

Targeting KRAS in Pancreatic Ductal Adenocarcinoma: The Long Road to Cure
 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
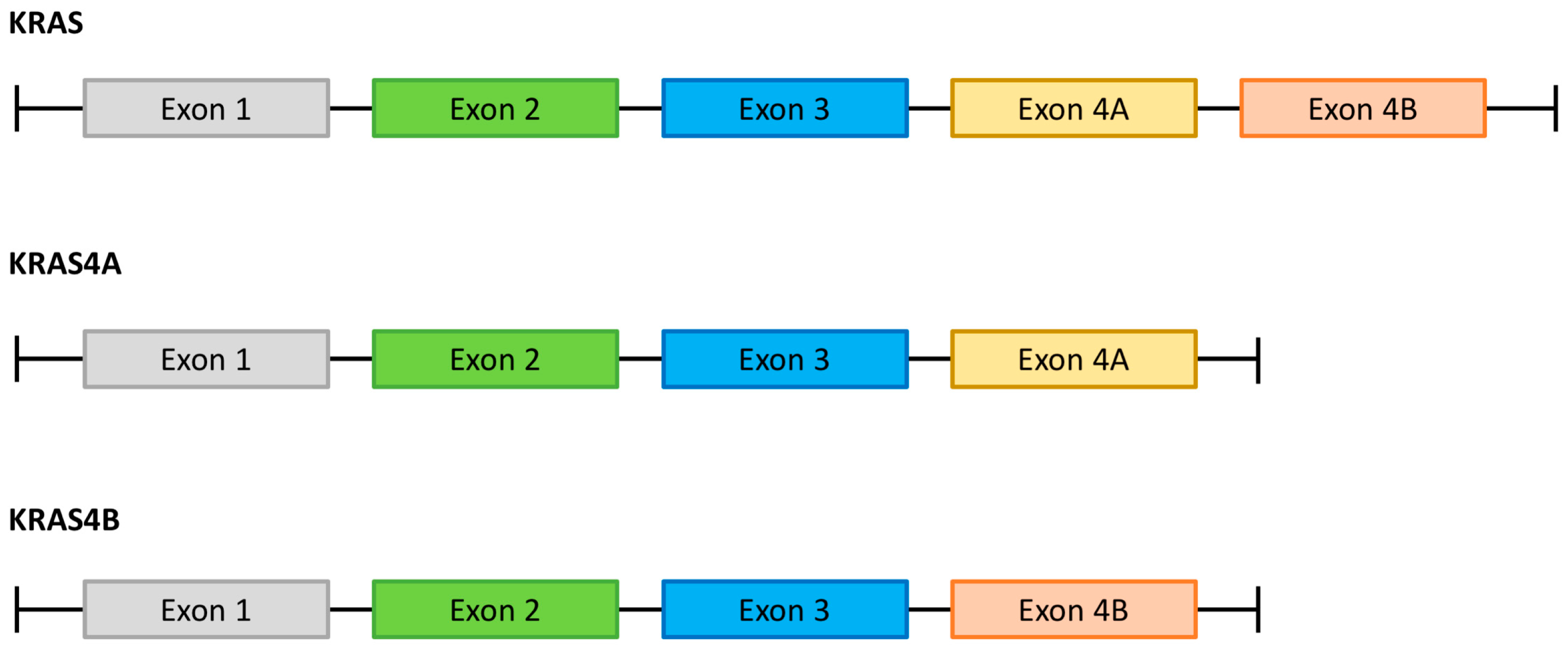
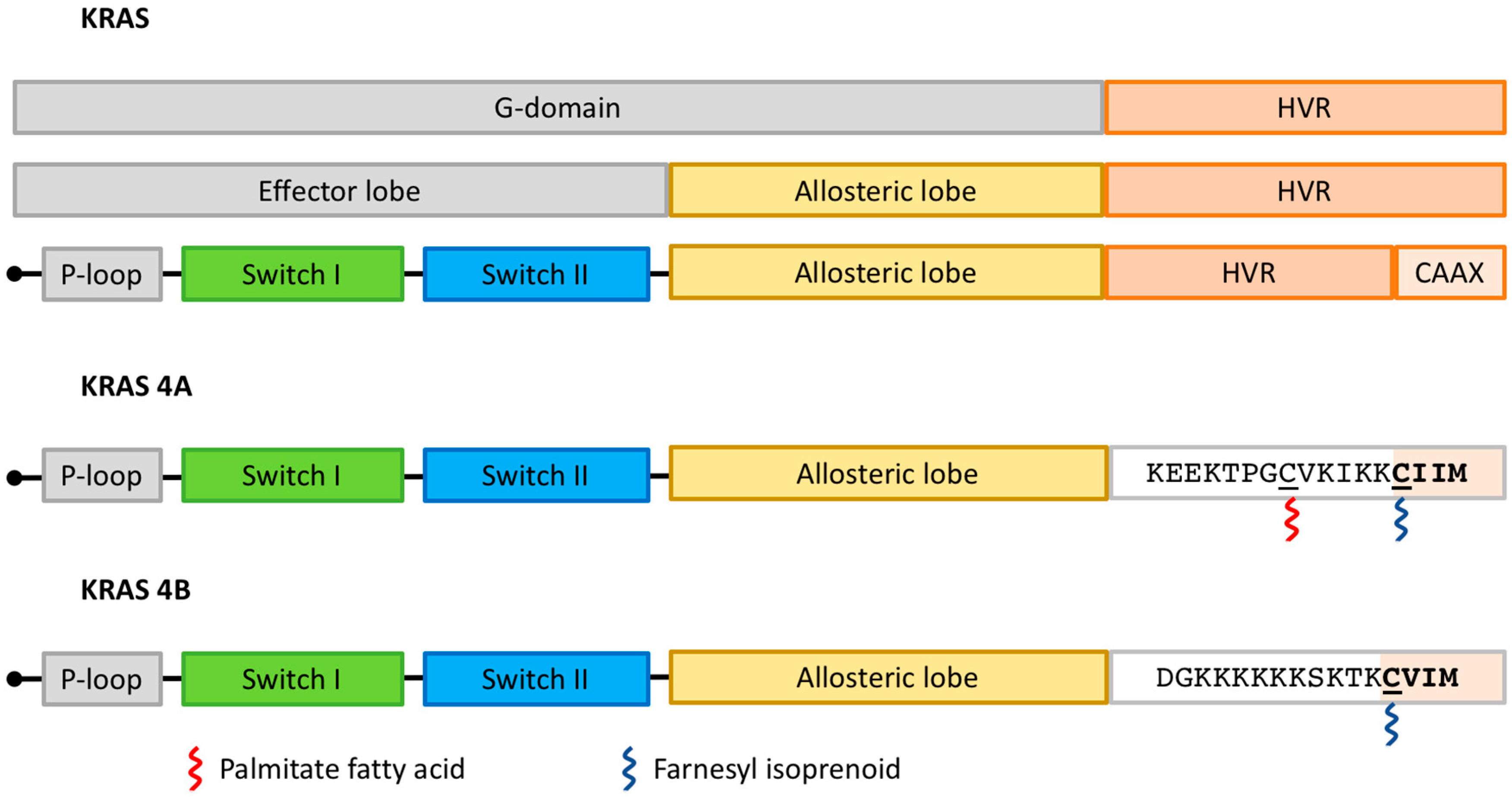
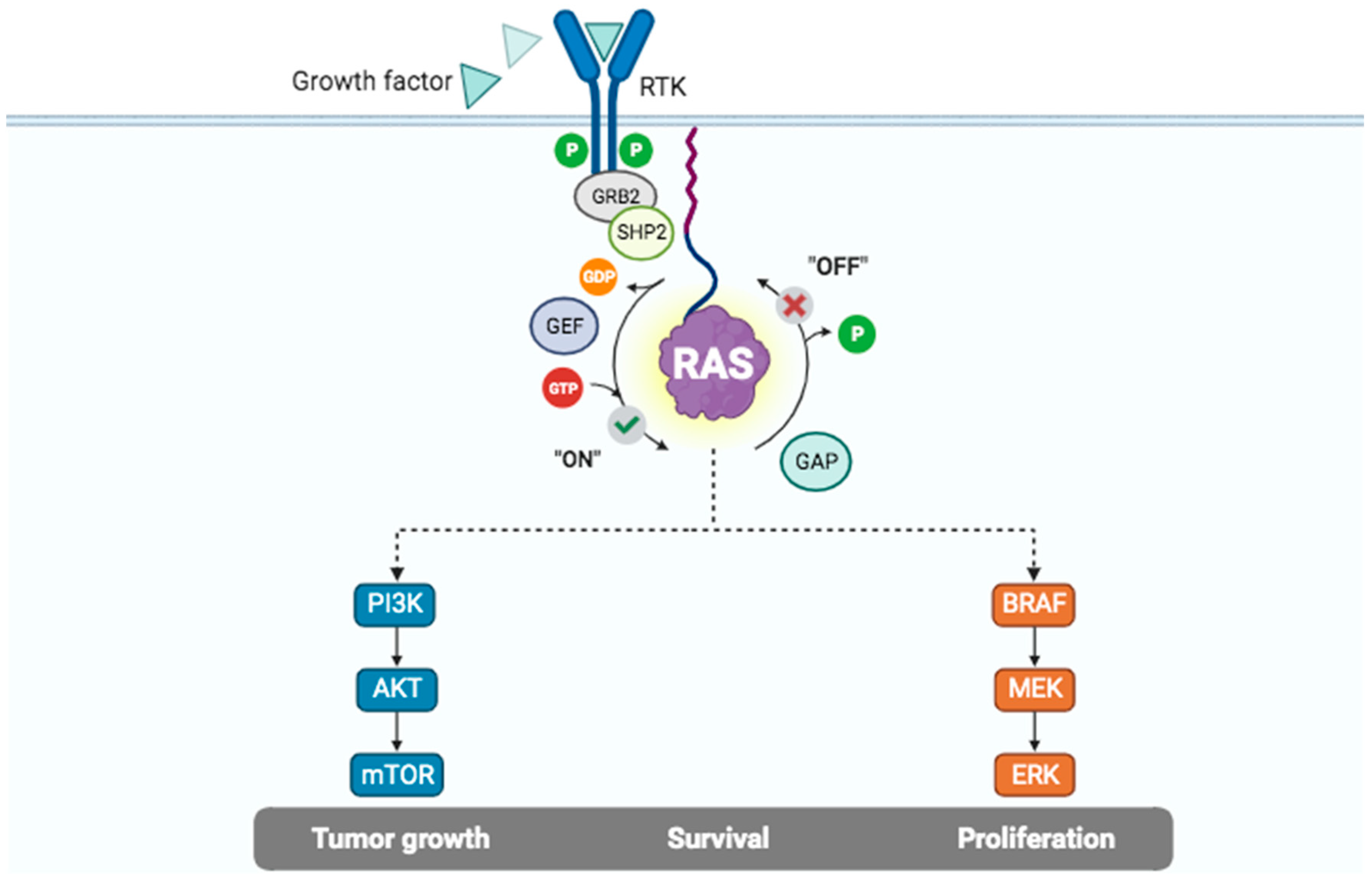
2. KRAS as a Member of Intracellular Signaling Pathways
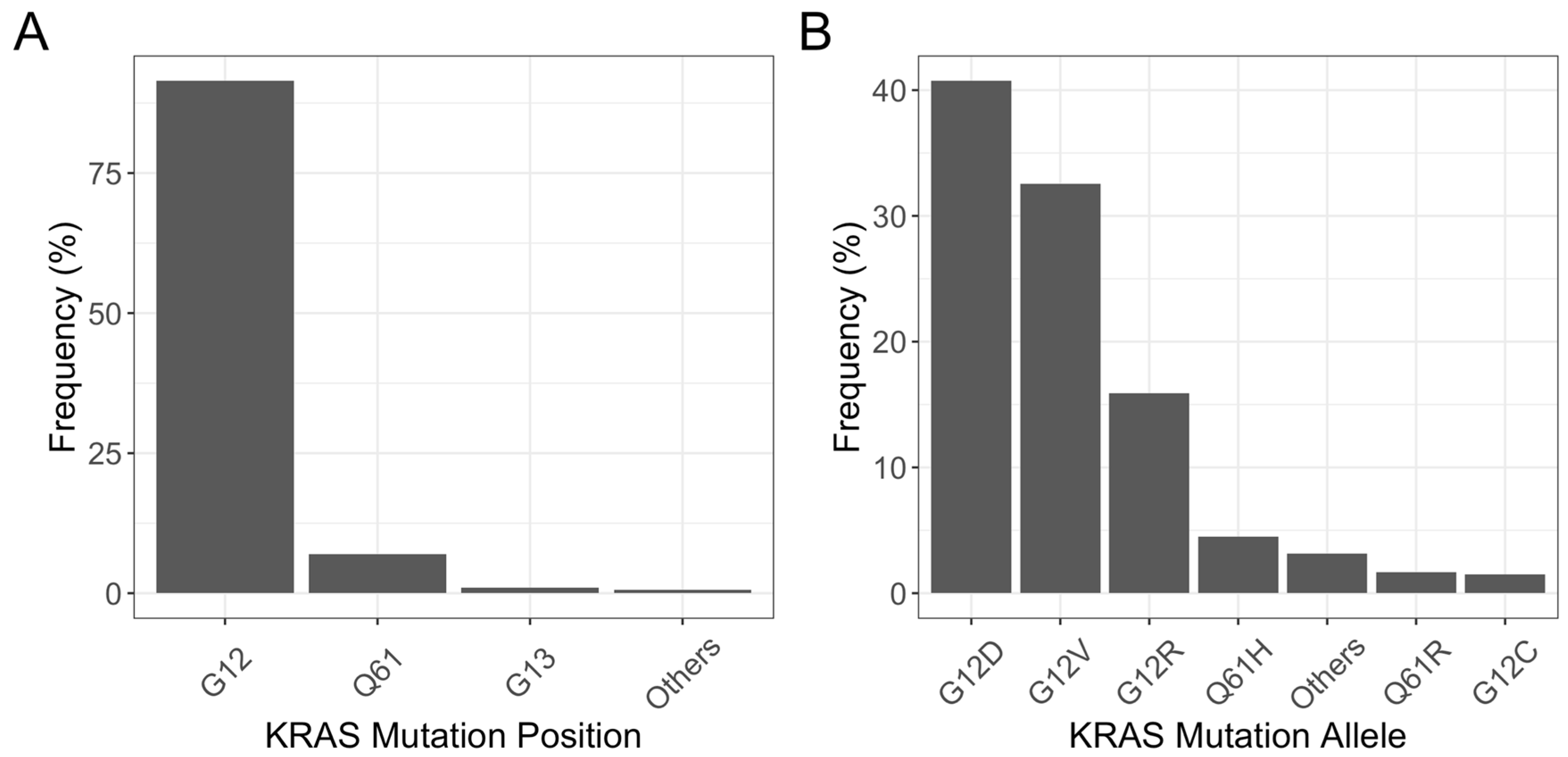
3. Frequency of KRAS Mutations in PDAC
4. Prognostic and Predictive Value of KRAS Mutation in PDAC
5. Targeting KRAS
5.1. Targeting KRAS Post-Translational Modifications and Trafficking
5.2. Targeting KRAS Oligomerization
5.3. Targeting Upstream Components of RAS Signaling Pathways
5.3.1. EGFR
5.3.2. SOS1
5.3.3. SHP2
5.4. Targeting Downstream Components of RAS Signaling Pathways
5.4.1. RAFs
5.4.2. MEK1/2
5.4.3. ERK1/2
5.5. Direct KRAS Inhibitors
5.5.1. Allele Specific RAS Inhibitors
G12C Inhibitors
G12D Inhibitors
Inhibitors of Other KRAS Mutant Isoforms
5.5.2. Pan-RAS (Non-Specific) Inhibitors
6. Targeting Metabolic Reprogramming
7. Targeting KRAS and Co-Occurring Genetic Alterations
7.1. CDKN2A
7.2. PIK3CA/AKT/mTOR
8. KRAS Vaccine
9. Interfering with mRNA
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef] [PubMed]
- de Jesus, V.H.F.; Guedes Camandaroba, M.P.; Spina Donadio, M.D.; Cabral, A.; Pimentel Muniz, T.; de Moura Leite, L.; Ferreira Sant’Ana, L.; Luiz da Costa, W., Jr.; Curado, M.P.; Coimbra, F.J.F. Clinico-pathological features and survival of patients with malignant exocrine pancreatic neoplasms: The AC Camargo Cancer Center experience. J. Surg. Oncol. 2019, 119, 71–78. [Google Scholar] [CrossRef] [PubMed]
- de Jesus, V.H.F.; da Costa, W.L., Jr.; Claro, L.C.L.; Coimbra, F.J.F.; Dettino, A.L.A.; Riechelmann, R.P.; Curado, M.P. Disparities in access to health care system as determinant of survival for patients with pancreatic cancer in the State of São Paulo, Brazil. Sci. Rep. 2021, 11, 6346. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Melisi, D.; Macarulla, T.; Cid, R.P.; Chandana, S.R.; De La Fouchardière, C.; Dean, A.; Kiss, I.; Lee, W.J.; Goetze, T.O.; et al. NALIRIFOX versus nab-paclitaxel and gemcitabine in treatment-naive patients with metastatic pancreatic ductal adenocarcinoma (NAPOLI 3): A randomised, open-label, phase 3 trial. Lancet 2023. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- McCormick, F. A brief history of RAS and the RAS Initiative. Adv. Cancer Res. 2021, 153, 1–27. [Google Scholar] [CrossRef]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef]
- Goitre, L.; Trapani, E.; Trabalzini, L.; Retta, S.F. The Ras Superfamily of Small GTPases: The Unlocked Secrets. In Ras Signaling: Methods and Protocols; Trabalzini, L., Retta, S.F., Eds.; Humana Press: Totowa, NJ, USA, 2014; pp. 1–18. [Google Scholar]
- My Cancer Genome. HRAS. 2023. Available online: https://www.mycancergenome.org/content/gene/hras/ (accessed on 8 June 2023).
- My Cancer Genome. NRAS. 2023. Available online: https://www.mycancergenome.org/content/alteration/nras-mutation/ (accessed on 8 June 2023).
- Rásó, E. Splice variants of RAS—Translational significance. Cancer Metastasis Rev. 2020, 39, 1039–1049. [Google Scholar] [CrossRef]
- Pantsar, T. The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 2019, 18, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Kiel, C.; Matallanas, D.; Kolch, W. The Ins and Outs of RAS Effector Complexes. Biomolecules 2021, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Nuevo-Tapioles, C.; Philips, M.R. The role of KRAS splice variants in cancer biology. Front. Cell Dev. Biol. 2022, 10, 1033348. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.D.; Lopes, M.S.; Zhou, M.; Court, H.; Ponce, O.; Fiordalisi, J.J.; Gierut, J.J.; Cox, A.D.; Haigis, K.M.; Philips, M.R. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. USA 2015, 112, 779–784. [Google Scholar] [CrossRef]
- Marshall, C.B.; KleinJan, F.; Gebregiworgis, T.; Lee, K.-Y.; Fang, Z.; Eves, B.J.; Liu, N.F.; Gasmi-Seabrook, G.M.C.; Enomoto, M.; Ikura, M. NMR in integrated biophysical drug discovery for RAS: Past, present, and future. J. Biomol. NMR 2020, 74, 531–554. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Luo, J. KRAS mutation in pancreatic cancer. Semin. Oncol. 2021, 48, 10–18. [Google Scholar] [CrossRef]
- Muratcioglu, S.; Jang, H.; Gursoy, A.; Keskin, O.; Nussinov, R. PDEδ Binding to Ras Isoforms Provides a Route to Proper Membrane Localization. J. Phys. Chem. B 2017, 121, 5917–5927. [Google Scholar] [CrossRef]
- Whaby, M.; Wallon, L.; Mazzei, M.; Khan, I.; Teng, K.W.; Koide, S.; O’bryan, J.P. Mutations in the α4-α5 allosteric lobe of RAS do not significantly impair RAS signaling or self-association. J. Biol. Chem. 2022, 298, 102661. [Google Scholar] [CrossRef]
- Filchtinski, D.; Sharabi, O.; Rüppel, A.; Vetter, I.R.; Herrmann, C.; Shifman, J.M. What Makes Ras an Efficient Molecular Switch: A Computational, Biophysical, and Structural Study of Ras-GDP Interactions with Mutants of Raf. J. Mol. Biol. 2010, 399, 422–435. [Google Scholar] [CrossRef]
- Bandaru, P.; Kondo, Y.; Kuriyan, J. The Interdependent Activation of Son-of-Sevenless and Ras. Cold Spring Harb. Perspect. Med. 2018, 9, a031534. [Google Scholar] [CrossRef]
- Lowenstein, E.; Daly, R.; Batzer, A.; Li, W.; Margolis, B.; Lammers, R.; Ullrich, A.; Skolnik, E.; Bar-Sagi, D.; Schlessinger, J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992, 70, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Asmamaw, M.D.; Shi, X.-J.; Zhang, L.-R.; Liu, H.-M. A comprehensive review of SHP2 and its role in cancer. Cell. Oncol. 2022, 45, 729–753. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.L.; von Delft, F.; Brennan, P.E. Targeting the Small GTPase Superfamily through Their Regulatory Proteins. Angew. Chem. Int. Ed. 2019, 59, 6342–6366. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef]
- Hennig, A.; Markwart, R.; Esparza-Franco, M.A.; Ladds, G.; Rubio, I. Ras activation revisited: Role of GEF and GAP systems. Biol. Chem. 2015, 396, 831–848. [Google Scholar] [CrossRef]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef]
- Kim, H.J.; Na Lee, H.; Jeong, M.S.; Jang, S.B. Oncogenic KRAS: Signaling and Drug Resistance. Cancers 2021, 13, 5599. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Windon, A.L.; Loaiza-Bonilla, A.; Jensen, C.E.; Randall, M.; Morrissette, J.J.D.; Shroff, S.G. A KRAS wild type mutational status confers a survival advantage in pancreatic ductal adenocarcinoma. J. Gastrointest. Oncol. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Philip, P.A.; Azar, I.; Xiu, J.; Hall, M.J.; Hendifar, A.E.; Lou, E.; Hwang, J.J.; Gong, J.; Feldman, R.; Ellis, M.; et al. Molecular Characterization of KRAS Wild-type Tumors in Patients with Pancreatic Adenocarcinoma. Clin. Cancer Res. 2022, 28, 2704–2714. [Google Scholar] [CrossRef]
- Ardalan, B.; Ciner, A.; Baca, Y.; Darabi, S.; Kasi, A.; Lou, E.; Azqueta, J.I.; Xiu, J.; Nabhan, C.; Shields, A.F.; et al. Not all treated KRAS-mutant pancreatic adenocarcinomas are equal: KRAS G12D and survival outcome. J. Clin. Oncol. 2023, 41, 4020. [Google Scholar] [CrossRef]
- Ardalan, B.; Ciner, A.; Baca, Y.; Darabi, S.; Kasi, A.; Lou, E.; Azqueta, J.I.; Xiu, J.; Nabhan, C.; Shields, A.F.; et al. Prognostic indicators of KRAS G12X mutations in pancreatic cancer. J. Clin. Oncol. 2023, 41, 735. [Google Scholar] [CrossRef]
- Ciner, A.; Ardalan, B.; Baca, Y.; Darabi, S.; Kasi, A.; Lou, E.; Azqueta, J.I.; Xiu, J.; Nabhan, C.; Shields, A.F.; et al. KRAS G12C-mutated pancreatic cancer: Clinical outcomes based on chemotherapeutic regimen. J. Clin. Oncol. 2023, 41, 4150. [Google Scholar] [CrossRef]
- Suzuki, T.; Masugi, Y.; Inoue, Y.; Hamada, T.; Tanaka, M.; Takamatsu, M.; Arita, J.; Kato, T.; Kawaguchi, Y.; Kunita, A.; et al. KRAS variant allele frequency, but not mutation positivity, associates with survival of patients with pancreatic cancer. Cancer Sci. 2022, 113, 3097–3109. [Google Scholar] [CrossRef]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Öllinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.J.; Ho, L.; Ranganathan, S.; Abbruzzese, J.L.; Alpaugh, R.K.; Beard, M.; Lewis, N.L.; McLaughlin, S.; Rogatko, A.; Perez-Ruixo, J.J.; et al. Phase II and Pharmacodynamic Study of the Farnesyltransferase Inhibitor R115777 as Initial Therapy in Patients With Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2003, 21, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.S.; McCoy, S.; Whitehead, R.P.; Iqbal, S.; Wade, J.L.; Giguere, J.K.; Abbruzzese, J.L. A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: A Southwest oncology group (SWOG 9924) study. Investig. New Drugs 2005, 23, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; van de Velde, H.; Karasek, P.; Oettle, H.; Vervenne, W.; Szawlowski, A.; Schoffski, P.; Post, S.; Verslype, C.; Neumann, H.; et al. Phase III Trial of Gemcitabine Plus Tipifarnib Compared With Gemcitabine Plus Placebo in Advanced Pancreatic Cancer. J. Clin. Oncol. 2004, 22, 1430–1438. [Google Scholar] [CrossRef]
- Ledford, H. Cancer researchers revisit ‘failed’ clinical trials. Nature 2013. [Google Scholar] [CrossRef]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.-K. K- and N-Ras Are Geranylgeranylated in Cells Treated with Farnesyl Protein Transferase Inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef]
- Kazi, A.; Xiang, S.; Yang, H.; Chen, L.; Kennedy, P.; Ayaz, M.; Fletcher, S.; Cummings, C.; Lawrence, H.R.; Beato, F.; et al. Dual Farnesyl and Geranylgeranyl Transferase Inhibitor Thwarts Mutant KRAS-Driven Patient-Derived Pancreatic Tumors. Clin. Cancer Res. 2019, 25, 5984–5996. [Google Scholar] [CrossRef]
- Spencer-Smith, R.; Li, L.; Prasad, S.; Koide, A.; Koide, S.; O’Bryan, J.P. Targeting the α4-α5 interface of RAS results in multiple levels of inhibition. Small GTPases 2019, 10, 378–387. [Google Scholar] [CrossRef]
- Khan, I.; Marelia-Bennet, C.; Lefler, J.; Zuberi, M.; Denbaum, E.; Koide, A.; Connor, D.M.; Broome, A.-M.; Pécot, T.; Timmers, C.; et al. Targeting the KRAS α4-α5 allosteric interface inhibits pancreatic cancer tumorigenesis. Small GTPases 2021, 13, 114–127. [Google Scholar] [CrossRef]
- Oliveira-Cunha, M.; Newman, W.G.; Siriwardena, A.K. Epidermal Growth Factor Receptor in Pancreatic Cancer. Cancers 2011, 3, 1513–1526. [Google Scholar] [CrossRef]
- Forster, T.; Huettner, F.J.; Springfeld, C.; Loehr, M.; Kalkum, E.; Hackbusch, M.; Hackert, T.; Diener, M.K.; Probst, P. Cetuximab in Pancreatic Cancer Therapy: A Systematic Review and Meta-Analysis. Oncology 2019, 98, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Halfdanarson, T.R.; Foster, N.R.; Kim, G.P.; Meyers, J.P.; Smyrk, T.C.; McCullough, A.E.; Ames, M.M.; Jaffe, J.P.; Alberts, S.R. A Phase II Randomized Trial of Panitumumab, Erlotinib, and Gemcitabine Versus Erlotinib and Gemcitabine in Patients with Untreated, Metastatic Pancreatic Adenocarcinoma: North Central Cancer Treatment Group Trial N064B (Alliance). Oncologist 2019, 24, 589-e160. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib Plus Gemcitabine Compared With Gemcitabine Alone in Patients With Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Hammel, P.; Huguet, F.; van Laethem, J.L.; Goldstein, D.; Glimelius, B.; Artru, P.; Borbath, I.; Bouché, O.; Shannon, J.; André, T.; et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine With or Without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA 2016, 315, 1844–1853. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Waldschmidt, D.T.; Stahl, M.; Reinacher-Schick, A.; Freiberg-Richter, J.; Fischer von Weikersthal, L.; Kaiser, F.; Kanzler, S.; Frickhofen, N.; Seufferlein, T.; et al. Afatinib plus gemcitabine versus gemcitabine alone as first-line treatment of metastatic pancreatic cancer: The randomised, open-label phase II ACCEPT study of the Arbeitsgemeinschaft Internistische Onkologie with an integrated analysis of the ‘burden of therapy’ method. Eur. J. Cancer 2021, 146, 95–106. [Google Scholar] [PubMed]
- Propper, D.; Davidenko, I.; Bridgewater, J.; Kupcinskas, L.; Fittipaldo, A.; Hillenbach, C.; Klughammer, B.; Ducreux, M. Phase II, randomized, biomarker identification trial (MARK) for erlotinib in patients with advanced pancreatic carcinoma. Ann. Oncol. 2014, 25, 1384–1390. [Google Scholar] [CrossRef]
- Qin, S.; Bai, Y.; Wang, Z.; Chen, Z.; Xu, R.; Xu, J.; Zhang, H.; Chen, J.; Yuan, Y.; Liu, T.; et al. Nimotuzumab combined with gemcitabine versus gemcitabine in K-RAS wild-type locally advanced or metastatic pancreatic cancer: A prospective, randomized-controlled, double-blinded, multicenter, and phase III clinical trial. J. Clin. Oncol. 2022, 40, LBA4011. [Google Scholar] [CrossRef]
- Schultheis, B.; Reuter, D.; Ebert, M.P.; Siveke, J.; Kerkhoff, A.; Berdel, W.E.; Hofheinz, R.; Behringer, D.M.; Schmidt, W.E.; Goker, E.; et al. Gemcitabine combined with the monoclonal antibody nimotuzumab is an active first-line regimen inKRAS wildtype patients with locally advanced or metastatic pancreatic cancer: A multicenter, randomized phase IIb study. Ann. Oncol. 2017, 28, 2429–2435. [Google Scholar] [CrossRef]
- Boriack-Sjodin, P.A.; Margarit, S.M.; Bar-Sagi, D.; Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Zhang, J.; Nussinov, R. Inhibitors of Ras-SOS Interactions. ChemMedChem 2016, 11, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Cancer: The Ras renaissance. Nature 2015, 520, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Jeng, H.-H.; Taylor, L.J.; Bar-Sagi, D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat. Commun. 2012, 3, 1168. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Gort, E.; Pant, S.; Lolkema, M.; Sebastian, M.; Scheffler, M.; Hwang, J.; Dünzinger, U.; Riemann, K.; Kitzing, T.; et al. 524P A phase I, open-label, dose-escalation trial of BI 1701963 in patients (pts) with KRAS mutated solid tumours: A snapshot analysis. Ann. Oncol. 2021, 32, S591–S592. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1–KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2020, 11, 142–157. [Google Scholar] [CrossRef]
- Zhou, C.; Fan, Z.; Zhou, Z.; Li, Y.; Cui, R.; Liu, C.; Zhou, G.; Diao, X.; Jiang, H.; Zheng, M.; et al. Discovery of the First-in-Class Agonist-Based SOS1 PROTACs Effective in Human Cancer Cells Harboring Various KRAS Mutations. J. Med. Chem. 2022, 65, 3923–3942. [Google Scholar] [CrossRef]
- Mohi, M.G.; Neel, B.G. The role of Shp2 (PTPN11) in cancer. Curr. Opin. Genet. Dev. 2007, 17, 23–30. [Google Scholar] [CrossRef]
- Song, Y.; Zhao, M.; Wu, Y.; Yu, B.; Liu, H.-M. A multifunctional cross-validation high-throughput screening protocol enabling the discovery of new SHP2 inhibitors. Acta Pharm. Sin. B 2020, 11, 750–762. [Google Scholar] [CrossRef]
- Quintana, E.; Schulze, C.J.; Myers, D.R.; Choy, T.J.; Mordec, K.; Wildes, D.; Shifrin, N.T.; Belwafa, A.; Koltun, E.S.; Gill, A.L.; et al. Allosteric Inhibition of SHP2 Stimulates Antitumor Immunity by Transforming the Immunosuppressive Environment. Cancer Res. 2020, 80, 2889–2902. [Google Scholar] [CrossRef]
- Dance, M.; Montagner, A.; Salles, J.P.; Yart, A.; Raynal, P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal. 2008, 20, 453–459. [Google Scholar] [CrossRef]
- Sun, J.; Lu, S.; Ouyang, M.; Lin, L.-J.; Zhuo, Y.; Liu, B.; Chien, S.; Neel, B.G.; Wang, Y. Antagonism between binding site affinity and conformational dynamics tunes alternative cis-interactions within Shp2. Nat. Commun. 2013, 4, 2037. [Google Scholar] [CrossRef] [PubMed]
- Hanafusa, H.; Torii, S.; Yasunaga, T.; Matsumoto, K.; Nishida, E. Shp2, an SH2-containing Protein-tyrosine Phosphatase, Positively Regulates Receptor Tyrosine Kinase Signaling by Dephosphorylating and Inactivating the Inhibitor Sprouty. J. Biol. Chem. 2004, 279, 22992–22995. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nature 2018, 20, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.L.; Haderk, F.; Bivona, T.G. Allosteric SHP2 inhibitors in cancer: Targeting the intersection of RAS, resistance, and the immune microenvironment. Curr. Opin. Chem. Biol. 2021, 62, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Brana, I.; Shapiro, G.; Johnson, M.L.; Yu, H.A.; Robbrecht, D.; Tan, D.S.-W.; Siu, L.L.; Minami, H.; Steeghs, N.; Hengelage, T.; et al. Initial results from a dose finding study of TNO155, a SHP2 inhibitor, in adults with advanced solid tumors. J. Clin. Oncol. 2021, 39, 3005. [Google Scholar] [CrossRef]
- McKean, M.; Barve, M.; Hong, D.; Parikh, A.; Rosen, E.; Yang, J.; Picard, R.; Yi, J.; Brail, L.; Vecchio, D.; et al. Preliminary results from FLAGSHP-1: A Phase I dose escalation study of ERAS-601, a potent SHP2 inhibitor, in patients with previously treated advanced or metastatic solid tumors. Eur. J. Cancer 2022, 174, S34. [Google Scholar] [CrossRef]
- Bendell, J.; Ulahannan, S.; Koczywas, M.; Brahmer, J.; Capasso, A.; Eckhardt, S.; Gordon, M.; McCoach, C.; Nagasaka, M.; Ng, K.; et al. Intermittent dosing of RMC-4630, a potent, selective inhibitor of SHP2, combined with the MEK inhibitor cobimetinib, in a phase 1b/2 clinical trial for advanced solid tumors with activating mutations of RAS signaling. Eur. J. Cancer 2020, 138, S8–S9. [Google Scholar] [CrossRef]
- Ou, S.; Koczywas, M.; Ulahannan, S.; Janne, P.; Pacheco, J.; Burris, H.; McCoach, C.; Wang, J.; Gordon, M.; Haura, E.; et al. A12 The SHP2 Inhibitor RMC-4630 in Patients with KRAS-Mutant Non-Small Cell Lung Cancer: Preliminary Evaluation of a First-in-Man Phase 1 Clinical Trial. J. Thorac. Oncol. 2020, 15, S15–S16. [Google Scholar] [CrossRef]
- Liu, C.; Lu, H.; Wang, H.; Loo, A.; Zhang, X.; Yang, G.; Kowal, C.; Delach, S.; Wang, Y.; Goldoni, S.; et al. Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling. Clin. Cancer Res. 2021, 27, 342–354. [Google Scholar] [CrossRef]
- Theard, P.L.; Sheffels, E.; Sealover, N.E.; Linke, A.J.; Pratico, D.J.; Kortum, R.L. Marked synergy by vertical inhibition of EGFR signaling in NSCLC spheroids shows SOS1 is a therapeutic target in EGFR-mutated cancer. Elife 2020, 9, e58204. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Cowzer, D.; Zameer, M.; Conroy, M.; Kolch, W.; Duffy, A.G. Targeting KRAS in Pancreatic Cancer. J. Pers. Med. 2022, 12, 1870. [Google Scholar] [CrossRef]
- Desai, J.; Gan, H.; Barrow, C.; Jameson, M.; Atkinson, V.; Haydon, A.; Millward, M.; Begbie, S.; Brown, M.; Markman, B.; et al. Phase I, Open-Label, Dose-Escalation/Dose-Expansion Study of Lifirafenib (BGB-283), an RAF Family Kinase Inhibitor, in Patients With Solid Tumors. J. Clin. Oncol. 2020, 38, 2140–2150. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, T.; Shin, S.; Han, S.-W.; Kim, J.-S.; Kim, Y.; Yoo, C.; Lee, D.; Ahn, C.; Kim, S.; et al. 529P A phase Ib trial of belvarafenib in combination with cobimetinib in patients (pts) with RAS- or RAF- mutated (m) solid tumors: Updated safety data and indication-specific efficacy results. Ann. Oncol. 2021, 32, S595. [Google Scholar] [CrossRef]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.-P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.-Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Bodoky, G.; Timcheva, C.; Spigel, D.R.; La Stella, P.J.; Ciuleanu, T.E.; Pover, G.; Tebbutt, N.C. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Investig. New Drugs 2011, 30, 1216–1223. [Google Scholar] [CrossRef]
- Ko, A.H.; Bekaii-Saab, T.; Van Ziffle, J.; Mirzoeva, O.M.; Joseph, N.M.; Talasaz, A.; Kuhn, P.; Tempero, M.A.; Collisson, E.A.; Kelley, R.K.; et al. A Multicenter, Open-Label Phase II Clinical Trial of Combined MEK plus EGFR Inhibition for Chemotherapy-Refractory Advanced Pancreatic Adenocarcinoma. Clin. Cancer Res. 2016, 22, 61–68. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Baker, N.M.; Miermont, A.M.; Thurman, R.D.; Pierobon, M.; Tran, T.H.; Anderson, A.O.; Waters, A.M.; Diehl, J.N.; Papke, B.; et al. Atypical KRAS(G12R) Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov. 2020, 10, 104–123. [Google Scholar] [CrossRef]
- Diehl, A.C.; Hannan, L.M.; Zhen, D.B.; Coveler, A.L.; King, G.; A Cohen, S.; Harris, W.P.; Shankaran, V.; Wong, K.M.; Green, S.; et al. KRAS Mutation Variants and Co-occurring PI3K Pathway Alterations Impact Survival for Patients with Pancreatic Ductal Adenocarcinomas. Oncologist 2022, 27, 1025–1033. [Google Scholar] [CrossRef]
- Kenney, C.; Kunst, T.; Webb, S.; Christina, D., Jr.; Arrowood, C.; Steinberg, S.M.; Mettu, N.B.; Kim, E.J.; Rudloff, U. Phase II study of selumetinib, an orally active inhibitor of MEK1 and MEK2 kinases, in KRAS(G12R)-mutant pancreatic ductal adenocarcinoma. Investig. New Drugs 2021, 39, 821–828. [Google Scholar] [CrossRef]
- Ardalan, B.; Azqueta, J.; Sleeman, D. Cobimetinib Plus Gemcitabine: An Active Combination in KRAS G12R-Mutated Pancreatic Ductal Adenocarcinoma Patients in Previously Treated and Failed Multiple Chemotherapies. J. Pancreat. Cancer 2021, 7, 65–70. [Google Scholar] [CrossRef]
- Grierson, P.M.; Tan, B.; Pedersen, K.S.; Park, H.; Suresh, R.; A Amin, M.; A Trikalinos, N.; Knoerzer, D.; Kreider, B.; Reddy, A.; et al. Phase Ib Study of Ulixertinib Plus Gemcitabine and Nab-Paclitaxel in Patients with Metastatic Pancreatic Adenocarcinoma. Oncologist 2022, 28, e115–e123. [Google Scholar] [CrossRef]
- Wang, J.; Johnson, M.; Barve, M.; Pelster, M.; Chen, X.; Li, Z.; Gordon, J.; Reiss, M.; Pai, S.; Falchook, G.; et al. Preliminary results from HERKULES-1: A phase 1b/2, open-label, multicenter study of ERAS-007, an oral ERK1/2 inhibitor, in patients with advanced or metastatic solid tumors. Eur. J. Cancer 2022, 174, S80–S81. [Google Scholar] [CrossRef]
- Punekar, S.R.; Velcheti, V.; Neel, B.G.; Wong, K.K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 2022, 19, 637–655. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. The KRAS crowd targets its next cancer mutations. Nat. Rev. Drug Discov. 2023, 22, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.S.; Yaeger, R.; Spira, A.I.; Pelster, M.S.; Sabari, J.K.; Hafez, N.; Barve, M.; Velastegui, K.; Yan, X.; Shetty, A.; et al. Adagrasib in Advanced Solid Tumors Harboring a KRAS(G12C) Mutation. J. Clin. Oncol. 2023, 26, 10–200. [Google Scholar]
- Lindsay, C.R.; Blackhall, F.H. Direct Ras G12C inhibitors: Crossing the rubicon. Br. J. Cancer 2019, 121, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Asimgil, H.; Ertetik, U.; Çevik, N.C.; Ekizce, M.; Doğruöz, A.; Gökalp, M.; Arık-Sever, E.; Istvanffy, R.; Friess, H.; Ceyhan, G.O.; et al. Targeting the undruggable oncogenic KRAS: The dawn of hope. J. Clin. Investig. 2022, 7. [Google Scholar] [CrossRef]
- Salem, M.E.; El-Refai, S.M.; Sha, W.; Puccini, A.; Grothey, A.; George, T.J.; Hwang, J.J.; O’Neil, B.; Barrett, A.S.; Kadakia, K.C.; et al. Landscape of KRAS(G12C), Associated Genomic Alterations, and Interrelation With Immuno-Oncology Biomarkers in KRAS-Mutated Cancers. JCO Precis. Oncol. 2022, 6, e2100245. [Google Scholar] [CrossRef]
- Christensen, J.G.; Olson, P.; Briere, T.; Wiel, C.; Bergo, M.O. Targeting Kras(g12c)-mutant cancer with a mutation-specific inhibitor. J. Intern. Med. 2020, 288, 183–191. [Google Scholar] [CrossRef]
- Ostrem, J.M.L.; Shokat, K.M. Targeting KRAS G12C with Covalent Inhibitors. Ann. Rev. Cancer Biol. 2022, 6, 49–64. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Chandhoke, A.S.; Yu, Q.; Małachowska, B.; Kuljanin, M.; Gikandi, A.; Stańczak, M.; Gableske, S.; Jedrychowski, M.P.; Scott, D.A.; et al. Defining and Targeting Adaptations to Oncogenic KRAS(G12C) Inhibition Using Quantitative Temporal Proteomics. Cell Rep. 2020, 30, 4584–4599.e4. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Oya, Y.; Mitsudomi, T. Is adagrasib just another sotorasib?-or, should we differentiate their usage according to patients’ clinical presentation? Transl. Lung Cancer Res. 2023, 12, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Satake, H.; George, T.J.; Yaeger, R.; Hollebecque, A.; Garrido-Laguna, I.; Schuler, M.; Burns, T.F.; Coveler, A.L.; Flchook, G.S.; et al. Sotorasib in KRAS p.G12C-Mutated Advanced Pancreatic Cancer. N. Engl. J. Med. 2023, 388, 33–43. [Google Scholar] [CrossRef]
- Murciano-Goroff, Y.R.; Heist, R.S.; Kuboki, Y.; Koyama, T.; Ammakkanavar, N.R.; Hollebecque, A.; Patnaik, A.; Shimizu, T.; Spira, A.I.; Nagasaka, M.; et al. Abstract CT028: A first-in-human phase 1 study of LY3537982, a highly selective and potent KRAS G12C inhibitor in patients with KRAS G12C-mutant advanced solid tumors. Cancer Res 2023, 83, CT028. [Google Scholar] [CrossRef]
- Sacher, A.; LoRusso, P.; Patel, M.R.; Miller, W.H.; Garralda, E.; Forster, M.D.; Santoro, A.; Falcon, A.; Kim, T.W.; Paz-Ares, L.; et al. Single-Agent Divarasib (GDC-6036) in Solid Tumors with a KRAS G12C Mutation. New Engl. J. Med. 2023, 389, 710–721. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. The KRAS-G12C inhibitor: Activity and resistance. Cancer Gene Ther. 2021, 29, 875–878. [Google Scholar] [CrossRef]
- Bhamidipati, D.; Subbiah, V. Cracking KRAS(G12C) across all solid tumors: The new kid on the block for tissue-agnostic precision medicine. ESMO Open 2023, 8, 101591. [Google Scholar] [CrossRef]
- Blaquier, J.B.; Cardona, A.F.; Recondo, G. Resistance to KRAS(G12C) Inhibitors in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 787585. [Google Scholar] [CrossRef]
- Arbour, K.C.; Lito, P. Expanding the Arsenal of Clinically Active KRAS G12C Inhibitors. J. Clin. Oncol. 2022, 40, 2609–2611. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Murciano-Goroff, Y.R.; Xue, J.Y.; Ang, A.; Lucas, J.; Mai, T.T.; Da Cruz Paula, A.F.; Saiki, A.Y.; Mohn, D.; Achanta, P.; et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 2021, 599, 679–683. [Google Scholar] [CrossRef]
- Yaeger, R.; Solit, D.B. Overcoming Adaptive Resistance to KRAS Inhibitors Through Vertical Pathway Targeting. Clin. Cancer Res. 2020, 26, 1538–1540. [Google Scholar] [CrossRef]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef]
- Xiaolun, W.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar]
- Hallin, J.; Bowcut, V.; Calinisan, A.; Briere, D.M.; Hargis, L.; Engstrom, L.D.; Laguer, J.; Medwid, J.; Vanderpool, D.; Lifset, E.; et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat. Med. 2022, 28, 2171–2182. [Google Scholar] [CrossRef]
- Kemp, S.B.; Cheng, N.; Markosyan, N.; Sor, R.; Kim, I.-K.; Hallin, J.; Shoush, J.; Quinones, L.; Brown, N.V.; Bassett, J.B.; et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2022, 13, 298–311. [Google Scholar] [CrossRef]
- Mahadevan, K.K.; McAndrews, K.M.; LeBleu, V.S.; Yang, S.; Lyu, H.; Li, B.; Sockwell, A.M.; Kirtley, M.L.; Morse, S.J.; Moreno Diaz, B.A.; et al. Oncogenic Kras (G12D) specific non-covalent inhibitor reprograms tumor microenvironment to prevent and reverse early pre-neoplastic pancreatic lesions and in combination with immunotherapy regresses advanced PDAC in a CD8 (+) T cells dependent manner. bioRxiv 2023. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, T.; Yoshinari, T.; Nishizono, Y.; Tasaki, M.; Inamura, K.; Ishioka, H.; Suzuki, A.; Osaki, F.; Yamanaka, Y.; Hayakawa, M. Abstract 5735: Novel KRAS G12D degrader ASP3082 demonstrates in vivo, dose-dependent KRAS degradation, KRAS pathway inhibition, and antitumor efficacy in multiple KRAS G12D-mutated cancer models. Cancer Res 2023, 83, 5735. [Google Scholar] [CrossRef]
- Han, J.M.; Jung, H.J. Cyclophilin A/CD147 Interaction: A Promising Target for Anticancer Therapy. Int. J. Mol. Sci. 2022, 23, 9341. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhai, Q.; Bharadwaj, U.; Wang, H.; Li, F.; Fisher, W.E.; Chen, C.; Yao, Q. Cyclophilin A is overexpressed in human pancreatic cancer cells and stimulates cell proliferation through CD147. Cancer 2006, 106, 2284–2294. [Google Scholar] [CrossRef] [PubMed]
- Knox, J.E.; Jiang, J.; Burnett, G.L.; Liu, Y.; Weller, C.E.; Wang, Z.; McDowell, L.; Steele, S.L.; Chin, S.; Chou, K.J.; et al. Abstract 3596: RM-036, a first-in-class, orally-bioavailable, Tri-Complex covalent KRASG12D(ON) inhibitor, drives profound anti-tumor activity in KRASG12D mutant tumor models. Cancer Res. 2022, 82, 3596. [Google Scholar] [CrossRef]
- Jiang, L.; Menard, M.; Weller, C.; Wang, Z.; Burnett, L.; Aronchik, I.; Steele, S.; Flagella, M.; Zhao, R.; Evans, J.W.W.; et al. Abstract 526: RMC-9805, a first-in-class, mutant-selective, covalent and oral KRASG12D(ON) inhibitor that induces apoptosis and drives tumor regression in preclinical models of KRASG12D cancers. Cancer Res. 2023, 83, 526. [Google Scholar] [CrossRef]
- Menard, M.J.; Chow, C.; Chen, K.; Blaj, C.; Shifrin, N.T.; Courtney, H.; Nasholm, N.M.; Pham, A.; Kumamoto, A.; Ganesh, S.; et al. Abstract 3475: RMC-9805, a first-in-class, mutant-selective, covalent and orally bioavailable KRASG12D(ON) inhibitor, promotes cancer-associated neoantigen recognition and synergizes with immunotherapy in preclinical models. Cancer Res. 2023, 83, 3475. [Google Scholar] [CrossRef]
- Zhang, Z.; Guiley, K.Z.; Shokat, K.M. Chemical acylation of an acquired serine suppresses oncogenic signaling of K-Ras(G12S). Nat. Chem. Biol. 2022, 18, 1177–1183. [Google Scholar] [CrossRef]
- Zhang, Z.; Morstein, J.; Ecker, A.K.; Guiley, K.Z.; Shokat, K.M. Chemoselective Covalent Modification of K-Ras(G12R) with a Small Molecule Electrophile. J. Am. Chem. Soc. 2022, 144, 15916–15921. [Google Scholar] [CrossRef]
- Pharma, J. Jacobio Announces Annual Results of 2021, R&D Investment Increases 83%. 2022. Available online: http://www.jacobiopharma.com/en/news/Jacobio-Announces-Annual-Results-of-2021-R%26D-Investment-Increases-83%25 (accessed on 1 August 2023).
- Akhave, N.S.; Biter, A.B.; Hong, D.S. The Next Generation of KRAS Targeting: Reasons for Excitement and Concern. Mol. Cancer Ther. 2022, 21, 1645–1651. [Google Scholar] [CrossRef]
- Kim, D.; Herdeis, L.; Rudolph, D.; Zhao, Y.; Böttcher, J.; Vides, A.; Ayala-Santos, C.I.; Pourfarjam, Y.; Cuevas-Navarro, A.; Xue, J.Y.; et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature 2023, 619, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Foote, J.B.; Mattox, T.E.; Keeton, A.B.; Purnachandra, G.N.; Maxuitenko, Y.; Chen, X.; Valiyaveettil, J.; Buchsbaum, D.J.; Piazza, G.A.; El-Rayes, B.F. Abstract 4140: Oncogenic KRAS inhibition with ADT-007 primes T cell responses in pancreatic ductal adenocarcinoma. Cancer Res. 2023, 83, 4140. [Google Scholar] [CrossRef]
- Bery, N.; Miller, A.; Rabbitts, T. A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nature 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Lim, S.M.; Hanif, E.A.M.; Chin, S.-F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Minerva Anestesiol. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, C.; Yang, G.; Zhao, B.; Wang, W. The role of autophagy in pancreatic cancer progression. Biochim. Biophys. Acta (BBA) Rev. Cancer 2021, 1876, 188592. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Minerva Anestesiol. 2011, 25, 717–729. [Google Scholar] [CrossRef]
- Yang, A.; Kimmelman, A.C. Inhibition of autophagy attenuates pancreatic cancer growth independent of TP53/TRP53 status. Autophagy 2014, 10, 1683–1684. [Google Scholar] [CrossRef]
- Kim, M.-J.; Woo, S.-J.; Yoon, C.-H.; Lee, J.-S.; An, S.; Choi, Y.-H.; Hwang, S.-G.; Yoon, G.; Lee, S.-J. Involvement of Autophagy in Oncogenic K-Ras-induced Malignant Cell Transformation. J. Biol. Chem. 2011, 286, 12924–12932. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with p53 Alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-P.; Hu, L.-F.; Zheng, H.-F.; Mao, C.-J.; Hu, W.-D.; Xiong, K.-P.; Wang, F.; Liu, C.-F. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.-C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; A Liotta, L.; et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef] [PubMed]
- AlMasri, S.S.; Zenati, M.S.; Desilva, A.; Nassour, I.; Boone, B.A.; Singhi, A.D.; Bartlett, D.L.; Liotta, L.A.; Espina, V.; Loughran, P.; et al. Encouraging long-term survival following autophagy inhibition using neoadjuvant hydroxychloroquine and gemcitabine for high-risk patients with resectable pancreatic carcinoma. Cancer Med. 2021, 10, 7233–7241. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and Pharmacodynamic Study of Autophagy Inhibition Using Hydroxychloroquine in Patients With Metastatic Pancreatic Adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel with or without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef]
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef]
- Fei, N.; Wen, S.; Ramanathan, R.; Hogg, M.E.; Zureikat, A.H.; Lotze, M.T.; Bahary, N.; Singhi, A.D.; Zeh, H.J.; Boone, B.A. SMAD4 loss is associated with response to neoadjuvant chemotherapy plus hydroxychloroquine in patients with pancreatic adenocarcinoma. Clin. Transl. Sci. 2021, 14, 1822–1829. [Google Scholar] [CrossRef]
- Solitro, A.R.; MacKeigan, J.P.; Lushington, G.H.; Chaguturu, R.; Bosc, D.; Jakhlal, J.; Deprez, B.; Deprez-Poulain, R.; Chai, C.L.; Mátyus, P.; et al. Leaving the lysosome behind: Novel developments in autophagy inhibition. Futur. Med. Chem. 2016, 8, 73–86. [Google Scholar] [CrossRef]
- Xavier, C.B.; Marchetti, K.R.; Castria, T.B.; Jardim, D.L.F.; Fernandes, G.S. Trametinib and Hydroxychloroquine (HCQ) Combination Treatment in KRAS-Mutated Advanced Pancreatic Adenocarcinoma: Detailed Description of Two Cases. J. Gastrointest. Cancer 2020, 52, 374–380. [Google Scholar] [CrossRef]
- Stalnecker, C.A.; Grover, K.R.; Edwards, A.C.; Coleman, M.F.; Yang, R.; DeLiberty, J.M.; Papke, B.; Goodwin, C.M.; Pierobon, M.; Petricoin, E.F.; et al. Concurrent Inhibition of IGF1R and ERK Increases Pancreatic Cancer Sensitivity to Autophagy Inhibitors. Cancer Res. 2021, 82, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Lin, H.; Yu, J.; Yu, J.; Hu, Z. Autophagy and the Immune Response. Autophagy Biol. Dis. 2019, 1206, 595–634. [Google Scholar] [CrossRef]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I; Springer: Berlin/Heidelberg, Germany, 2020; Volume 581, ISBN 0000007587. [Google Scholar]
- Khan, R.; Panja, S.; Ding, L.; Tang, S.; Tang, W.; Kapoor, E.; Bennett, R.G.; Oupický, D. Polymeric Chloroquine as an Effective Antimigration Agent in the Treatment of Pancreatic Cancer. Mol. Pharm. 2022, 19, 4631–4643. [Google Scholar] [CrossRef] [PubMed]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2020, 124, 333–344. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Hong, D.S.; Vandross, A.L.; Psoinos, C.M.; Brennan, D.M.; Sherman, M.L.; Ruiz-Soto, R.; Reu, F.J.; Weekes, C.D. A phase 1/2 study of DCC-3116 as a single agent and in combination with trametinib in patients with advanced or metastatic solid tumors with RAS or RAF mutations. J. Clin. Oncol. 2022, 40 (Suppl. S16). [Google Scholar] [CrossRef]
- Liang, S.; Li, X.; Gao, C.; Zhang, L. microRNA-based autophagy inhibition as targeted therapy in pancreatic cancer. Biomed. Pharmacother. 2020, 132, 110799. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Hong, J.; Iacobuzio-Donahue, C.A. The pancreatic cancer genome revisited. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, B.A.; Wang, H.; Witkiewicz, A.K.; Marshall, J.L.; He, A.R.; Vail, P.; Knudsen, E.S.; Pishvaian, M.J. A Phase I Study of Ribociclib Plus Everolimus in Patients with Metastatic Pancreatic Adenocarcinoma Refractory to Chemotherapy. J. Pancreat. Cancer 2020, 6, 45–54. [Google Scholar] [CrossRef]
- Baghdadi, T.A.; Halabi, S.; Garrett-Mayer, E.; Mangat, P.K.; Ahn, E.R.; Sahai, V.; Alvarez, R.H.; Kim, E.S.; Yost, K.J.; Rygiel, A.L.; et al. Palbociclib in Patients With Pancreatic and Biliary Cancer With CDKN2A Alterations: Results From the Targeted Agent and Profiling Utilization Registry Study. JCO Precis. Oncol. 2019, 3, 1–8. [Google Scholar]
- ClinicalTrials.gov. A Study of Abemaciclib (LY2835219) Alone or in Combination with Other Agents in Participants with Previously Treated Pancreatic Ductal Adenocarcinoma. 2019. Available online: https://classic.clinicaltrials.gov/ct2/show/results/NCT02981342?view=results2023 (accessed on 1 August 2023).
- Willobee, B.A.; Gaidarski, A.A.; Dosch, A.R.; Castellanos, J.A.; Dai, X.; Mehra, S.; Messaggio, F.; Srinivasan, S.; VanSaun, M.N.; Nagathihalli, N.S.; et al. Combined Blockade of MEK and CDK4/6 Pathways Induces Senescence to Improve Survival in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2021, 20, 1246–1256. [Google Scholar] [CrossRef]
- Kato, S.; Adashek, J.J.; Shaya, J.; Okamura, R.; Jimenez, R.E.; Lee, S.; Sicklick, J.K.; Kurzrock, R. Concomitant MEK and Cyclin Gene Alterations: Implications for Response to Targeted Therapeutics. Clin. Cancer Res. 2021, 27, 2792–2797. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Ford, J.M.; O’Dwyer, P.J.; Shapiro, G.I.; McShane, L.M.; Freidlin, B.; O’Cearbhaill, R.E.; George, S.; Glade-Bender, J.; Lyman, G.H.; et al. National Cancer Institute Combination Therapy Platform Trial with Molecular Analysis for Therapy Choice (ComboMATCH). Clin. Cancer Res. 2023, 29, 1412–1422. [Google Scholar] [CrossRef]
- Goodwin, C.M.; Waters, A.M.; Klomp, J.E.; Javaid, S.; Bryant, K.L.; Stalnecker, C.A.; Drizyte-Miller, K.; Papke, B.; Yang, R.; Amparo, A.M.; et al. Combination Therapies with CDK4/6 Inhibitors to Treat KRAS-Mutant Pancreatic Cancer. Cancer Res. 2023, 83, 141–157. [Google Scholar] [CrossRef]
- Raybould, A.L.; Burgess, B.; Urban, C.; Naim, R.; Lee, M.S.; McRee, A.J. A phase Ib trial of ERK inhibition with ulixertinib combined with palbociclib in patients (Pts) with advanced solid tumors. J. Clin. Oncol. 2021, 39, 3103. [Google Scholar] [CrossRef]
- Ulahannan, S.V.; Spigel, D.R.; Lee, M.S.; Fakih, M.; Grierson, P.; Christenson, E.; Chiorean, E.G.; Outlaw, D.A.; Khan, G.; Atreya, C.E.; et al. Preliminary results from ERAS-007 plus palbociclib (palbo) in patients (pts) with KRAS/NRAS mutant (m) colorectal cancer (CRC) or KRASm pancreatic ductal adenocarcinoma (PDAC) in HERKULES-3 study: A phase 1b/2 study of agents targeting the mitogen-activated protein kinase (MAPK) pathway in pts with advanced gastrointestinal malignancies (GI cancers). J. Clin. Oncol. 2023, 41, 3558. [Google Scholar] [CrossRef]
- Mortazavi, M.; Moosavi, F.; Martini, M.; Giovannetti, E.; Firuzi, O. Prospects of targeting PI3K/AKT/mTOR pathway in pancreatic cancer. Crit. Rev. Oncol. 2022, 176, 103749. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Sanchez, C.; Spitrzer, D.; Plambeck-Suess, S.; Gibbs, J.; Hawkins, W.G.; Denardo, D.; Gao, F.; Pufahl, R.A.; Lockhart, A.C.; et al. Synergistic Effects of Concurrent Blockade of PI3K and MEK Pathways in Pancreatic Cancer Preclinical Models. PLoS ONE 2013, 8, e77243. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grilley-Olson, J.E.; Bedard, P.L.; Fasolo, A.; Cornfeld, M.; Cartee, L.; Razak, A.R.A.; Stayner, L.-A.; Wu, Y.; Greenwood, R.; Singh, R.; et al. A phase Ib dose-escalation study of the MEK inhibitor trametinib in combination with the PI3K/mTOR inhibitor GSK2126458 in patients with advanced solid tumors. Investig. New Drugs 2016, 34, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.; McDonough, S.; Philip, P.A.; Cardin, D.; Wang-Gillam, A.; Hui, L.; Tejani, M.A.; Seery, T.E.; Dy, I.A.; Al Baghdadi, T.; et al. Effect of Selumetinib and MK-2206 vs Oxaliplatin and Fluorouracil in Patients With Metastatic Pancreatic Cancer After Prior Therapy: SWOG S1115 Study Randomized Clinical Trial. JAMA Oncol. 2017, 3, 516–522. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Patnaik, A.; Papadopoulos, K.P.; Rasco, D.W.; Becerra, C.R.; Allred, A.J.; Orford, K.; Aktan, G.; Ferron-Brady, G.; Ibrahim, N.; et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother. Pharmacol. 2015, 75, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Gounder, M.; Rodon, J.; Janku, F.; Lolkema, M.P.; Stephenson, J.J.; Bedard, P.L.; Schuler, M.; Sessa, C.; LoRusso, P.; et al. Phase Ib Study of Combination Therapy with MEK Inhibitor Binimetinib and Phosphatidylinositol 3-Kinase Inhibitor Buparlisib in Patients with Advanced Solid Tumors with RAS/RAF Alterations. Oncologist 2020, 25, e160–e169. [Google Scholar] [CrossRef] [PubMed]
- Ciuffreda, L.; Del Curatolo, A.; Falcone, I.; Conciatori, F.; Bazzichetto, C.; Cognetti, F.; Corbo, V.; Scarpa, A.; Milella, M. Lack of growth inhibitory synergism with combined MAPK/PI3K inhibition in preclinical models of pancreatic cancer. Ann. Oncol. 2017, 28, 2896–2898. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, Y.; Qian, L.; Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 2021, 14, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Chapman, P.B.; Feilchenfeldt, J.; Brennan, M.F.; Capanu, M.; Gansukh, B.B.; Jacobs, G.B.; Levin, A.B.; Neville, D.; Kelsen, D.P.; et al. Targeting Mutated K-ras in Pancreatic Adenocarcinoma Using an Adjuvant Vaccine. Am. J. Clin. Oncol. 2011, 34, 321–325. [Google Scholar] [CrossRef]
- Wedén, S.; Klemp, M.; Gladhaug, I.P.; Møller, M.; Eriksen, J.A.; Gaudernack, G.; Buanes, T. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int. J. Cancer 2011, 128, 1120–1128. [Google Scholar] [CrossRef]
- Gjertsen, M.K.; Buanes, T.; Rosseland, A.R.; Bakka, A.; Gladhaug, I.; Søreide, O.; Eriksen, J.A.; Møller, M.; Baksaas, I.; Lothe, R.A.; et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int. J. Cancer 2001, 92, 441–450. [Google Scholar] [CrossRef]
- Muscarella, P.; Bekaii-Saab, T.; McIntyre, K.; Rosemurgy, A.; Ross, S.B.; Richards, D.A.; Fisher, W.E.; Flynn, P.J.; Mattson, A.; Coeshott, C.; et al. A Phase 2 Randomized Placebo-Controlled Adjuvant Trial of GI-4000, a Recombinant Yeast Expressing Mutated RAS Proteins in Patients with Resected Pancreas Cancer. J. Pancreat. Cancer 2021, 7, 8–19. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 2020, 65, 14–20. [Google Scholar] [CrossRef]
- Kijima, H.; Yamazaki, H.; Nakamura, M.; Scanlon, K.J.; Osamura, R.Y.; Ueyama, Y. Ribozyme against mutant K-ras mRNA suppresses tumor growth of pancreatic cancer. Int. J. Oncol. 2004, 24, 559–564. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef]
- Lentsch, E.; Li, L.; Pfeffer, S.; Ekici, A.B.; Taher, L.; Pilarsky, C.; Grützmann, R. CRISPR/Cas9-Mediated Knock-Out of KrasG12D Mutated Pancreatic Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 5706. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, L.; Lang, J.; Cheng, K.; Wang, Y.; Li, X.; Shi, J.; Wang, Y.; Nie, G. A CRISPR-Cas13a system for efficient and specific therapeutic targeting of mutant KRAS for pancreatic cancer treatment. Cancer Lett. 2018, 431, 171–181. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Adagrasib (MTRX 849) | Sotorasib (AMG 510) |
|---|---|---|
| Dose | 600 mg twice a day | 960 mg once a day |
| Half-life (hours) | 23 | 5.5 ± 1.8 |
| Study | KRYSTAL-1 | CodeBreaK100 |
| Phase | II | I/II |
| Sample size | 21 | 38 |
| ORR (%) | 33.3 | 21.0 |
| Median TTR (months) | 1.4 # | 1.5 |
| DCR (%) | 81.0 | 84.0 |
| DOR (%) | 5.3 # | 5.7 |
| Median PFS (months) | 5.4 | 4.0 |
| Median OS (months) | 8.0 | 6.9 |
| Grades 3–4 toxicities (%) | ||
| Fatigue | 6.3 # | 2.3 |
| AST increase | 3.2 # | 2.3 |
| ALT increase | - | 4.7 |
| QT prolongation | 6.3 # | - |
| Diarrhea | 1.6 # | 3.9 |
| Vomiting | 1.6 # | 3.9 |
| Anemia | 1.6 # | 4.7 |
| Drug | Setting | Phase | Clinical Trial Identifier |
|---|---|---|---|
| KRAS Inhibitors—G12C | |||
| HBI-2438 | Advanced solid tumors with KRAS G12C mutation | I | NCT05485974 |
| Adagrasib | Advanced PDAC with KRAS G12C mutation | I | NCT05634525 |
| RMC-6291 | Advanced solid tumors with KRAS G12C mutation | I | NCT05462717 |
| BPI-421286 | Advanced solid tumors with KRAS G12C mutation | I | NCT05315180 |
| D-1553 (Garsorasib) | Advanced solid tumors with KRAS G12C mutation | I | NCT04585035 |
| LY3537982 | Advanced solid tumors with KRAS G12C mutation | I | NCT04956640 |
| BI 1,823,911 (alone or in combination with other agents) | Advanced solid tumors with KRAS G12C mutation | I | NCT04973163 |
| JAB-21822 | Advanced solid tumors with KRAS G12C mutation | I/II | NCT05002270 |
| JNJ-74699157 | Advanced solid tumors wih KRAS G12C mutation | I | NCT04006301 |
| JDQ443 (alone or in combination with other agents) | Advanced solid tumors with KRAS G12C mutation | I | NCT04699188 |
| LY3537982 (alone or in combination with other agents) | Advanced solid tumors with KRAS G12C mutation | I/II | NCT04956640 |
| MK-1084 (alone or in combination with other agents) | Advanced solid tumors with KRAS G12C mutation | I | NCT05067283 |
| Adagrasib (alone or in combination with other agents | Advanced solid tumors with KRAS G12C mutation | I/II | NCT03785249 |
| Sotorasib (alone or in combination with other agents) | Advanced solid tumors with KRAS G12C mutation | Ib/II | NCT04185883 |
| KRAS inhibitor—G12D | |||
| MTRX1133 | Advanced solid tumors with KRAS G12D mutation | I/II | NCT05737706 |
| HRS-4642 | Advanced solid tumors with KRAS G12D mutation | I | NCT05533463 |
| RMC-9805 | Advanced solid tumors with KRAS G12D mutation | I | NCT06040541 |
| Pan-RAS inhibitor | |||
| RMC-6236 | Advanced solid tumors with KRAS mutation (except G12C) | I/Ib | NCT05379985 |
| KRAS degrader | |||
| ASP3082 | Advanced solid tumors with KRAS G12D mutation | I | NCT05382559 |
| SOS1 inhibitor | |||
| MRTX0902 (alone or in combination with adagrasib) | Advanced solid tumors with mutations in MAPK genes | I/II | NCT05578092 |
| BI 1701963 (alone or in combination with trametinib) | Advanced solid tumors with KRAS mutation | I | NCT04111458 |
| SPH2 inhibitor | |||
| RMC-4630 | Advanced solid tumors with MAPK alteration | I | NCT03634982 |
| RMC-4630 + LY3214996 | Advanced solid tumors with KRAS mutation | I/Ib | NCT04916236 |
| JAB-3068 | Advanced solid tumors | I/IIa | NCT03565003 |
| JAB-3312 | Advanced solid tumors | I | NCT04121286 |
| JAB-3312 | Advanced solid tumors | I | NCT04045496 |
| GDC-1971 + Atezolizumab | Advanced solid tumors | I | NCT05487235 |
| KRASi + anti-EGFR | |||
| JAB-21822 + Cetuximab | Advanced solid tumors with KRAS G12C | Ib/II | NCT05194995 |
| KRASi + SOS1i | |||
| Adagrasib + BI 1701963 | Advanced solid tumors with KRAS G12C | I/Ib | NCT04975256 (KRISTAL-14) |
| KRASi + SPH2i | |||
| Adagrasib + TNO155 | Advanced solid tumors with KRAS G12C | I/II | NCT04330664 (KRISTAL-2) |
| JAB-21822 + JAB-3312 | Advanced solid tumors with KRAS G12C | I/IIa | NCT05288205 |
| Autophagy inhibitors | |||
| DCC-3116 (ULK1/2 inhibitor) | Advanced solid tumors with RAS/BRAF mutation | I/II | NCT04892017 |
| Hydroxychloroquine + Trametinibe | Advanced PDAC | II | NCT05518110 (PaTcH) |
| Hydroxychloroquine + Trametinibe | Advanced PDAC | I | NCT03825289 (THREAD) |
| Hydroxychloroquine + Binimetinib | Advanced PDAC with KRAS mutation | I | NCT04132505 |
| MEK inhibitors | |||
| IMM-1-104 | Advanced solid tumors with RAS mutation | I/IIa | NCT05585320 |
| Adoptive cell therapy | |||
| Mutant KRAS G12V-specific TCR transduced T cell therapy | Advanced PDAC with KRAS G12V mutation | I/II | NCT04146298 |
| Mutant KRAS G12V-specific TCR transduced T cell therapy | Advanced PDAC with KRAS G12V mutation | I/II | NCT03190941 |
| Mutant KRAS G12D-specific TCR transduced T cell therapy | Advanced PDAC with KRAS G12D mutation | I/II | NCT03745326 |
| Vaccine | |||
| ELI-002 | Solid tumors with mutations in KRAS/NRAS (G12R/D) and minimal residual disease (ctDNA or CA 19-9 +) | I/II | NCT05726864 (AMPLIFY-7P) |
| ELI-002 | Solid tumors with mutations in KRAS/NRAS (G12R/D) and minimal residual disease (ctDNA or CA 19-9 +) | I | NCT04853017 (AMPLIFY-201) |
| mRNA-5671/V941 (alone or in combination with Pembrolizumab) | Advanced solid tumors with KRAS mutation | I | NCT03948763 (V941-001) |
| Mutant KRAS-Targeted Long Peptide Vaccine Combined + Nivolumab + Ipilimumab | Resected PDAC with KRAS mutation | I | NCT04117087 |
| Small interfering RNA | |||
| iExosomes | Advanced PDAC with KRAS G12D mutation | I | NCT03608631 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Jesus, V.H.F.; Mathias-Machado, M.C.; de Farias, J.P.F.; Aruquipa, M.P.S.; Jácome, A.A.; Peixoto, R.D. Targeting KRAS in Pancreatic Ductal Adenocarcinoma: The Long Road to Cure. Cancers 2023, 15, 5015. https://doi.org/10.3390/cancers15205015
de Jesus VHF, Mathias-Machado MC, de Farias JPF, Aruquipa MPS, Jácome AA, Peixoto RD. Targeting KRAS in Pancreatic Ductal Adenocarcinoma: The Long Road to Cure. Cancers. 2023; 15(20):5015. https://doi.org/10.3390/cancers15205015
Chicago/Turabian Stylede Jesus, Victor Hugo Fonseca, Maria Cecília Mathias-Machado, João Paulo Fogacci de Farias, Marcelo Porfirio Sunagua Aruquipa, Alexandre A. Jácome, and Renata D’Alpino Peixoto. 2023. "Targeting KRAS in Pancreatic Ductal Adenocarcinoma: The Long Road to Cure" Cancers 15, no. 20: 5015. https://doi.org/10.3390/cancers15205015
APA Stylede Jesus, V. H. F., Mathias-Machado, M. C., de Farias, J. P. F., Aruquipa, M. P. S., Jácome, A. A., & Peixoto, R. D. (2023). Targeting KRAS in Pancreatic Ductal Adenocarcinoma: The Long Road to Cure. Cancers, 15(20), 5015. https://doi.org/10.3390/cancers15205015