

PDE3A Is a Highly Expressed Therapy Target in Myxoid Liposarcoma
 , ,
, ,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort and Selection Criteria
2.2. RNA Extraction and Sequencing
2.3. Processing of Sequencing Data
2.4. BAM File QC Metrics
2.5. Transcriptome Reference Data Fetching and Processing
2.6. Identification of DEG between LPS Subtypes
2.7. LPS-Subtype-Specific Pathway Analysis
2.8. Quantitative Real-Time PCR of LPS Tissue RNA
2.9. Immunohistochemical Staining
2.10. Cell Culture
2.11. Western Blotting
2.12. Cell Viability and Cytotoxicity Experiments
2.13. Statistical Analysis
3. Results
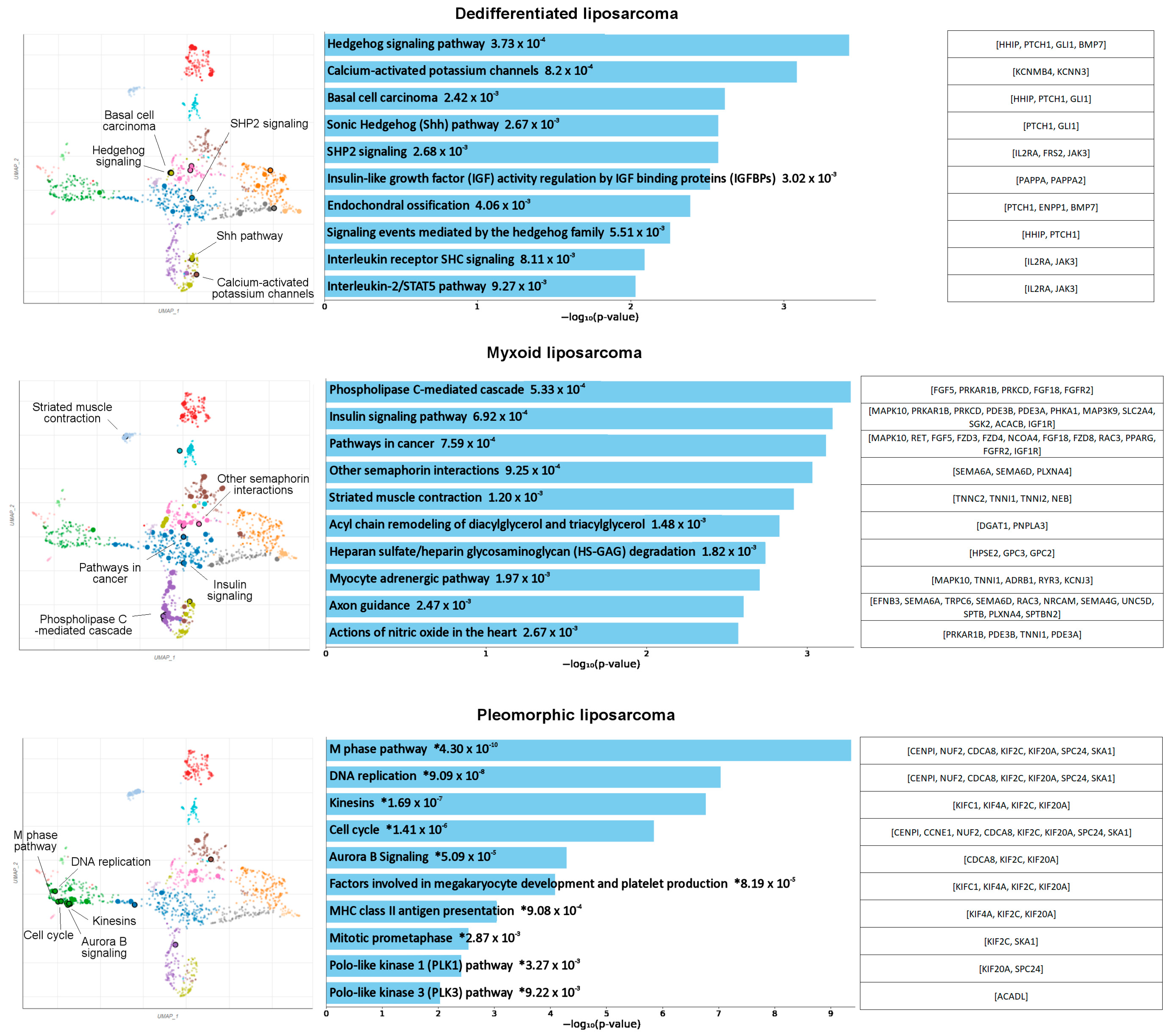
3.1. Identification of Potentially Targetable Subtype-Specific Genes and Pathways
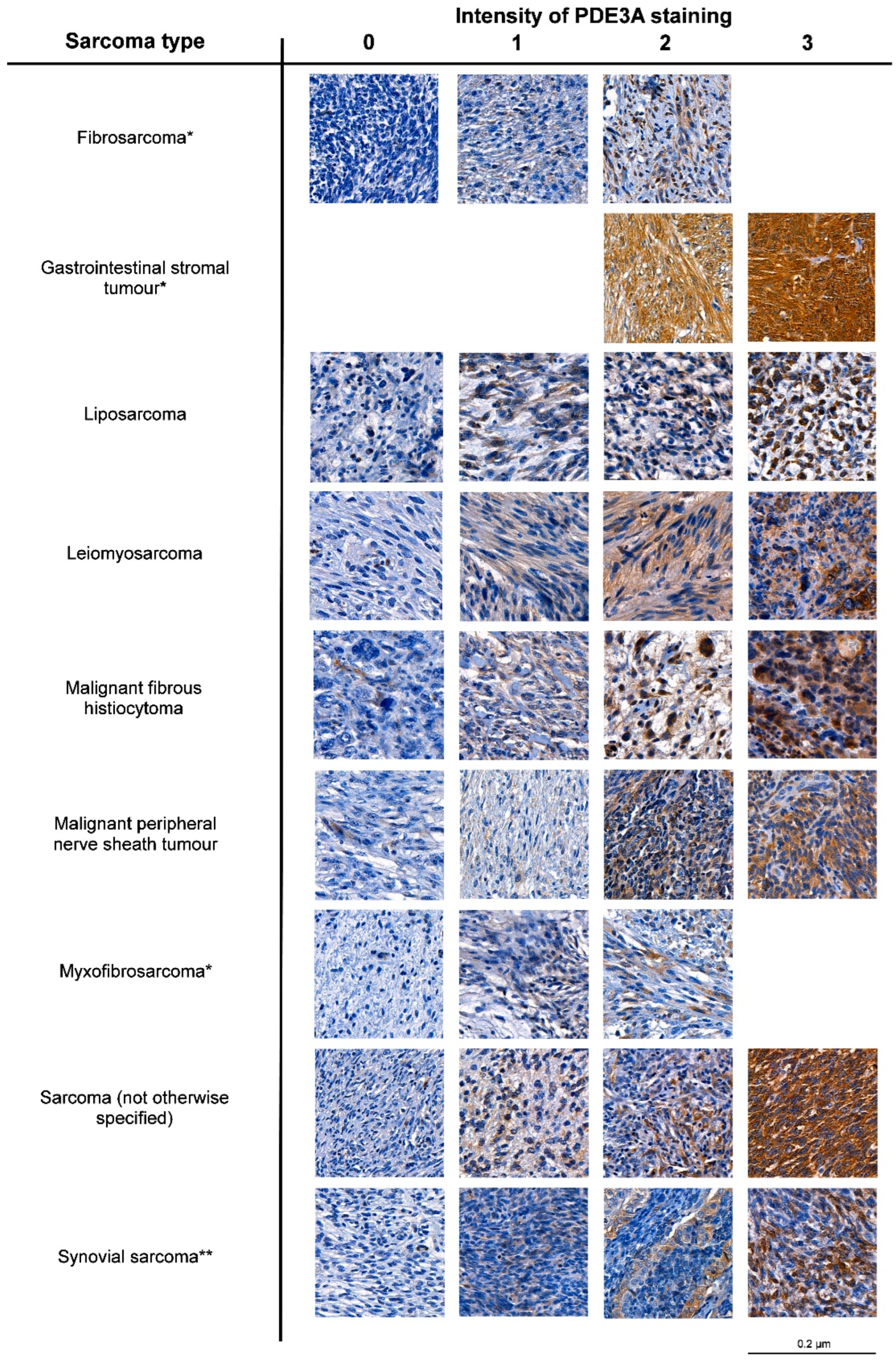
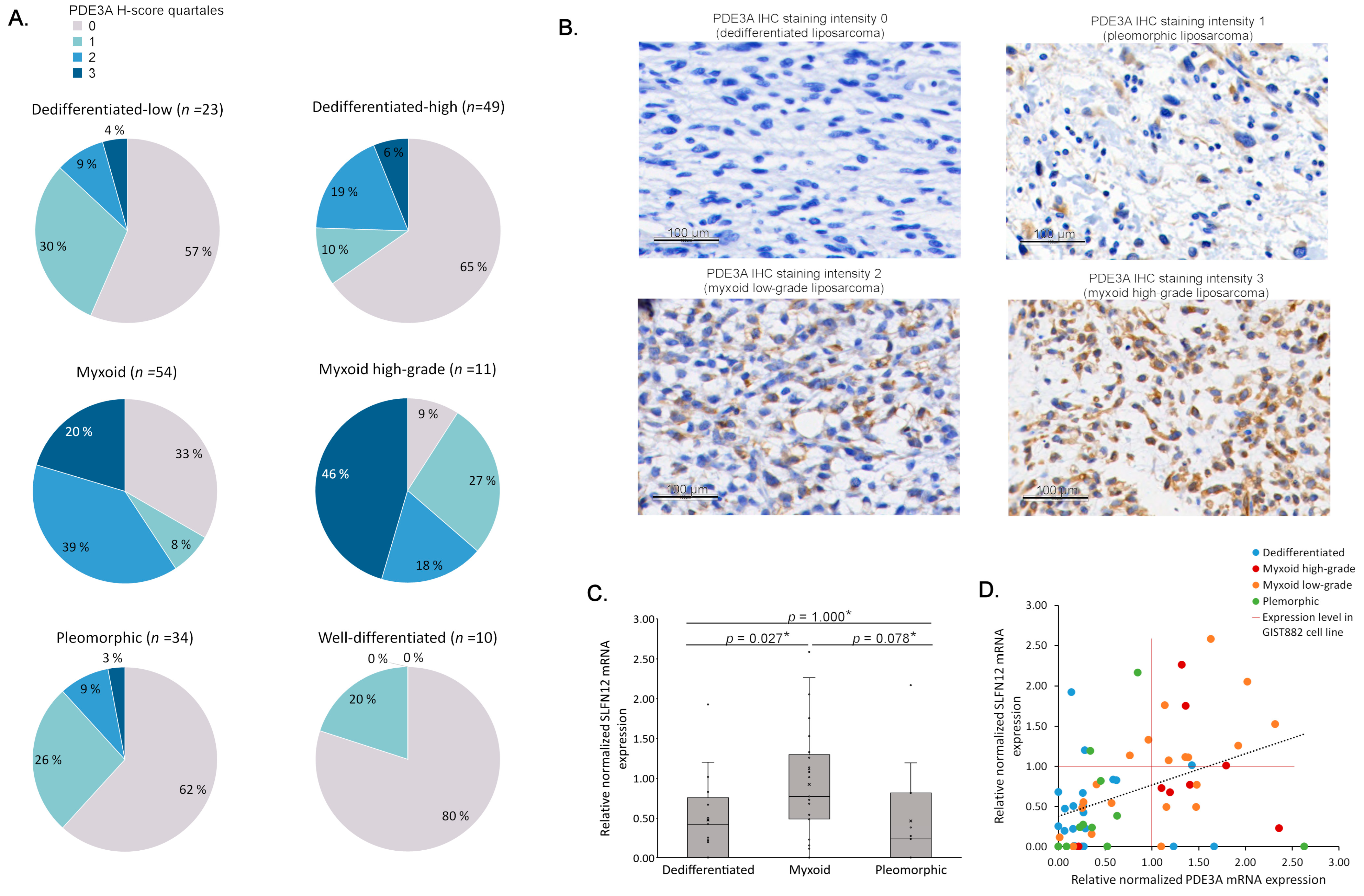
3.2. PDE3A mRNA and Protein Expression Is Frequent in LPS
3.3. Elevated PDE3A and SLFN12 Expression Is Typical for MLPS
3.4. PDE3A- and SLFN12-Coexpressing LPS Cell Lines Are Sensitive to PDE3A Modulators
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Soft Tissue and Bone Tumours, 5th ed.; WHO Classification of Tumours; International Agency for Research IARC: Lyon, France, 2020; Volume 3. [Google Scholar]
- Lee, A.T.J.; Thway, K.; Huang, P.H.; Jones, R.L. Clinical and Molecular Spectrum of Liposarcoma. J. Clin. Oncol. 2018, 36, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Manji, G.A.; Schwartz, G.K. Managing Liposarcomas: Cutting Through the Fat. J. Oncol. Pract. 2016, 12, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Knebel, C.; Lenze, U.; Pohlig, F.; Lenze, F.; Harrasser, N.; Suren, C.; Breitenbach, J.; Rechl, H.; von Eisenhart-Rothe, R.; Muhlhofer, H.M.L. Prognostic factors and outcome of Liposarcoma patients: A retrospective evaluation over 15 years. BMC Cancer 2017, 17, 410. [Google Scholar] [CrossRef] [PubMed]
- Kooby, D.A.; Antonescu, C.R.; Brennan, M.F.; Singer, S. Atypical lipomatous tumor/well-differentiated liposarcoma of the extremity and trunk wall: Importance of histological subtype with treatment recommendations. Ann. Surg. Oncol. 2004, 11, 78–84. [Google Scholar] [CrossRef]
- Gronchi, A.; Collini, P.; Miceli, R.; Valeri, B.; Renne, S.L.; Dagrada, G.; Fiore, M.; Sanfilippo, R.; Barisella, M.; Colombo, C.; et al. Myogenic differentiation and histologic grading are major prognostic determinants in retroperitoneal liposarcoma. Am. J. Surg. Pathol. 2015, 39, 383–393. [Google Scholar] [CrossRef]
- Gahvari, Z.; Parkes, A. Dedifferentiated Liposarcoma: Systemic Therapy Options. Curr. Treat. Options Oncol. 2020, 21, 15. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef]
- Demetri, G.D.; Schoffski, P.; Grignani, G.; Blay, J.Y.; Maki, R.G.; Van Tine, B.A.; Alcindor, T.; Jones, R.L.; D’Adamo, D.R.; Guo, M.; et al. Activity of Eribulin in Patients With Advanced Liposarcoma Demonstrated in a Subgroup Analysis From a Randomized Phase III Study of Eribulin Versus Dacarbazine. J. Clin. Oncol. 2017, 35, 3433–3439. [Google Scholar] [CrossRef]
- Nishio, J.; Nakayama, S.; Nabeshima, K.; Yamamoto, T. Biology and Management of Dedifferentiated Liposarcoma: State of the Art and Perspectives. J. Clin. Med. 2021, 10, 3230. [Google Scholar] [CrossRef]
- De Graaff, M.A.; Malu, S.; Guardiola, I.; Kruisselbrink, A.B.; de Jong, Y.; Corver, W.E.; Gelderblom, H.; Hwu, P.; Nielsen, T.O.; Lazar, A.J.; et al. High-Throughput Screening of Myxoid Liposarcoma Cell Lines: Survivin Is Essential for Tumor Growth. Transl. Oncol. 2017, 10, 546–554. [Google Scholar] [CrossRef]
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for Soft Tissue Sarcoma: Current Status and Future Perspectives. Adv. Ther. 2016, 33, 1055–1071. [Google Scholar] [CrossRef] [PubMed]
- Schoffski, P.; Chawla, S.; Maki, R.G.; Italiano, A.; Gelderblom, H.; Choy, E.; Grignani, G.; Camargo, V.; Bauer, S.; Rha, S.Y.; et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: A randomised, open-label, multicentre, phase 3 trial. Lancet 2016, 387, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Grosso, F.; Sanfilippo, R.; Virdis, E.; Piovesan, C.; Collini, P.; Dileo, P.; Morosi, C.; Tercero, J.C.; Jimeno, J.; D’Incalci, M.; et al. Trabectedin in myxoid liposarcomas (MLS): A long-term analysis of a single-institution series. Ann. Oncol. 2009, 20, 1439–1444. [Google Scholar] [CrossRef]
- Barretina, J.; Taylor, B.S.; Banerji, S.; Ramos, A.H.; Lagos-Quintana, M.; Decarolis, P.L.; Shah, K.; Socci, N.D.; Weir, B.A.; Ho, A.; et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 2010, 42, 715–721. [Google Scholar] [CrossRef]
- Berthold, R.; Isfort, I.; Erkut, C.; Heinst, L.; Grunewald, I.; Wardelmann, E.; Kindler, T.; Aman, P.; Grunewald, T.G.P.; Cidre-Aranaz, F.; et al. Fusion protein-driven IGF-IR/PI3K/AKT signals deregulate Hippo pathway promoting oncogenic cooperation of YAP1 and FUS-DDIT3 in myxoid liposarcoma. Oncogenesis 2022, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, M.; Cyra, M.; Isfort, I.; Jeiler, B.; Kruger, A.; Grunewald, I.; Steinestel, K.; Altvater, B.; Rossig, C.; Hafner, S.; et al. Phosphatidylinositol-3-kinase (PI3K)/Akt Signaling is Functionally Essential in Myxoid Liposarcoma. Mol. Cancer Ther. 2019, 18, 834–844. [Google Scholar] [CrossRef]
- Trautmann, M.; Menzel, J.; Bertling, C.; Cyra, M.; Isfort, I.; Steinestel, K.; Elges, S.; Grunewald, I.; Altvater, B.; Rossig, C.; et al. FUS-DDIT3 Fusion Protein-Driven IGF-IR Signaling is a Therapeutic Target in Myxoid Liposarcoma. Clin. Cancer Res. 2017, 23, 6227–6238. [Google Scholar] [CrossRef]
- Anderson, W.J.; Jo, V.Y. Pleomorphic liposarcoma: Updates and current differential diagnosis. Semin. Diagn. Pathol. 2019, 36, 122–128. [Google Scholar] [CrossRef]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef]
- Palmer, D.; Maurice, D.H. Dual expression and differential regulation of phosphodiesterase 3A and phosphodiesterase 3B in human vascular smooth muscle: Implications for phosphodiesterase 3 inhibition in human cardiovascular tissues. Mol. Pharmacol. 2000, 58, 247–252. [Google Scholar] [CrossRef]
- Chen, J.; Liu, N.; Huang, Y.; Wang, Y.; Sun, Y.; Wu, Q.; Li, D.; Gao, S.; Wang, H.W.; Huang, N.; et al. Structure of PDE3A-SLFN12 complex and structure-based design for a potent apoptosis inducer of tumor cells. Nat. Commun. 2021, 12, 6204. [Google Scholar] [CrossRef] [PubMed]
- An, R.; Liu, J.; He, J.; Wang, F.; Zhang, Q.; Yu, Q. PDE3A inhibitor anagrelide activates death signaling pathway genes and synergizes with cell death-inducing cytokines to selectively inhibit cancer cell growth. Am. J. Cancer Res. 2019, 9, 1905–1921. [Google Scholar] [PubMed]
- Lewis, T.A.; de Waal, L.; Wu, X.; Youngsaye, W.; Wengner, A.; Kopitz, C.; Lange, M.; Gradl, S.; Ellermann, M.; Lienau, P.; et al. Optimization of PDE3A Modulators for SLFN12-Dependent Cancer Cell Killing. ACS Med. Chem. Lett. 2019, 10, 1537–1542. [Google Scholar] [CrossRef] [PubMed]
- Pulkka, O.P.; Gebreyohannes, Y.K.; Wozniak, A.; Mpindi, J.P.; Tynninen, O.; Icay, K.; Cervera, A.; Keskitalo, S.; Murumagi, A.; Kulesskiy, E.; et al. Anagrelide for Gastrointestinal Stromal Tumor. Clin. Cancer Res. 2019, 25, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Garvie, C.W.; Wu, X.; Papanastasiou, M.; Lee, S.; Fuller, J.; Schnitzler, G.R.; Horner, S.W.; Baker, A.; Zhang, T.; Mullahoo, J.P.; et al. Structure of PDE3A-SLFN12 complex reveals requirements for activation of SLFN12 RNase. Nat. Commun. 2021, 12, 4375. [Google Scholar] [CrossRef]
- Li, D.; Chen, J.; Ai, Y.; Gu, X.; Li, L.; Che, D.; Jiang, Z.; Li, L.; Chen, S.; Huang, H.; et al. Estrogen-Related Hormones Induce Apoptosis by Stabilizing Schlafen-12 Protein Turnover. Mol. Cell 2019, 75, 1103–1116 e9. [Google Scholar] [CrossRef]
- De Waal, L.; Lewis, T.A.; Rees, M.G.; Tsherniak, A.; Wu, X.; Choi, P.S.; Gechijian, L.; Hartigan, C.; Faloon, P.W.; Hickey, M.J.; et al. Identification of cancer-cytotoxic modulators of PDE3A by predictive chemogenomics. Nat. Chem. Biol. 2016, 12, 102–108. [Google Scholar] [CrossRef]
- Ai, Y.; He, H.; Chen, P.; Yan, B.; Zhang, W.; Ding, Z.; Li, D.; Chen, J.; Ma, Y.; Cao, Y.; et al. An alkaloid initiates phosphodiesterase 3A-schlafen 12 dependent apoptosis without affecting the phosphodiesterase activity. Nat. Commun. 2020, 11, 3236. [Google Scholar] [CrossRef]
- Yan, B.; Ding, Z.; Zhang, W.; Cai, G.; Han, H.; Ma, Y.; Cao, Y.; Wang, J.; Chen, S.; Ai, Y. Multiple PDE3A modulators act as molecular glues promoting PDE3A-SLFN12 interaction and induce SLFN12 dephosphorylation and cell death. Cell Chem. Biol. 2022, 29, 958–969 e5. [Google Scholar] [CrossRef]
- Lee, S.; Hoyt, S.; Wu, X.; Garvie, C.; McGaunn, J.; Shekhar, M.; Totzl, M.; Rees, M.G.; Cherniack, A.D.; Meyerson, M.; et al. Velcrin-induced selective cleavage of tRNA(Leu)(TAA) by SLFN12 causes cancer cell death. Nat. Chem. Biol. 2022, 19, 301–310. [Google Scholar] [CrossRef]
- Salmikangas, S.; Bohling, T.; Merikoski, N.; Jagdeo, J.; Sampo, M.; Vesterinen, T.; Sihto, H. Tensin2 Is a Novel Diagnostic Marker in GIST, Associated with Gastric Location and Non-Metastatic Tumors. Cancers 2022, 14, 3212. [Google Scholar] [CrossRef]
- Sundqvist, B.; Kilpinen, S.; Bohling, T.; Koljonen, V.; Sihto, H. Activation of Oncogenic and Immune-Response Pathways Is Linked to Disease-Specific Survival in Merkel Cell Carcinoma. Cancers 2022, 14, 3591. [Google Scholar] [CrossRef] [PubMed]
- R: A Language and Environment for Statistical Computing, version 4.1.2; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 1 November 2021).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Tange, O. GNU Parallel: The command-line power tool. USENIX Mag 2011, 36, 42. [Google Scholar]
- Graubert, A.; Aguet, F.; Ravi, A.; Ardlie, K.G.; Getz, G. RNA-SeQC 2: Efficient RNA-seq quality control and quantification for large cohorts. Bioinformatics 2021, 37, 3048–3050. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Kilpinen, S.; Autio, R.; Ojala, K.; Iljin, K.; Bucher, E.; Sara, H.; Pisto, T.; Saarela, M.; Skotheim, R.I.; Bjorkman, M.; et al. Systematic bioinformatic analysis of expression levels of 17,330 human genes across 9783 samples from 175 types of healthy and pathological tissues. Genome Biol. 2008, 9, R139. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Huang, R.; Grishagin, I.; Wang, Y.; Zhao, T.; Greene, J.; Obenauer, J.C.; Ngan, D.; Nguyen, D.T.; Guha, R.; Jadhav, A.; et al. The NCATS BioPlanet—An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front. Pharmacol. 2019, 10, 445. [Google Scholar] [CrossRef]
- Agaram, N.P.; Zhang, L.; Sung, Y.S.; Singer, S.; Stevens, T.; Prieto-Granada, C.N.; Bishop, J.A.; Wood, B.A.; Swanson, D.; Dickson, B.C.; et al. GLI1-amplifications expand the spectrum of soft tissue neoplasms defined by GLI1 gene fusions. Mod. Pathol. 2019, 32, 1617–1626. [Google Scholar] [CrossRef]
- Tap, W.D.; Eilber, F.C.; Ginther, C.; Dry, S.M.; Reese, N.; Barzan-Smith, K.; Chen, H.W.; Wu, H.; Eilber, F.R.; Slamon, D.J.; et al. Evaluation of well-differentiated/de-differentiated liposarcomas by high-resolution oligonucleotide array-based comparative genomic hybridization. Genes Chromosomes Cancer 2011, 50, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Asmann, Y.W.; Erickson-Johnson, M.R.; Oliveira, J.L.; Zhang, H.; Moura, R.D.; Lazar, A.J.; Lev, D.; Bill, K.; Lloyd, R.V.; et al. High-resolution genomic mapping reveals consistent amplification of the fibroblast growth factor receptor substrate 2 gene in well-differentiated and dedifferentiated liposarcoma. Genes Chromosomes Cancer 2011, 50, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Saada-Bouzid, E.; Burel-Vandenbos, F.; Ranchere-Vince, D.; Birtwisle-Peyrottes, I.; Chetaille, B.; Bouvier, C.; Chateau, M.C.; Peoc’h, M.; Battistella, M.; Bazin, A.; et al. Prognostic value of HMGA2, CDK4, and JUN amplification in well-differentiated and dedifferentiated liposarcomas. Mod. Pathol. 2015, 28, 1404–1414. [Google Scholar] [CrossRef] [PubMed]
- Creytens, D.; Van Gorp, J.; Speel, E.J.; Ferdinande, L. Characterization of the 12q amplicons in lipomatous soft tissue tumors by multiplex ligation-dependent probe amplification-based copy number analysis. Anticancer Res. 2015, 35, 1835–1842. [Google Scholar] [PubMed]
- Renner, M.; Wolf, T.; Meyer, H.; Hartmann, W.; Penzel, R.; Ulrich, A.; Lehner, B.; Hovestadt, V.; Czwan, E.; Egerer, G.; et al. Integrative DNA methylation and gene expression analysis in high-grade soft tissue sarcomas. Genome Biol. 2013, 14, r137. [Google Scholar] [CrossRef]
- Hemminger, J.A.; Toland, A.E.; Scharschmidt, T.J.; Mayerson, J.L.; Guttridge, D.C.; Iwenofu, O.H. Expression of cancer-testis antigens MAGEA1, MAGEA3, ACRBP, PRAME, SSX2, and CTAG2 in myxoid and round cell liposarcoma. Mod. Pathol. 2014, 27, 1238–1245. [Google Scholar] [CrossRef]
- May, C.D.; Garnett, J.; Ma, X.; Landers, S.M.; Ingram, D.R.; Demicco, E.G.; Al Sannaa, G.A.; Vu, T.; Han, L.; Zhang, Y.; et al. AXL is a potential therapeutic target in dedifferentiated and pleomorphic liposarcomas. BMC Cancer 2015, 15, 901. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Li, L.; Nian, Y.; Chen, Y.; Shen, R.; Ma, X. Pregnancy-associated plasma protein-A (PAPPA) promotes breast cancer progression. Bioengineered 2022, 13, 291–307. [Google Scholar] [CrossRef]
- Hong, Q.; Li, R.; Zhang, Y.; Gu, K. Fibrillin 2 gene knockdown inhibits invasion and migration of lung cancer cells. Cell Mol. Biol. 2020, 66, 190–196. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, J.; Yang, C.; Wei, P.; Yang, M.; Han, H.; Chen, H.D.; Yue, T.; Xiao, S.; Chen, X.; et al. METTL1 promotes neuroblastoma development through m(7)G tRNA modification and selective oncogenic gene translation. Biomark. Res. 2022, 10, 68. [Google Scholar] [CrossRef]
- Shen, Z.; Feng, X.; Fang, Y.; Li, Y.; Li, Z.; Zhan, Y.; Lin, M.; Li, G.; Ding, Y.; Deng, H. POTEE drives colorectal cancer development via regulating SPHK1/p65 signaling. Cell Death Dis. 2019, 10, 863. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Smolen, G.A.; Zhang, J.; Wittner, B.; Schott, B.J.; Brachtel, E.; Ramaswamy, S.; Maheswaran, S.; Haber, D.A. A developmentally regulated inducer of EMT, LBX1, contributes to breast cancer progression. Genes Dev. 2009, 23, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Fan, T.; Shi, Z.; Jiang, H. CDCA2 Promotes HCC Cells Development via AKT-mTOR Pathway. Anal. Cell. Pathol. 2022, 2022, 9912254. [Google Scholar] [CrossRef]
- Zhao, Z.Q.; Wu, X.J.; Cheng, Y.H.; Zhou, Y.F.; Ma, X.M.; Zhang, J.; Heng, X.Y.; Feng, F. TROAP regulates cell cycle and promotes tumor progression through Wnt/beta-Catenin signaling pathway in glioma cells. CNS Neurosci. Ther. 2021, 27, 1064–1076. [Google Scholar] [CrossRef]
- Mullard, M.; Cade, M.; Morice, S.; Dupuy, M.; Danieau, G.; Amiaud, J.; Renault, S.; Lezot, F.; Brion, R.; Thepault, R.A.; et al. Sonic Hedgehog Signature in Pediatric Primary Bone Tumors: Effects of the GLI Antagonist GANT61 on Ewing’s Sarcoma Tumor Growth. Cancers 2020, 12, 3438. [Google Scholar] [CrossRef] [PubMed]
- Niyaz, M.; Khan, M.S.; Mudassar, S. Hedgehog Signaling: An Achilles’ Heel in Cancer. Transl. Oncol. 2019, 12, 1334–1344. [Google Scholar] [CrossRef]
- Zuniga, L.; Cayo, A.; Gonzalez, W.; Vilos, C.; Zuniga, R. Potassium Channels as a Target for Cancer Therapy: Current Perspectives. OncoTargets Ther. 2022, 15, 783–797. [Google Scholar] [CrossRef]
- Owusu Obeng, E.; Rusciano, I.; Marvi, M.V.; Fazio, A.; Ratti, S.; Follo, M.Y.; Xian, J.; Manzoli, L.; Billi, A.M.; Mongiorgi, S.; et al. Phosphoinositide-Dependent Signaling in Cancer: A Focus on Phospholipase C Isozymes. Int. J. Mol. Sci. 2020, 21, 2581. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Yin, J.; Zhang, J.; Jiang, Y. Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: A challenge for cancer therapy. J. Hematol. Oncol. 2020, 13, 64. [Google Scholar] [CrossRef]
- Vandenberghe, P.; Hague, P.; Hockman, S.C.; Manganiello, V.C.; Demetter, P.; Erneux, C.; Vanderwinden, J.M. Phosphodiesterase 3A: A new player in development of interstitial cells of Cajal and a prospective target in gastrointestinal stromal tumors (GIST). Oncotarget 2017, 8, 41026–41043. [Google Scholar] [CrossRef]
- Wu, X.; Schnitzler, G.R.; Gao, G.F.; Diamond, B.; Baker, A.R.; Kaplan, B.; Williamson, K.; Westlake, L.; Lorrey, S.; Lewis, T.A.; et al. Mechanistic insights into cancer cell killing through interaction of phosphodiesterase 3A and schlafen family member 12. J. Biol. Chem. 2020, 295, 3431–3446. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.; Socci, N.D.; Ambrosini, G.; Sambol, E.; Decarolis, P.; Wu, Y.; O’Connor, R.; Maki, R.; Viale, A.; Sander, C.; et al. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma. Cancer Res. 2007, 67, 6626–6636. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dedifferentiated | Myxoid | Pleomorphic | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Symbol | logFC | p Value | Symbol | logFC | p Value | Symbol | logFC | p Value | |
| 1 | GLI1 | 4.019 | 8.5674 × 10−12 | POTEE | 6.724 | 2.4549 × 10−34 | TROAP | 2.374 | 3.1991 × 10−12 |
| 2 | HHIP | 3.981 | 2.8013 × 10−11 | GNAT3 | 6.010 | 2.6398 × 10−24 | HAS1 | 2.117 | 0.000052 |
| 3 | B4 GALNT1 | 3.322 | 3.8753 × 10−13 | SHANK2 | 5.408 | 4.2321 × 10−34 | SPC24 | 2.022 | 7.1793 × 10−8 |
| 4 | PAPPA2 | 2.856 | 5.1590 × 10−7 | SLC17A8 | 5.106 | 7.1407 × 10−20 | CDCA2 | 1.958 | 8.3866 × 10−9 |
| 5 | LRRC4B | 2.828 | 4.0311 × 10−7 | LBX1 | 4.757 | 3.4411 × 10−25 | CNKSR2 | 1.948 | 0.00015 |
| 6 | FBN2 | 2.731 | 3.983 × 10−11 | UNC5D | 4.666 | 6.53067 × 10−24 | UPK3BL1 | 1.944 | 0.0015 |
| 7 | ENTPD2 | 2.672 | 1.5089 × 10−8 | RASGEF1C | 4.491 | 1.2908 × 10−22 | TMEM170B | 1.858 | 0.000035 |
| 8 | PTCH1 | 2.566 | 5.0399 × 10−11 | SPATA22 | 4.387 | 4.4755 × 10−19 | DEPDC1 | 1.853 | 1.1343 × 10−6 |
| 9 | METTL1 | 2.530 | 9.3422 × 10−14 | KLHDC8A | 4.303 | 8.3055 × 10−19 | KIF2C | 1.835 | 7.3270 × 10−7 |
| 10 | COLGALT2 | 2.499 | 3.7948 × 10−9 | AMN | 4.159 | 1.3435 × 10−20 | DLGAP5 | 1.827 | 1.0800 × 10−8 |
| 11 | PI15 | 2.458 | 1.2388 × 10−7 | POU3F3 | 4.087 | 5.5783 × 10−18 | C18orf54 | 1.824 | 2.1967 × 10−7 |
| 12 | AVIL | 2.457 | 2.0786 × 10−14 | CTAG2 | 4.074 | 7.8181 × 10−12 | CCNE1 | 1.799 | 0.000078 |
| 13 | GALNT17 | 2.333 | 1.3743 × 10−8 | NPW | 4.034 | 3.4685 × 10−16 | TNFAIP8L3 | 1.741 | 0.00057 |
| 14 | HMGA2 | 2.308 | 0.000032 | CSMD1 | 3.980 | 4.3107 × 10−19 | SLC6A8 | 1.722 | 0.00080 |
| 15 | IRAK3 | 2.307 | 1.3883 × 10−14 | ADAMTS19 | 3.970 | 3.8458 × 10−16 | ACAN | 1.714 | 0.0017 |
| 16 | PAPPA | 2.263 | 3.7620 × 10−9 | SOX1 | 3.928 | 8.7166 × 10−15 | KIF14 | 1.707 | 5.75845 × 10−7 |
| 17 | SLC35E3 | 2.240 | 6.6762 × 10−15 | SIM1 | 3.927 | 4.2415 × 10−23 | KIF20A | 1.691 | 0.00013 |
| 18 | ATP23 | 2.231 | 4.6716 × 10−9 | GIPR | 3.855 | 2.2921 × 10−18 | RFX8 | 1.682 | 0.00035 |
| 19 | YEATS4 | 2.226 | 7.2431 × 10−13 | NELL1 | 3.803 | 1.4580 × 10−15 | CDCA5 | 1.681 | 7.7032 × 10−6 |
| 20 | PRRT2 | 2.195 | 6.2120 × 10−10 | DPP10 | 3.783 | 3.4701 × 10−14 | DPP4 | 1.622 | 0.00039 |
| 21 | GRIN2D | 2.188 | 1.8108 × 10−7 | MYH15 | 3.777 | 8.4681 × 10−19 | NUF2 | 1.621 | 1.2021 × 10−6 |
| 22 | NTN1 | 2.168 | 4.9020 × 10−8 | COL23A1 | 3.745 | 1.7877 × 10−18 | CEP55 | 1.617 | 0.000017 |
| 23 | TUBB3 | 2.142 | 1.5008 × 10−6 | TTPA | 3.743 | 1.6634 × 10−13 | ACADL | 1.615 | 0.0019 |
| 24 | C1QL1 | 2.102 | 8.84882 × 10−6 | KCNJ3 | 3.683 | 2.1530 × 10−15 | RNF112 | 1.612 | 0.00061 |
| 25 | MDM2 | 2.100 | 1.1237 × 10−11 | BMPR1B | 3.542 | 4.8406 × 10−19 | GFRA2 | 1.606 | 0.000083 |
| Sarcoma Type | PDE3A Intensity | Total | ||||
|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | |||
| Fibrosarcoma | N | 7 | 2 | 1 | 0 | 10 |
| % | 70 | 20 | 10 | 0 | ||
| Gastrointestinal stromal tumor | N | 0 | 0 | 8 | 19 | 27 |
| % | 0 | 0 | 29.6 | 70.4 | ||
| Liposarcoma | N | 46 | 24 | 14 | 3 | 87 |
| % | 52.9 | 27.6 | 16.1 | 3.4 | ||
| Leiomyosarcoma | N | 19 | 17 | 17 | 6 | 59 |
| % | 32.2 | 28.8 | 28.8 | 10.2 | ||
| Malignant fibrohistiocytoma | N | 133 | 67 | 19 | 3 | 222 |
| % | 59.9 | 30.2 | 8.6 | 1.4 | ||
| Malignant peripheral nerve sheath tumor | N | 11 | 4 | 3 | 2 | 20 |
| % | 55 | 20 | 15 | 10 | ||
| Myxofibrosarcoma | N | 27 | 14 | 7 | 0 | 48 |
| % | 56.3 | 29.2 | 14.6 | 0 | ||
| Sarcoma (not otherwise specified) | N | 14 | 12 | 5 | 2 | 33 |
| % | 42.2 | 36.4 | 15.2 | 6.1 | ||
| Synovial sarcoma | N | 29 | 4 | 3 | 1 | 37 |
| % | 78.4 | 10.8 | 8.1 | 2.7 | ||
| Total | N | 286 | 144 | 77 | 36 | 543 |
| % | 52.7 | 26.5 | 14.2 | 6.6 | ||
| Total Cases | PDE3A H-Score | p Value | ||
|---|---|---|---|---|
| Low | High | |||
| All cases | 181 | 123 (68.0%) | 58 (32.0%) | |
| Sex | 0.016 | |||
| Male | 95 | 57 (60.0%) | 38 (40.0%) | |
| Female | 86 | 66 (76.7%) | 20 (23.3%) | |
| Histological subtype | <0.001 * | |||
| Well-differentiated | 10 | 10 (100%) | 0 (0%) | |
| Dedifferentiated | 72 | 57 (79.2%) | 15 (20.8%) | |
| Myxoid | 65 | 26 (40.0%) | 39 (60.0%) | |
| Pleomorphic | 34 | 30 (88.2%) | 4 (11.8%) | |
| Tumor site | 0.060 * | |||
| Limb | 83 | 50 (60.2%) | 33 (39.8%) | |
| Retroperitoneal | 48 | 39 (81.3%) | 9 (18.8%) | |
| Trunk | 36 | 25 (69.4%) | 11 (30.6%) | |
| Abdomen | 11 | 8 (72.7%) | 3 (27.3%) | |
| Head | 3 | 1 (33.3%) | 2 (66.7%) | |
| Age at the time of diagnosis | 0.070 | |||
| Median (range in years) | 59 | 60 (20−92) | 54 (22−88) | |
| Tumor size | 0.532 | |||
| Median (range in cm3) | 10 | 10 (1.5−40) | 9.25 (2.2−30) | |
| Data not available | 17 | 8 | ||
| Metastasis at diagnosis | 0.271 | |||
| Present | 8 | 4 (2.2%) | 4 (2.2%) | |
| Not present | 173 | 119 (65.7%) | 54 (29.8%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toivanen, K.; Kilpinen, S.; Ojala, K.; Merikoski, N.; Salmikangas, S.; Sampo, M.; Böhling, T.; Sihto, H. PDE3A Is a Highly Expressed Therapy Target in Myxoid Liposarcoma. Cancers 2023, 15, 5308. https://doi.org/10.3390/cancers15225308
Toivanen K, Kilpinen S, Ojala K, Merikoski N, Salmikangas S, Sampo M, Böhling T, Sihto H. PDE3A Is a Highly Expressed Therapy Target in Myxoid Liposarcoma. Cancers. 2023; 15(22):5308. https://doi.org/10.3390/cancers15225308
Chicago/Turabian StyleToivanen, Kirsi, Sami Kilpinen, Kalle Ojala, Nanna Merikoski, Sami Salmikangas, Mika Sampo, Tom Böhling, and Harri Sihto. 2023. "PDE3A Is a Highly Expressed Therapy Target in Myxoid Liposarcoma" Cancers 15, no. 22: 5308. https://doi.org/10.3390/cancers15225308
APA StyleToivanen, K., Kilpinen, S., Ojala, K., Merikoski, N., Salmikangas, S., Sampo, M., Böhling, T., & Sihto, H. (2023). PDE3A Is a Highly Expressed Therapy Target in Myxoid Liposarcoma. Cancers, 15(22), 5308. https://doi.org/10.3390/cancers15225308