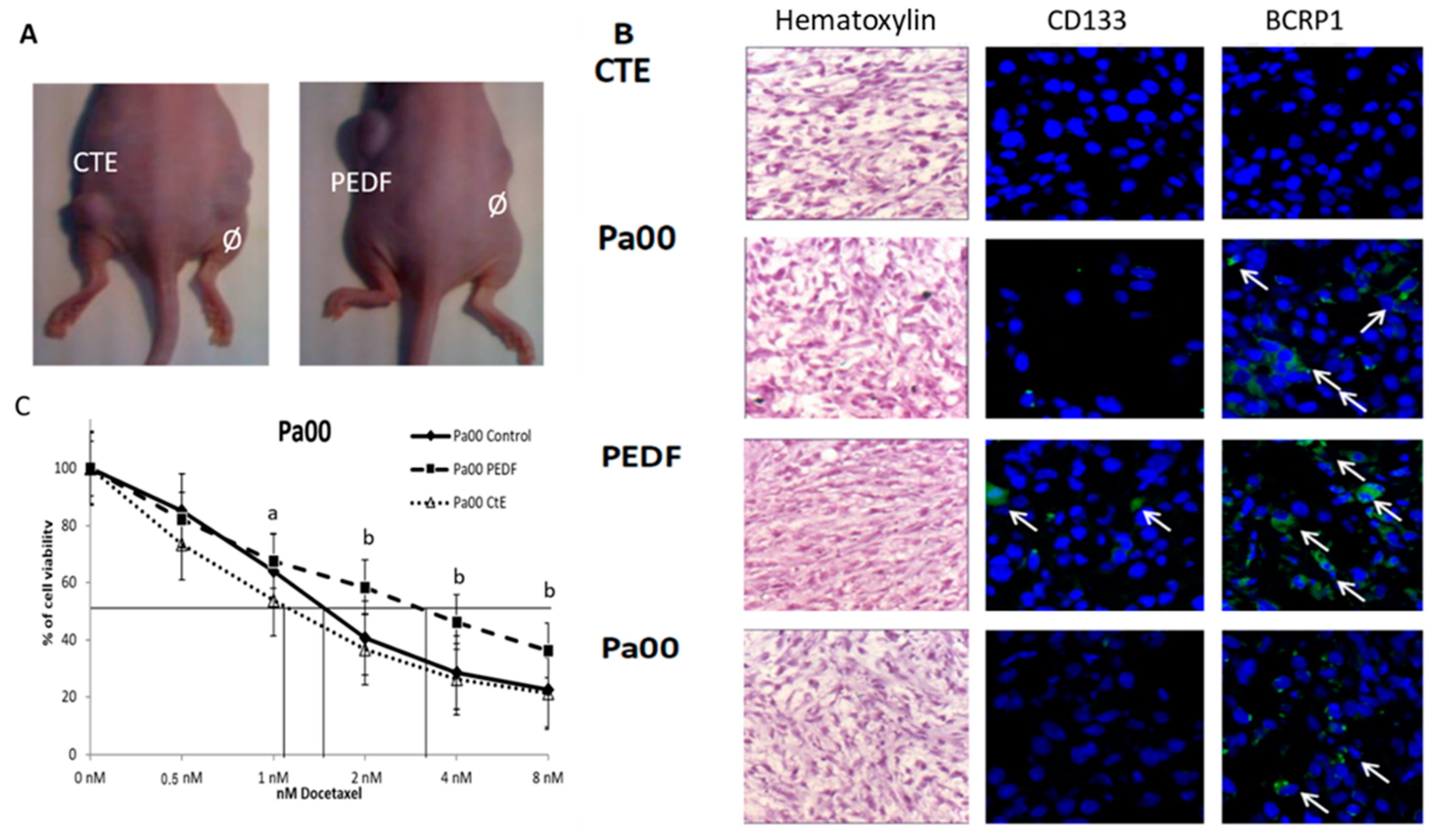
2.3. Cter-PEDF Counteracts the Effects of Native PEDF, Decreasing the Resistance in Tumors and Inducing Anoikis and Depleting CSC
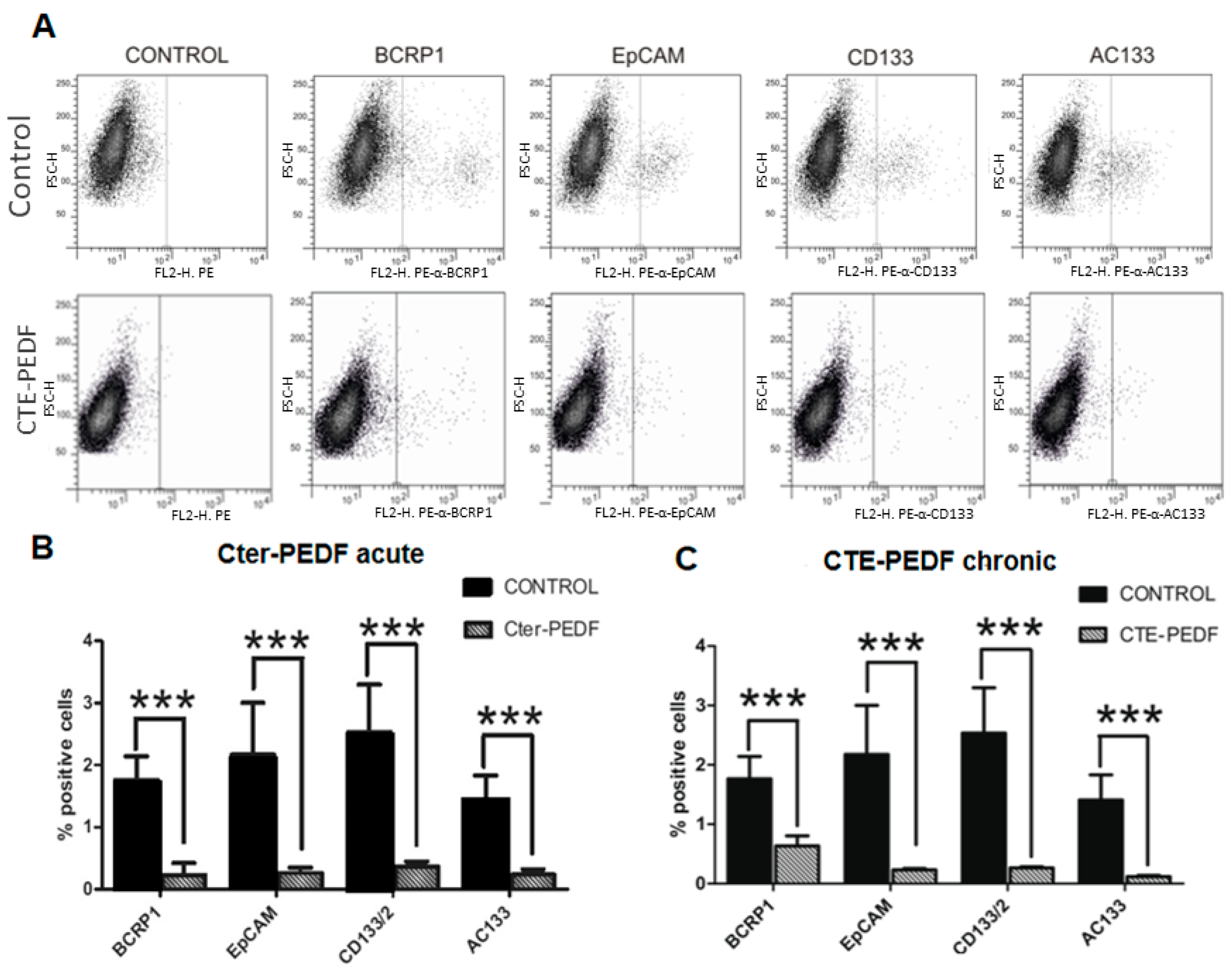
2.4. CTE-PEDF and Cter-PEDF Depletes Percentage of CSC Expressing Markers
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Staining with DDAO and Sorting
4.3. Cytometry Assay
4.4. Gene Expression
4.5. Xenografts
4.6. PEDF and Cter-PEDF, CTE-PEDF Production
4.7. Treatment with PEDF and CTE-PEDF
4.8. Treatment with Radiotherapy and CTE-PEDF
4.9. Dose–Response Curve
4.10. MTT Assay
4.11. Methyl Purple Assay
4.12. Histology
4.13. Analysis of Cell Morphology
4.14. Immnuno Assays
4.15. Measurements of the Xenograft Tumors
4.16. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AC133 | Glycosylated epitope of the pentaspan membrane protein CD133, originally identified as a marker for CD34 + hematopoietic stem and progenitor cells. |
| BCRP1 | Breast cancer resistance protein (BCRP1), also known as placenta-specific ATP-binding cassette (ABC) protein (ABCP) or ABC G-subfamily member 2 (ABCG2). |
| CD133 | CD133 antigen, also known as prominin-1, is a glycoprotein that in humans is encoded by the PROM1 gene. It has been proposed to act as an organizer of cell membrane topology. |
| CM-sph | Mammospheres. |
| CSC | Cancer stem cells. |
| CTA-PEDF | Carboxy-terminal fragment of PEDF (Ala227). |
| CTE-PEDF | Carboxy-terminal fragment of PEDF (Glu227). |
| DDAO | Near-infrared (NIR) red fluorescent probe with excitation wavelength (600–650 nm) and long emission wavelength (λem = 656 nm). DIV: days in vitro. |
| ECM | Extracellular matrix. |
| EpCAM | Epithelial cell adhesion molecule, also known as CD326. |
| GADPH | Glyceraldehyde 3-phosphate dehydrogenase. |
| LT+ | Long-term label-retaining cells. |
| MDA-MB-231 | Triple-Negative Breast Cancer Cell Line. |
| MCF7 | Epithelial cell line from breast tissue of a patient with metastatic adenocarcinoma. |
| p21 | p21 Cip1 (alternatively p21 Waf1), also known as cyclin-dependent kinase inhibitor 1 or CDK-interacting protein 1. |
| PEDF | Pigment Epithelium-Derived Factor. |
| PEDFr | PEDF receptor. |
| PKA | Protein kinase A (EC 2.7.11.11). |
| Tert | Telomerase protein. |
| TIC | Tumor-initiating cells. |
| EMT | Epithelial–mesenchymal transition. |
| TROP2 | Transmembrane glycoprotein encoded by Tacstd2 gene overexpressed in cancers. |
References
- Hulka, B.S.; Moorman, P.G. Breast cancer: Hormones and other risk factors. Maturitas 2008, 61, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Trujillo-Martínez, M.; Gómez-Flores-Ramos, L.; Sánchez-Zamorano, L.M.; Reynoso-Noverón, N.; Grimaldo, L.; Albavera-Hernández, C.; Flores-Luna, L. Pharmacogenetics in breast cancer: Implications of cytochrome p450 genes for disease-free survival in young women. Rev. Senol. Patol. Mamar. J. Breast Sci. 2021, 35, 269–284. [Google Scholar] [CrossRef]
- Pilleron, S.; Soerjomataram, I.; Charvat, H.; Chokunonga, E.; Somdyala, N.I.M.; Wabinga, H.; Korir, A.; Bray, F.; Jemal, A.; Maxwell Parkin, D. Cancer incidence in older adults in selected regions of sub-Saharan Africa, 2008–2012. Int. J. Cancer 2019, 144, 1824–1833. [Google Scholar] [CrossRef]
- Ferlay, J.; Forman, D.; Mathers, C.D.; Bray, F. Breast and cervical cancer in 187 countries between 1980 and 2010. Lancet 2012, 379, 1390–1391. [Google Scholar] [CrossRef] [PubMed]
- Leone, J.P.; Vallejo, C.T.; Hassett, M.J.; Leone, J.; Graham, N.; Tayob, N.; Freedman, R.A.; Tolaney, S.M.; Leone, B.A.; Winer, E.P.; et al. Factors associated with late risks of breast cancer-specific mortality in the SEER registry. Breast Cancer Res. Treat. 2021, 189, 203–212. [Google Scholar] [CrossRef]
- Leone, J.P.; Leone, B.A.; Tayob, N.; Hassett, M.J.; Leone, J.; Freedman, R.A.; Tolaney, S.M.; Winer, E.P.; Vallejo, C.T.; Lin, N.U. Twenty-year risks of breast cancer-specific mortality for stage III breast cancer in the surveillance, epidemiology, and end results registry. Breast Cancer Res. Treat. 2021, 187, 843–852. [Google Scholar] [CrossRef]
- Ma, L.; Liu, T.; Jin, Y.; Wei, J.; Yang, Y.; Zhang, H. ABCG2 is required for self-renewal and chemoresistance of CD133-positive human colorectal cancer cells. Tumour Biol. 2016, 37, 12889–12896. [Google Scholar] [CrossRef]
- Liu, C.; Liu, L.; Chen, X.; Cheng, J.; Zhang, H.; Shen, J.; Shan, J.; Xu, Y.; Yang, Z.; Lai, M.; et al. Sox9 regulates self-renewal and tumorigenicity by promoting symmetrical cell division of cancer stem cells in hepatocellular carcinoma. Hepatology 2016, 64, 117–129. [Google Scholar] [CrossRef]
- Moore, N.; Houghton, J.; Lyle, S. Slow-Cycling Therapy-Resistant Cancer Cells. Stem Cells Dev. 2012, 21, 1822–1830. [Google Scholar] [CrossRef]
- Srinivasan, T.; Walters, J.; Bu, P.; Than, E.B.; Tung, K.-L.; Chen, K.-Y.; Panarelli, N.; Milsom, J.; Augenlicht, L.; Lipkin, S.M.; et al. NOTCH Signaling Regulates Asymmetric Cell Fate of Fast- and Slow-Cycling Colon Cancer-Initiating Cells. Cancer Res. 2016, 76, 3411–3421. [Google Scholar] [CrossRef]
- Sánchez-Danés, A.; Larsimont, J.-C.C.; Liagre, M.; Muñoz-Couselo, E.; Lapouge, G.; Brisebarre, A.; Dubois, C.; Suppa, M.; Sukumaran, V.; del Marmol, V.; et al. A slow-cycling LGR5 tumour population mediates basal cell carcinoma relapse after therapy. Nature 2018, 562, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Castillejo, C.; Sánchez-Sánchez, F.; Andreu-Agulló, C.; Ferrón, S.R.S.R.; Aroca-Aguilar, J.D.D.; Sánchez, P.; Mira, H.; Escribano, J.; Fariñas, I. Pigment epithelium-derived factor is a niche signal for neural stem cell renewal. Nat. Neurosci. 2006, 9, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Castro-Garcia, P.; Díaz-Moreno, M.; Gil-Gas, C.; Fernández-Gómez, F.J.; Honrubia-Gómez, P.; Álvarez-Simón, C.B.; Sánchez-Sánchez, F.; Cano, J.C.C.; Almeida, F.; Blanco, V.; et al. Defects in subventricular zone pigmented epithelium-derived factor niche signaling in the senescence-accelerated mouse prone-8. FASEB J. 2015, 29, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Becerra, S.P.; Sagasti, A.; Spinella, P.; Notario, V. Pigment epithelium-derived factor behaves like a noninhibitory serpin. Neurotrophic activity does not require the serpin reactive loop. J. Biol. Chem. 1995, 270, 25992–25999. [Google Scholar] [CrossRef]
- Harries, R.L.; Owen, S.; Ruge, F.; Morgan, M.; Li, J.; Zhang, Z.; Harding, K.G.; Torkington, J.; Jiang, W.G.; Cai, J. Impact of pigment epithelium-derived factor on colorectal cancer in vitro and in vivo. Oncotarget 2018, 9, 19192–19202. [Google Scholar] [CrossRef]
- Weidle, U.H.; Birzele, F.; Tiefenthaler, G. Potential of Protein-based Anti-metastatic Therapy with Serpins and Inter α-Trypsin Inhibitors. Cancer Genom. Proteom. 2018, 15, 225–238. [Google Scholar] [CrossRef]
- Belkacemi, L.; Zhang, S.X. Anti-tumor effects of pigment epithelium-derived factor (PEDF): Implication for cancer therapy: A mini-review. J. Exp. Clin. Cancer Res. 2016, 35, 4. [Google Scholar] [CrossRef]
- Konson, A.; Pradeep, S.; Seger, R. Phosphomimetic mutants of pigment epithelium-derived factor with enhanced antiangiogenic activity as potent anticancer agents. Cancer Res. 2010, 70, 6247–6257. [Google Scholar] [CrossRef]
- Taniwaki, T.; Becerra, S.P.; Chader, G.J.; Schwartz, J.P. Pigment Epithelium-Derived Factor Is a Survival Factor for Cerebellar Granule Cells in Culture. J. Neurochem. 1995, 64, 2509–2517. [Google Scholar] [CrossRef]
- Filleur, S.; Nelius, T.; De Riese, W.; Kennedy, R.C. Characterization of pedf: A multi-functional serpin family protein. J. Cell. Biochem. 2009, 106, 769–775. [Google Scholar] [CrossRef]
- Brook, N.; Gill, J.; Chih, H.J.; Francis, K.; Dharmarajan, A.; Chan, A.; Dass, C.R. Pigment epithelium-derived factor downregulation in oestrogen receptor positive breast cancer bone metastases is associated with menopause. Mol. Cell. Endocrinol. 2023, 559, 111792. [Google Scholar] [CrossRef] [PubMed]
- Manalo, K.B.; Choong, P.F.M.; Becerra, S.P.; Dass, C.R. Pigment epithelium-derived factor as an anticancer drug and new treatment methods following the discovery of its receptors: A patent perspective. Expert Opin. Ther. Pat. 2011, 21, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Yao, S.; Tian, H.; Jiao, P.; Yang, N.; Zhu, P.; Qin, S. Pigment epithelium-derived factor alleviates endothelial injury by inhibiting Wnt/β-catenin pathway. Lipids Health Dis. 2017, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Akiba, J.; Matsui, T.; Nakamura, K.; Hisamoto, T.; Abe, M.; Ikezono, Y.; Wada, F.; Iwamoto, H.; Nakamura, T.; et al. Pigment Epithelium-Derived Factor (PEDF) Prevents Hepatic Fat Storage, Inflammation, and Fibrosis in Dietary Steatohepatitis of Mice. Dig. Dis. Sci. 2017, 62, 1527–1536. [Google Scholar] [CrossRef]
- Andreu-Agulló, C.; Morante-Redolat, J.M.; Delgado, A.C.; Fariñas, I. Vascular niche factor PEDF modulates Notch-dependent stemness in the adult subependymal zone. Nat. Neurosci. 2009, 12, 1514–1523. [Google Scholar] [CrossRef]
- Pumiglia, K.; Temple, S. PEDF: Bridging neurovascular interactions in the stem cell niche. Nat. Neurosci. 2006, 9, 299–300. [Google Scholar] [CrossRef]
- Setoguchi, T.; Taga, T.; Kondo, T. Cancer stem cells persist in many cancer cell lines. Cell Cycle 2004, 3, 414–415. [Google Scholar] [CrossRef]
- Murase, M.; Kano, M.; Tsukahara, T.; Takahashi, A.; Torigoe, T.; Kawaguchi, S.; Kimura, S.; Wada, T.; Uchihashi, Y.; Kondo, T.; et al. Side population cells have the characteristics of cancer stem-like cells/cancer-initiating cells in bone sarcomas. Br. J. Cancer 2009, 101, 1425–1432. [Google Scholar] [CrossRef]
- Kondo, T.; Setoguchi, T.; Taga, T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl. Acad. Sci. USA 2004, 101, 781–786. [Google Scholar] [CrossRef]
- Tombran-Tink, J.; Aparicio, S.; Xu, X.; Tink, A.R.; Lara, N.; Sawant, S.; Barnstable, C.J.; Zhang, S.S.M. PEDF and the serpins: Phylogeny, sequence conservation, and functional domains. J. Struct. Biol. 2005, 151, 130–150. [Google Scholar] [CrossRef]
- Craword, S.E.; Fitchev, P.; Veliceasa, D.; Volpert, O.V. The many facets of PEDF in drug discovery and disease: A diamond in the rough or split personality disorder? Expert Opin. Drug Discov. 2013, 8, 769–792. [Google Scholar] [CrossRef] [PubMed]
- Raschke, S.; Eckardt, K.; Bjørklund Holven, K.; Jensen, J.; Eckel, J. Identification and Validation of Novel Contraction-Regulated Myokines Released from Primary Human Skeletal Muscle Cells. PLoS ONE 2013, 8, e62008. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Cheng, R.; Park, K.; Benyajati, S.; Moiseyev, G.; Sun, C.; Olson, L.E.; Yang, Y.; Eby, B.K.; Lau, K.; et al. Pigment epithelium-derived factor, a noninhibitory serine protease inhibitor, is renoprotective by inhibiting the Wnt pathway. Kidney Int. 2017, 91, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, N.I.; Volpert, O.V.; Jimenez, B. Pigment epithelium-derived factor as a multifunctional antitumor factor. J. Mol. Med. 2007, 85, 15–22. [Google Scholar] [CrossRef]
- Wang, X.; Xiu, P.; Wang, F.; Zhong, J.; Wei, H.; Xu, Z.; Liu, F.; Li, J. P18 peptide, a functional fragment of pigment epithelial-derived factor, inhibits angiogenesis in hepatocellular carcinoma via modulating VEGF/VEGFR2 signalling pathway. Oncol. Rep. 2017, 38, 755–766. [Google Scholar] [CrossRef]
- Tsai, T.-H.; Shih, S.-C.; Ho, T.-C.; Ma, H.-I.; Liu, M.-Y.; Chen, S.-L.; Tsao, Y.-P.; Tsai, T.H.; Shih, S.C.; Ho, T.C.; et al. Correction: Pigment Epithelium-Derived Factor 34-mer Peptide Prevents Liver Fibrosis and Hepatic Stellate Cell Activation through Down-Regulation of the PDGF Receptor. PLoS ONE 2014, 9, e108835. [Google Scholar] [CrossRef]
- Filleur, S.; Volz, K.; Nelius, T.; Mirochnik, Y.; Huang, H.; Zaichuk, T.A.; Aymerich, M.S.; Becerra, S.P.; Yap, R.; Veliceasa, D.; et al. Two functional epitopes of pigment epithelial-derived factor block angiogenesis and induce differentiation in prostate cancer. Cancer Res. 2005, 65, 5144–5152. [Google Scholar] [CrossRef]
- Becerra, S.P. Structure-function studies on PEDF. A noninhibitory serpin with neurotrophic activity. Adv. Exp. Med. Biol. 1997, 425, 223–237. [Google Scholar] [CrossRef]
- Kawahara, K.; Yoshida, T.; Maruno, T.; Oki, H.; Ohkubo, T.; Koide, T.; Kobayashi, Y. Spatiotemporal regulation of PEDF signaling by type I collagen remodeling. Proc. Natl. Acad. Sci. USA 2020, 117, 11450–11458. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Yamagishi, S.-I.; Sata, M. Structure-function relationships of PEDF. Curr. Mol. Med. 2010, 10, 302–311. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, F.; Aroca-Aguilar, J.-D.; Segura, I.; Ramírez-Castillejo, C.; Riese, H.H.; Coca-Prados, M.; Escribano, J. Expression and purification of functional recombinant human pigment epithelium-derived factor (PEDF) secreted by the yeast Pichia pastoris. J. Biotechnol. 2008, 134, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Castro-Garcia, P.; Gilgas, C.; Ferre-Fernandez, J.-J.; Sánchez-Sánchez, F.; Gil-Gas, C.; Honrubia-Gómez, P.; Belen Alvarez-Simón, C.; Ferré-Fernández, J.-J.J.-J.; Luis Sánchez-Sánchez, J.; Ma Garcia-Bueno, J.; et al. C-Terminal-PEDF Reduces IC50 Doses and Chemoresistant Population of CD133 and BCRP1-Positve Cancer Stem Like Cells. J. Anal. Oncol. 2013, 2, 195–208. [Google Scholar] [CrossRef]
- Honrubia-Gómez, P.; López-Garrido, M.-P.M.-P.; Gil-Gas, C.; Sánchez-Sánchez, J.; Alvarez-Simon, C.; Cuenca-Escalona, J.; Perez, A.F.A.F.; Arias, E.; Moreno, R.; Sánchez-Sánchez, F.; et al. Pedf derived peptides affect colorectal cancer cell lines resistance and tumour re-growth capacity. Oncotarget 2019, 10, 2973–2986. [Google Scholar] [CrossRef] [PubMed]
- Herpel, E.; Jensen, K.; Muley, T.; Warth, A.; Schnabel, P.A.; Meister, M.; Herth, F.J.F.; Dienemann, H.; Thomas, M.; Gottschling, S. The cancer stem cell antigens CD133, BCRP1/ABCG2 and CD117/c-KIT are not associated with prognosis in resected early-stage non-small cell lung cancer. Anticancer Res. 2011, 31, 4491–4500. [Google Scholar]
- Hu, J.; Li, J.; Yue, X.; Wang, J.; Liu, J.; Sun, L.; Kong, D. Expression of the cancer stem cell markers ABCG2 and OCT-4 in right-sided colon cancer predicts recurrence and poor outcomes. Oncotarget 2017, 8, 28463–28470. [Google Scholar] [CrossRef]
- Nicolazzo, C.; Raimondi, C.; Francescangeli, F.; Ceccarelli, S.; Trenta, P.; Magri, V.; Marchese, C.; Zeuner, A.; Gradilone, A.; Gazzaniga, P. EpCAM-Expressing Circulating Tumor Cells in Colorectal Cancer. Int. J. Biol. Markers 2017, 32, 415–420. [Google Scholar] [CrossRef]
- Xiang, D.; Shigdar, S.; Bean, A.G.; Bruce, M.; Yang, W.; Mathesh, M.; Wang, T.; Yin, W.; Tran, P.H.-L.; Shamaileh, H.A.; et al. Transforming doxorubicin into a cancer stem cell killer via EpCAM aptamer-mediated delivery. Theranostics 2017, 7, 4071–4086. [Google Scholar] [CrossRef]
- Ying, X.; Wu, J.; Meng, X.; Zuo, Y.; Xia, Q.; Chen, J.; Feng, Y.; Liu, R.; Li, L.; Huang, W. AC133 expression associated with poor prognosis in stage II colorectal cancer. Med. Oncol. 2013, 30, 356. [Google Scholar] [CrossRef]
- Lang, J.; Lan, X.; Liu, Y.; Jin, X.; Wu, T.; Sun, X.; Wen, Q.; An, R. Targeting cancer stem cells with an 131I-labeled anti-AC133 monoclonal antibody in human colorectal cancer xenografts. Nucl. Med. Biol. 2015, 42, 505–512. [Google Scholar] [CrossRef]
- Barrantes-Freer, A.; Renovanz, M.; Eich, M.; Braukmann, A.; Sprang, B.; Spirin, P.; Pardo, L.A.; Giese, A.; Kim, E.L. CD133 Expression Is Not Synonymous to Immunoreactivity for AC133 and Fluctuates throughout the Cell Cycle in Glioma Stem-Like Cells. PLoS ONE 2015, 10, e0130519. [Google Scholar] [CrossRef]
- Bourseau-Guilmain, E.; Béjaud, J.; Griveau, A.; Lautram, N.; Hindré, F.; Weyland, M.; Benoit, J.P.; Garcion, E. Development and characterization of immuno-nanocarriers targeting the cancer stem cell marker AC133. Int. J. Pharm. 2012, 423, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Livas, T.; Kyprianou, N. Anoikis and EMT: Lethal "Liaisons" during Cancer Progression. Crit. Rev. Oncog. 2016, 21, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Daniela da Silva, S.; Xu, B.; Maschietto, M.; Marchi, F.A.; Alkailani, M.I.; Bijian, K.; Xiao, D.; Alaoui-Jamali, M.A. Traf2 Cooperates with Focal Adhesion Signaling To Regulate Cancer Cell Susceptibility To Anoikis. Mol. Cancer Ther. 2018, 18, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Saharat, K.; Lirdprapamongkol, K.; Chokchaichamnankit, D.; Srisomsap, C.; Svasti, J.; Paricharttanakul, N.M. Tumor Susceptibility Gene 101 Mediates Anoikis Resistance of Metastatic Thyroid Cancer Cells. Cancer Genom. Proteom. 2018, 15, 473–483. [Google Scholar] [CrossRef]
- Du, S.; Miao, J.; Zhu, Z.; Xu, E.; Shi, L.; Ai, S.; Wang, F.; Kang, X.; Chen, H.; Lu, X.; et al. NADPH oxidase 4 regulates anoikis resistance of gastric cancer cells through the generation of reactive oxygen species and the induction of EGFR. Cell Death Dis. 2018, 9, 948. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef]
- Gilmore, A.P. Anoikis. Cell Death Differ. 2005, 12 (Suppl. S2), 1473–1477. [Google Scholar] [CrossRef]
- Fitzgerald, D.P.; Subramanian, P.; Deshpande, M.; Graves, C.; Gordon, I.; Qian, Y.; Snitkovsky, Y.; Liewehr, D.J.; Steinberg, S.M.; Paltán-Ortiz, J.D.; et al. Opposing effects of pigment epithelium-derived factor on breast cancer cell versus neuronal survival: Implication for brain metastasis and metastasis-induced brain damage. Cancer Res. 2012, 72, 144–153. [Google Scholar] [CrossRef]
- Cai, J.; Parr, C.; Watkins, G.; Jiang, W.G.; Boulton, M. Decreased pigment epithelium-derived factor expression in human breast cancer progression. Clin. Cancer Res. 2006, 12, 3510–3517. [Google Scholar] [CrossRef]
- Brook, N.; Brook, E.; Dass, C.R.; Chan, A.; Dharmarajan, A. Pigment Epithelium-Derived Factor and Sex Hormone-Responsive Cancers. Cancers 2020, 12, 3483. [Google Scholar] [CrossRef]
- Brook, N.; Gill, J.; Dharmarajan, A.; Chan, A.; Dass, C.R. NFκB-Mediated Mechanisms Drive PEDF Expression and Function in Pre- and Post-Menopausal Oestrogen Levels in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 5641. [Google Scholar] [CrossRef] [PubMed]
- Filiz, G.; Dass, C.R. Reduction in tumour cell invasion by pigment epithelium-derived factor is mediated by membrane type-1 matrix metalloproteinase downregulation. Pharmazie 2012, 67, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Abooshahab, R.; Hooshmand, K.; Salami, H.-A.; Dass, C.R. The Impact of Pigment-Epithelium-Derived Factor on MCF-7 Cell Metabolism in the Context of Glycaemic Condition. Pharmaceutics 2023, 15, 2140. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, P.; Deshpande, M.; Locatelli-Hoops, S.; Moghaddam-Taaheri, S.; Gutierrez, D.; Fitzgerald, D.P.; Guerrier, S.; Rapp, M.; Notario, V.; Becerra, S.P. Identification of pigment epithelium-derived factor protein forms with distinct activities on tumor cell lines. J. Biomed. Biotechnol. 2012, 2012, 425907. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Zhou, T.; Fang, S.; Jia, M.; Xu, Z.; Dai, Z.; Li, C.; Li, S.; Li, L.; Zhang, T.; et al. Pigment epithelium-derived factor (PEDF) inhibits breast cancer metastasis by down-regulating fibronectin. Breast Cancer Res. Treat. 2014, 148, 61–72. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, M.; Xu, P.; Yu, Y.; Ye, G.; Zhang, L.; Wu, A. Expression of pigment epithelium-derived factor is associated with a good prognosis and is correlated with epithelial-mesenchymal transition-related genes in infiltrating ductal breast carcinoma. Oncol. Lett. 2016, 11, 116–124. [Google Scholar] [CrossRef]
- Zhou, D.; Xu, P.; Zhang, M.; Ye, G.; Zhang, L. Inhibitory effects of pigment epithelium-derived factor on epithelial-mesenchymal transition, migration and invasion of breast cancer. Int. J. Clin. Exp. Pathol. 2017, 10, 10593–10602. [Google Scholar]
- Belkacemi, L.; Atkins, J.L.; Yang, L.; Gadgil, P.; Sater, A.K.; Chow, D.S.; Bose, R.N.; Zhang, S.X. Phosphaplatin anti-tumor effect enhanced by liposomes partly via an up-regulation of PEDF in breast cancer. Anticancer Res. 2018, 38, 623–646. [Google Scholar] [CrossRef]
- Tsuruhisa, S.; Matsui, T.; Koga, Y.; Sotokawauchi, A.; Yagi, M.; Yamagishi, S.-I. Pigment epithelium-derived factor inhibits advanced glycation end product-induced proliferation, VEGF and MMP-9 expression in breast cancer cells via interaction with laminin receptor. Oncol. Lett. 2021, 22, 629. [Google Scholar] [CrossRef]
- Jones, I.C.; Carnagarin, R.; Armstrong, J.; Lin, D.P.L.; Baxter-Holland, M.; Elahy, M.; Dass, C.R. Pigment Epithelium-Derived Factor: Inhibition of Phosphorylation of Insulin Receptor (IR)/IR Substrate (IRS), Osteogeneration from Adipocytes, and Increased Levels Due to Doxorubicin Exposure. Pharmaceutics 2023, 15, 1960. [Google Scholar] [CrossRef]
- Bhadada, S.V.; Goyal, B.R.; Patel, M.M. Angiogenic targets for potential disorders. Fundam. Clin. Pharmacol. 2011, 25, 29–47. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Cheng, S.Q.; Ji, H.F.; Wang, J.S.; Xu, H.T.; Zhang, G.Q.; Pang, D. Evaluation of protein pigment epithelium-derived factor (PEDF) and microvessel density (MVD) as prognostic indicators in breast cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, K.; Bar-Joseph, H.; Grossman, H.; Hasky, N.; Uri-Belapolsky, S.; Stemmer, S.M.; Chuderland, D.; Shalgi, R.; Ben-Aharon, I. Pigment epithelium-derived factor alleviates tamoxifen-induced endometrial hyperplasia. Mol. Cancer Ther. 2015, 14, 2840–2849. [Google Scholar] [CrossRef]
- Mao, Y.; Zhu, L.; Huang, Z.; Luo, C.; Zhou, T.; Li, L.; Wang, G.; Yang, Z.; Qi, W.; Yang, X.; et al. Stem-like tumor cells involved in heterogeneous vasculogenesis in breast cancer. Endocr. Relat. Cancer 2020, 27, 23–39. [Google Scholar] [CrossRef]
- Pedersen, R.N.; Esen, B.Ö.; Mellemkjær, L.; Christiansen, P.; Ejlertsen, B.; Lash, T.L.; Nørgaard, M.; Cronin-Fenton, D. The Incidence of Breast Cancer Recurrence 10–32 Years After Primary Diagnosis. JNCI J. Natl. Cancer Inst. 2022, 114, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Rumble, R.B.; Macrae, E.; Barton, D.L.; Connolly, H.K.; Dickler, M.N.; Fallowfield, L.; Fowble, B.; Ingle, J.N.; Jahanzeb, M.; et al. Endocrine Therapy for Hormone Receptor-Positive Metastatic Breast Cancer: American Society of Clinical Oncology Guideline. J. Clin. Oncol. 2016, 34, 3069–3103. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef]
- Martin, T.A.; Jiang, W.G. Evaluation of the expression of stem cell markers in human breast cancer reveals a correlation with clinical progression and metastatic disease in ductal carcinoma. Oncol. Rep. 2014, 31, 262–272. [Google Scholar] [CrossRef]
- Moghbeli, M.; Moghbeli, F.; Forghanifard, M.M.; Abbaszadegan, M.R. Cancer stem cell detection and isolation. Med. Oncol. 2014, 31, 69. [Google Scholar] [CrossRef]
- Kumar, S.; Nandi, A.; Singh, S.; Regulapati, R.; Li, N.; Tobias, J.W.; Siebel, C.W.; Blanco, M.A.; Klein-Szanto, A.J.; Lengner, C.; et al. Dll1+ quiescent tumor stem cells drive chemoresistance in breast cancer through NF-κB survival pathway. Nat. Commun. 2021, 12, 432. [Google Scholar] [CrossRef]
- Yin, J.; Park, G.; Kim, T.H.; Hong, J.H.; Kim, Y.-J.; Jin, X.; Kang, S.; Jung, J.-E.; Kim, J.-Y.J.H.; Yun, H.; et al. Pigment Epithelium-Derived Factor (PEDF) Expression Induced by EGFRvIII Promotes Self-renewal and Tumor Progression of Glioma Stem Cells. PLoS Biol. 2015, 13, e1002152. [Google Scholar] [CrossRef] [PubMed]
- Chi, F.; Jin, X.; Chen, L.; He, G.; Han, S. TRG16, targeted by miR-765, inhibits breast cancer stem cell-like properties via regulating the NF-κB pathway. Mol. Cell. Biochem. 2022, 477, 2801–2816. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wang, J.; Zhen, S.; Duan, Y.; Li, Q.; Liu, L. C1ql4 regulates breast cancer cell stemness and epithelial-mesenchymal transition through PI3K/AKT/NF-κB signaling pathway. Front. Oncol. 2023, 13, 1192482. [Google Scholar] [CrossRef]
- Sahoo, S.; Ashraf, B.; Duddu, A.S.; Biddle, A.; Jolly, M.K. Interconnected high-dimensional landscapes of epithelial–mesenchymal plasticity and stemness in cancer. Clin. Exp. Metastasis 2022, 39, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Bei, Y.; Tian, Q.; He, J.; Wang, R.; Wang, Q.; Sun, L.; Ke, J.; Xie, C.; Shen, P. PFKFB4 facilitates palbociclib resistance in oestrogen receptor-positive breast cancer by enhancing stemness. Cell Prolif. 2023, 56, e13337. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Zhao, L.; Huang, S.; Jiang, Q.; Wei, Y.; Wu, J.L.; Zhang, Z.; Li, Y. Small peptide LINC00511-133aa encoded by LINC00511 regulates breast cancer cell invasion and stemness through the Wnt/β-catenin pathway. Mol. Cell. Probes 2023, 69, 101913. [Google Scholar] [CrossRef]
- Al-Qadi, N.; Halim, A.; Mackie, H.; Sempere, L.; Moore, A. Exploring the effects of inhibiting miR-10b on breast cancer stemness and metastasis. J. Clin. Oncol. 2023, 41, e13084. [Google Scholar] [CrossRef]
- Li, F.; Niu, M.; Qin, K.; Guo, R.; Yi, Y.; Xu, J.; Li, L.; Xie, S.; Fu, M.; Wen, N.; et al. FBXL2 promotes E47 protein instability to inhibit breast cancer stemness and paclitaxel resistance. Oncogene 2023, 42, 339–350. [Google Scholar] [CrossRef]
- García Bueno, J.M.; Ocaña, A.; Castro-García, P.; Gil Gas, C.; Sánchez-Sánchez, F.; Poblet, E.; Serrano, R.; Calero, R.; Ramírez-Castillejo, C. An update on the biology of cancer stem cells in breast cancer. Clin. Transl. Oncol. 2008, 10, 786–793. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Y.; Fan, C.; Gao, P.; Wang, X.; Wei, G.; Wei, J. Estrogen promotes stemness and invasiveness of ER-positive breast cancer cells through Gli1 activation. Mol. Cancer 2014, 13, 137. [Google Scholar] [CrossRef]
- Rokkam, P.; Gugalavath, S.; Gift Kumar, D.K.; Vempati, R.K.; Malla, R.R. Prognostic Role of Hedgehog-GLI1 Signaling Pathway in Aggressive and Metastatic Breast Cancers. Curr. Drug Metab. 2020, 21, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Manore, S.G.; Doheny, D.L.; Wong, G.L.; Lo, H.W. IL-6/JAK/STAT3 Signaling in Breast Cancer Metastasis: Biology and Treatment. Front. Oncol. 2022, 12, 866014. [Google Scholar] [CrossRef]
- Huang, W.-C.; Hung, C.-M.; Wei, C.-T.; Chen, T.-M.; Chien, P.-H.; Pan, H.-L.; Lin, Y.-M.; Chen, Y.-J. Interleukin-6 expression contributes to lapatinib resistance through maintenance of stemness property in HER2-positive breast cancer cells. Oncotarget 2016, 7, 62352–62363. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.E. Animal Models for Studying Prevention and Treatment of Breast Cancer. In Animal Models for the Study of Human Disease; Academic Press: Cambridge, MA, USA, 2013; pp. 997–1018. [Google Scholar] [CrossRef]
- Fiegl, M.; Kaufmann, H.; Zojer, N.; Schuster, R.; Wiener, H.; Müllauer, L.; Roka, S.; Huber, H.; Drach, J. Malignant cell detection by fluorescence in situ hybridization (FISH) in effusions from patients with carcinoma. Hum. Pathol. 2000, 31, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Fiegl, M.; Massoner, A.; Haun, M.; Sturm, W.; Kaufmann, H.; Hack, R.; Krugmann, J.; Fritzer-Szekeres, M.; Grünewald, K.; Gastl, G.; et al. Sensitive detection of tumour cells in effusions by combining cytology and fluorescence in situ hybridisation (FISH). Br. J. Cancer 2004, 91, 558–563. [Google Scholar] [CrossRef]
- Xin, L.; Liu, C.; Liu, Y.; Mansel, R.E.; Ruge, F.; Davies, E.; Jiang, W.G.; Martin, T.A. SIKs suppress tumor function and regulate drug resistance in breast cancer. Am. J. Cancer Res. 2021, 11, 3537. [Google Scholar]
- Sato, M. Phenotypic screening using large-scale genomic libraries to identify drug targets for the treatment of cancer. Oncol. Lett. 2020, 19, 3617–3627. [Google Scholar] [CrossRef]
- Pernía, O.; Sastre-Perona, A.; Rodriguez-Antolín, C.; García-Guede, A.; Palomares-Bralo, M.; Rosas, R.; Sanchez-Cabrero, D.; Cruz, P.; Rodriguez, C.; Diestro, M.; et al. A Novel Role for the Tumor Suppressor Gene ITF2 in Tumorigenesis and Chemotherapy Response. Cancers 2020, 12, 786. [Google Scholar] [CrossRef]
- Shiokawa, D.; Sakai, H.; Ohata, H.; Miyazaki, T.; Kanda, Y.; Sekine, S.; Narushima, D.; Hosokawa, M.; Kato, M.; Suzuki, Y.; et al. Slow-Cycling Cancer Stem Cells Regulate Progression and Chemoresistance in Colon Cancer. Cancer Res. 2020, 80, 4451–4464. [Google Scholar] [CrossRef]
- Sánchez-Díez, M.; Alegría-Aravena, N.; López-Montes, M.; Quiroz-Troncoso, J.; González-Martos, R.; Menéndez-Rey, A.; Sánchez-Sánchez, J.L.; Pastor, J.M.; Ramírez-Castillejo, C. Implication of Different Tumor Biomarkers in Drug Resistance and Invasiveness in Primary and Metastatic Colorectal Cancer Cell Lines. Biomedicines 2022, 10, 1083. [Google Scholar] [CrossRef]
- Zhao, P.; Sun, J.; Huang, X.; Zhang, X.; Liu, X.; Liu, R.; Du, G.; Gan, W.; Yang, C.; Tang, Y.; et al. Targeting the KLF5-EphA2 axis can restrain cancer stemness and overcome chemoresistance in basal-like breast cancer. Int. J. Biol. Sci. 2023, 19, 1861. [Google Scholar] [CrossRef]
- Chen, P.; Tomschik, M.; Nelson, K.M.; Oakey, J.; Gatlin, J.C.; Levy, D.L. Nucleoplasmin is a limiting component in the scaling of nuclear size with cytoplasmic volume. J. Cell Biol. 2019, 218, 4063–4078. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yang, Z.; Su, Y.; Ma, R.; Du, X.; Gao, Y.; Li, Y. Loss of NPM2 expression is a potential immunohistochemical marker for malignant peritoneal mesothelioma: A single-center study of 92 cases. World J. Surg. Oncol. 2022, 20, 350. [Google Scholar] [CrossRef] [PubMed]
- Kai, J.D.; Cheng, L.H.; Li, B.F.; Kang, K.; Xiong, F.; Fu, J.C.; Wang, S. MYH9 is a novel cancer stem cell marker and prognostic indicator in esophageal cancer that promotes oncogenesis through the PI3K/AKT/mTOR axis. Cell Biol. Int. 2022, 46, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Liu, L.; Ashby, J.M.; Wu, D.; Chen, Y.; O’Neill, S.S.; Huang, S.; Wang, J.; Wang, G.; Cheng, D.; et al. Recruitment of KMT2C/MLL3 to DNA Damage Sites Mediates DNA Damage Responses and Regulates PARP Inhibitor Sensitivity in Cancer. Cancer Res. 2021, 81, 3358–3373. [Google Scholar] [CrossRef]
- Shah, M.Y.; Martinez-Garcia, E.; Phillip, J.M.; Chambliss, A.B.; Popovic, R.; Ezponda, T.; Small, E.C.; Will, C.; Phillip, M.P.; Neri, P.; et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 2016, 35, 5905–5915. [Google Scholar] [CrossRef]
- An, Y.; Ongkeko, W.M. ABCG2: The key to chemoresistance in cancer stem cells? Expert Opin. Drug Metab. Toxicol. 2009, 5, 1529–1542. [Google Scholar] [CrossRef]
- Farina, A.R.; Cappabianca, L.A.; Zelli, V.; Sebastiano, M.; Mackay, A.R. Mechanisms involved in selecting and maintaining neuroblastoma cancer stem cell populations, and perspectives for therapeutic targeting. World J. Stem Cells 2021, 13, 685–736. [Google Scholar] [CrossRef]
- Zhang, M.; Tombran-Tink, J.; Yang, S.; Zhang, X.; Li, X.; Barnstable, C.J. PEDF is an endogenous inhibitor of VEGF-R2 angiogenesis signaling in endothelial cells. Exp. Eye Res. 2021, 213, 108828. [Google Scholar] [CrossRef]
- Luo, C.; Yin, H.; Gao, T.; Ma, C.; Liu, J.; Zhang, T.; Xu, Z.; Wang, X.; Zhang, D.; Qi, W.; et al. PEDF inhibits lymphatic metastasis of nasopharyngeal carcinoma as a new lymphangiogenesis inhibitor. Cell Death Dis. 2021, 12, 295. [Google Scholar] [CrossRef]
- Carpino, G.; Cardinale, V.; Di Giamberardino, A.; Overi, D.; Donsante, S.; Colasanti, T.; Amato, G.; Mennini, G.; Franchitto, M.; Conti, F.; et al. Thrombospondin 1 and 2 along with PEDF inhibit angiogenesis and promote lymphangiogenesis in intrahepatic cholangiocarcinoma. J. Hepatol. 2021, 75, 1377–1386. [Google Scholar] [CrossRef]
- Abooshahab, R.; Al-Salami, H.; Dass, C.R. The increasing role of pigment epithelium-derived factor in metastasis: From biological importance to a promising target. Biochem. Pharmacol. 2021, 193, 114787. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Gonzalez-Molina, J.; Mendonça da Silva, J.; Fuller, B.; Selden, C. The extracellular fluid macromolecular composition differentially affects cell-substrate adhesion and cell morphology. Sci. Rep. 2019, 9, 8505. [Google Scholar] [CrossRef]
- Brook, N.; Dharmarajan, A.; Chan, A.; Dass, C.R. Potential therapeutic role for pigment epithelium-derived factor in post-menopausal breast cancer bone metastasis. J. Pharm. Pharmacol. 2023, 75, 873–885. [Google Scholar] [CrossRef]
- Abooshahab, R.; Hooshmand, K.; Luna, G.; Al-Salami, H.; Dass, C.R. Metabolomics Profiling Reveals the Role of PEDF in Triple-Negative Breast Cancer Cell MDA-MB-231 under Glycaemic Loading. Pharmaceutics 2023, 15, 543. [Google Scholar] [CrossRef]
- Shih, S.C.; Ho, T.C.; Chen, S.L.; Tsao, Y.P. Pigment epithelium-derived factor (PEDF) peptide promotes the expansion of hepatic stem/progenitor cells via ERK and STAT3-dependent signaling. Am. J. Transl. Res. 2017, 9, 1114. [Google Scholar]
- Koukourakis, M.I.; Simopoulos, C.; Polychronidis, A.; Perente, S.; Botaitis, S.; Giatromanolaki, A.; Sivridis, E. The effect of trastuzumab/docatexel combination on breast cancer angiogenesis: Dichotomus effect predictable by the HIFIα/VEGF pre-treatment status? Anticancer Res. 2003, 23, 1673–1680. [Google Scholar]
- Pham, T.L.; Kakazu, A.H.; He, J.; Nshimiyimana, R.; Petasis, N.A.; Jun, B.; Bazan, N.G.; Bazan, H.E.P. Elucidating the structure and functions of Resolvin D6 isomers on nerve regeneration with a distinctive trigeminal transcriptome. FASEB J. 2021, 35, e21775. [Google Scholar] [CrossRef]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef]
- Li, C.; Qiu, S.; Liu, X.; Guo, F.; Zhai, J.; Li, Z.; Deng, L.; Ge, L.; Qian, H.; Yang, L.; et al. Extracellular matrix-derived mechanical force governs breast cancer cell stemness and quiescence transition through integrin-DDR signaling. Signal Transduct. Target. Ther. 2023, 8, 247. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Gogoi, G.; Saikia, S.; Sharma, A.; Kalita, D.J.; Sarma, A.; Limaye, A.M.; Gaur, M.K.; Bhattacharyya, J.; Jaganathan, B.G. BMP4 enhances anoikis resistance and chemoresistance of breast cancer cells through canonical BMP signaling. J. Cell Commun. Signal. 2022, 16, 191–205. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G. Endothelial Progenitors in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1263, 85–115. [Google Scholar] [CrossRef] [PubMed]
- Ohmura-Kakutani, H.; Akiyama, K.; Maishi, N.; Ohga, N.; Hida, Y.; Kawamoto, T.; Iida, J.; Shindoh, M.; Tsuchiya, K.; Shinohara, N.; et al. Identification of Tumor Endothelial Cells with High Aldehyde Dehydrogenase Activity and a Highly Angiogenic Phenotype. PLoS ONE 2014, 9, e113910. [Google Scholar] [CrossRef] [PubMed]
- Meattini, I.; Becherini, C.; Boersma, L.; Kaidar-Person, O.; Marta, G.N.; Montero, A.; Offersen, B.V.; Aznar, M.C.; Belka, C.; Brunt, A.M.; et al. European Society for Radiotherapy and Oncology Advisory Committee in Radiation Oncology Practice consensus recommendations on patient selection and dose and fractionation for external beam radiotherapy in early breast cancer. Lancet Oncol. 2022, 23, e21–e31. [Google Scholar] [CrossRef]
- Hennequin, C.; Belkacémi, Y.; Bourgier, C.; Cowen, D.; Cutuli, B.; Fourquet, A.; Hannoun-Lévi, J.M.; Pasquier, D.; Racadot, S.; Rivera, S. Radiotherapy of breast cancer. Cancer/Radiotherapie 2022, 26, 221–230. [Google Scholar] [CrossRef]
- Troschel, F.M.; Palenta, H.; Borrmann, K.; Heshe, K.; Hua, S.H.; Yip, G.W.; Kiesel, L.; Eich, H.T.; Götte, M.; Greve, B. Knockdown of the prognostic cancer stem cell marker Musashi-1 decreases radio-resistance while enhancing apoptosis in hormone receptor-positive breast cancer cells via p21WAF1/CIP1. J. Cancer Res. Clin. Oncol. 2021, 147, 3299–3312. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- López-Otín, C.; Pietrocola, F.; Roiz-Valle, D.; Galluzzi, L.; Kroemer, G. Meta-hallmarks of aging and cancer. Cell Metab. 2023, 35, 12–35. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil-Gas, C.; Sánchez-Díez, M.; Honrubia-Gómez, P.; Sánchez-Sánchez, J.L.; Alvarez-Simón, C.B.; Sabater, S.; Sánchez-Sánchez, F.; Ramírez-Castillejo, C. Self-Renewal Inhibition in Breast Cancer Stem Cells: Moonlight Role of PEDF in Breast Cancer. Cancers 2023, 15, 5422. https://doi.org/10.3390/cancers15225422
Gil-Gas C, Sánchez-Díez M, Honrubia-Gómez P, Sánchez-Sánchez JL, Alvarez-Simón CB, Sabater S, Sánchez-Sánchez F, Ramírez-Castillejo C. Self-Renewal Inhibition in Breast Cancer Stem Cells: Moonlight Role of PEDF in Breast Cancer. Cancers. 2023; 15(22):5422. https://doi.org/10.3390/cancers15225422
Chicago/Turabian StyleGil-Gas, Carmen, Marta Sánchez-Díez, Paloma Honrubia-Gómez, Jose Luis Sánchez-Sánchez, Carmen B. Alvarez-Simón, Sebastia Sabater, Francisco Sánchez-Sánchez, and Carmen Ramírez-Castillejo. 2023. "Self-Renewal Inhibition in Breast Cancer Stem Cells: Moonlight Role of PEDF in Breast Cancer" Cancers 15, no. 22: 5422. https://doi.org/10.3390/cancers15225422
APA StyleGil-Gas, C., Sánchez-Díez, M., Honrubia-Gómez, P., Sánchez-Sánchez, J. L., Alvarez-Simón, C. B., Sabater, S., Sánchez-Sánchez, F., & Ramírez-Castillejo, C. (2023). Self-Renewal Inhibition in Breast Cancer Stem Cells: Moonlight Role of PEDF in Breast Cancer. Cancers, 15(22), 5422. https://doi.org/10.3390/cancers15225422