

GPR4 Knockout Attenuates Intestinal Inflammation and Forestalls the Development of Colitis-Associated Colorectal Cancer in Murine Models

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Dextran Sulfate Sodium (DSS)-Induced Colitis Mouse Model
2.3. Azoxymethane (AOM) and Dextran Sulfate Sodium (DSS)-Induced Colitis-Associated Colorectal Cancer Mouse Model (CAC)
2.4. Mouse Clinical Phenotype Scoring
2.5. Mouse Tissue Collection, Evaluation, and Processing
2.6. Histopathological Analysis
2.7. Tumor Necrotic Area Quantification
2.8. Immunohistochemistry
2.9. Microvessel Density Quantification
2.10. Tumor Proliferation Quantification
2.11. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.12. Statistical Analysis
3. Results
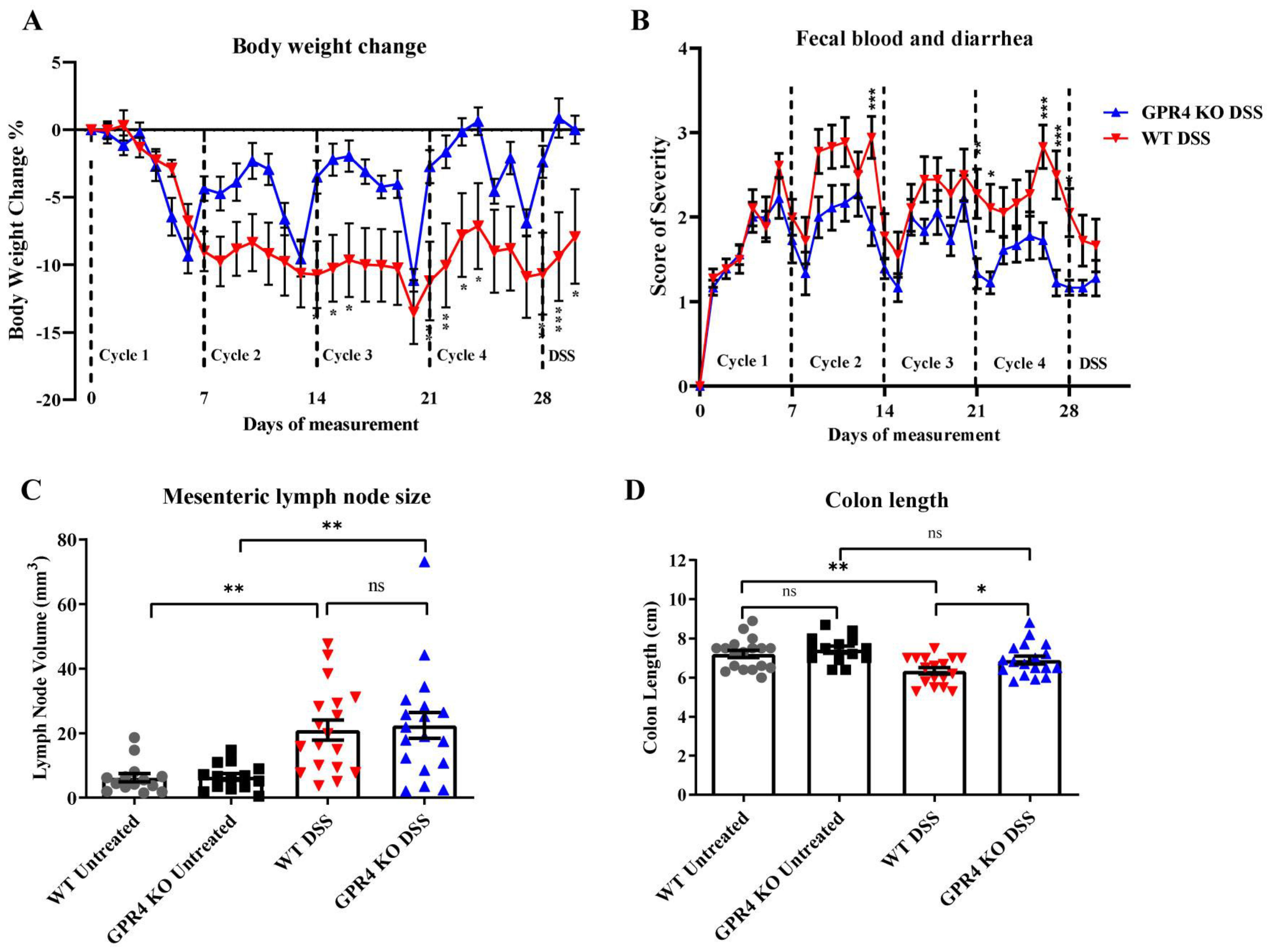
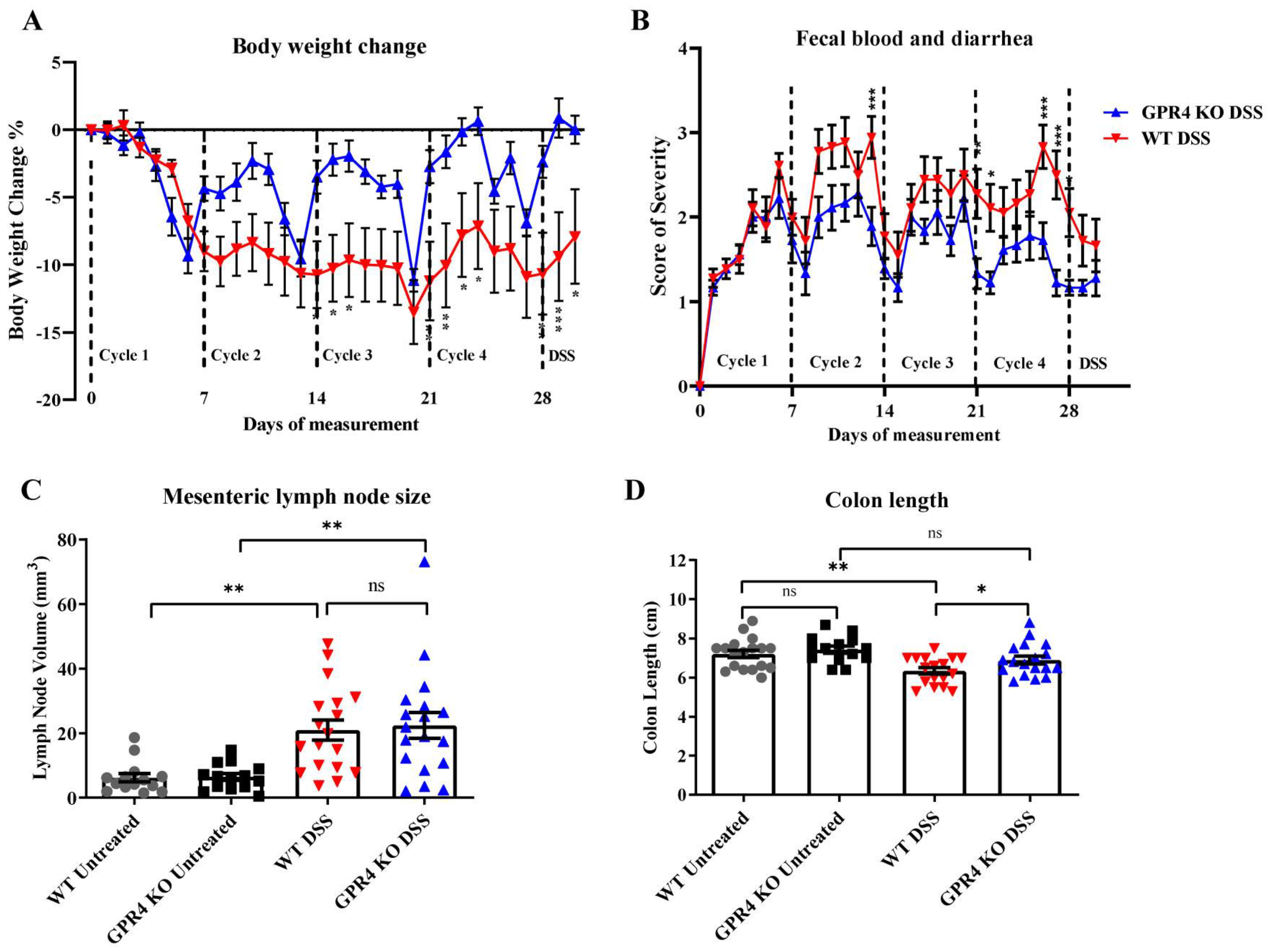
3.1. GPR4 Potentiates Intestinal Inflammation in the Chronic DSS-Induced Experimental Colitis Mouse Model
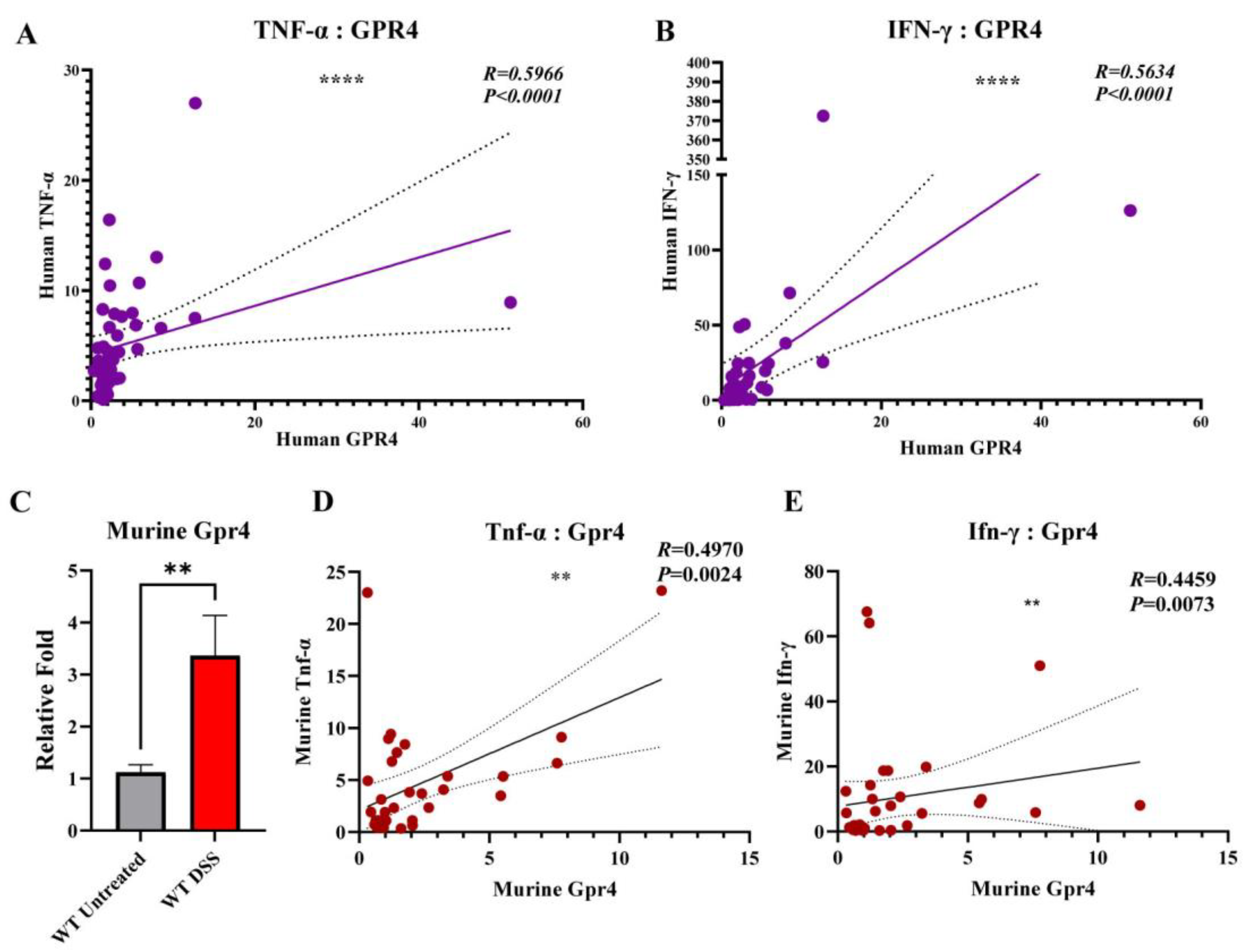
3.2. GPR4 Gene Expression Is Upregulated in Inflamed Colon Tissues and Positively Correlated with TNF-α and INF-γ Gene Expression
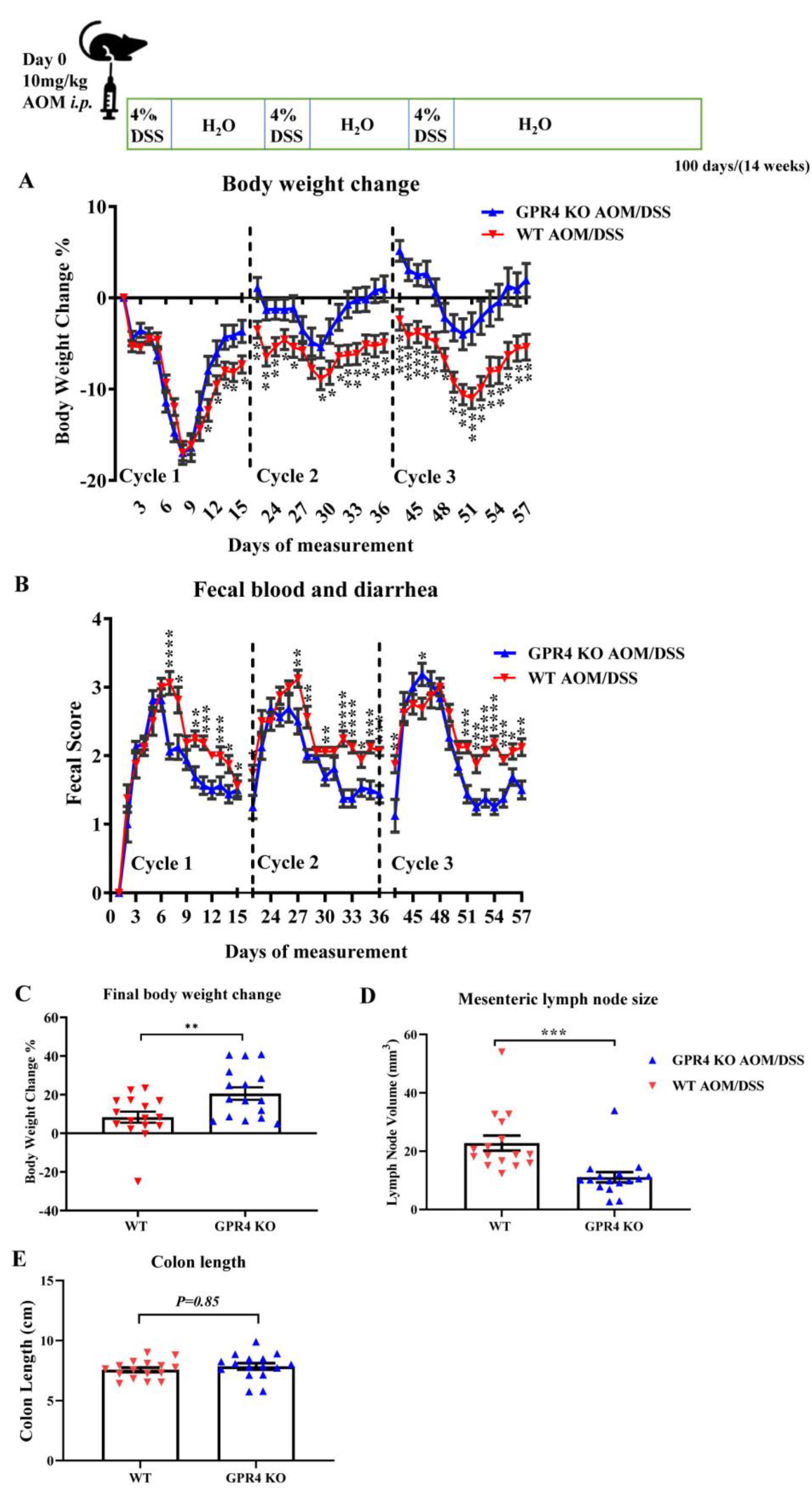
3.3. Genetic Deletion of GPR4 Reduces Disease Severity in CAC Induced by AOM/DSS in Mice
3.4. GPR4 Knockout Reduces Tumor Burden in the CAC Mouse Model
3.5. GPR4 Knockout Increases Necrosis and Cell Death and Decreases Cell Proliferation in the Tumors of AOM/DSS Mice
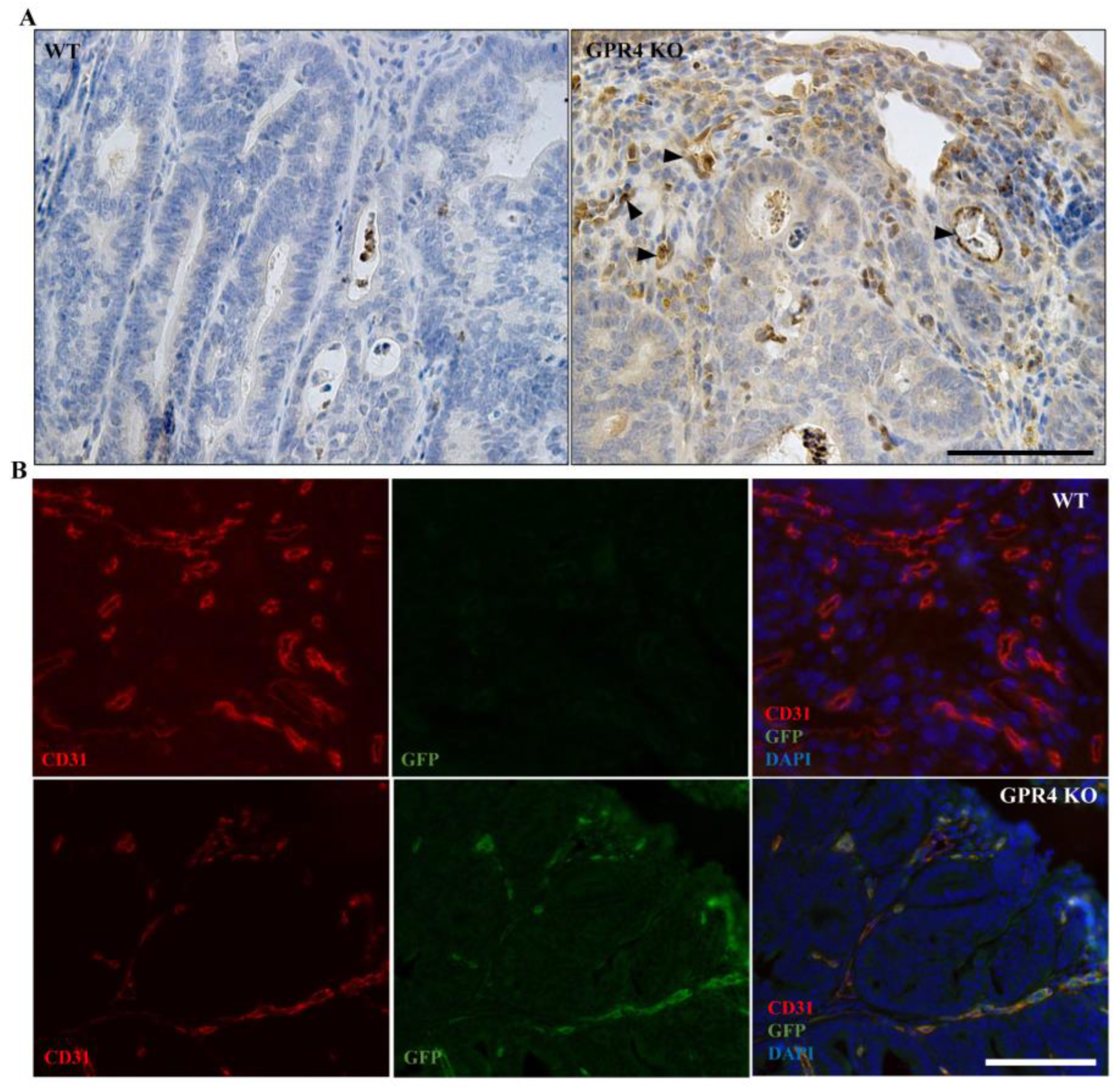
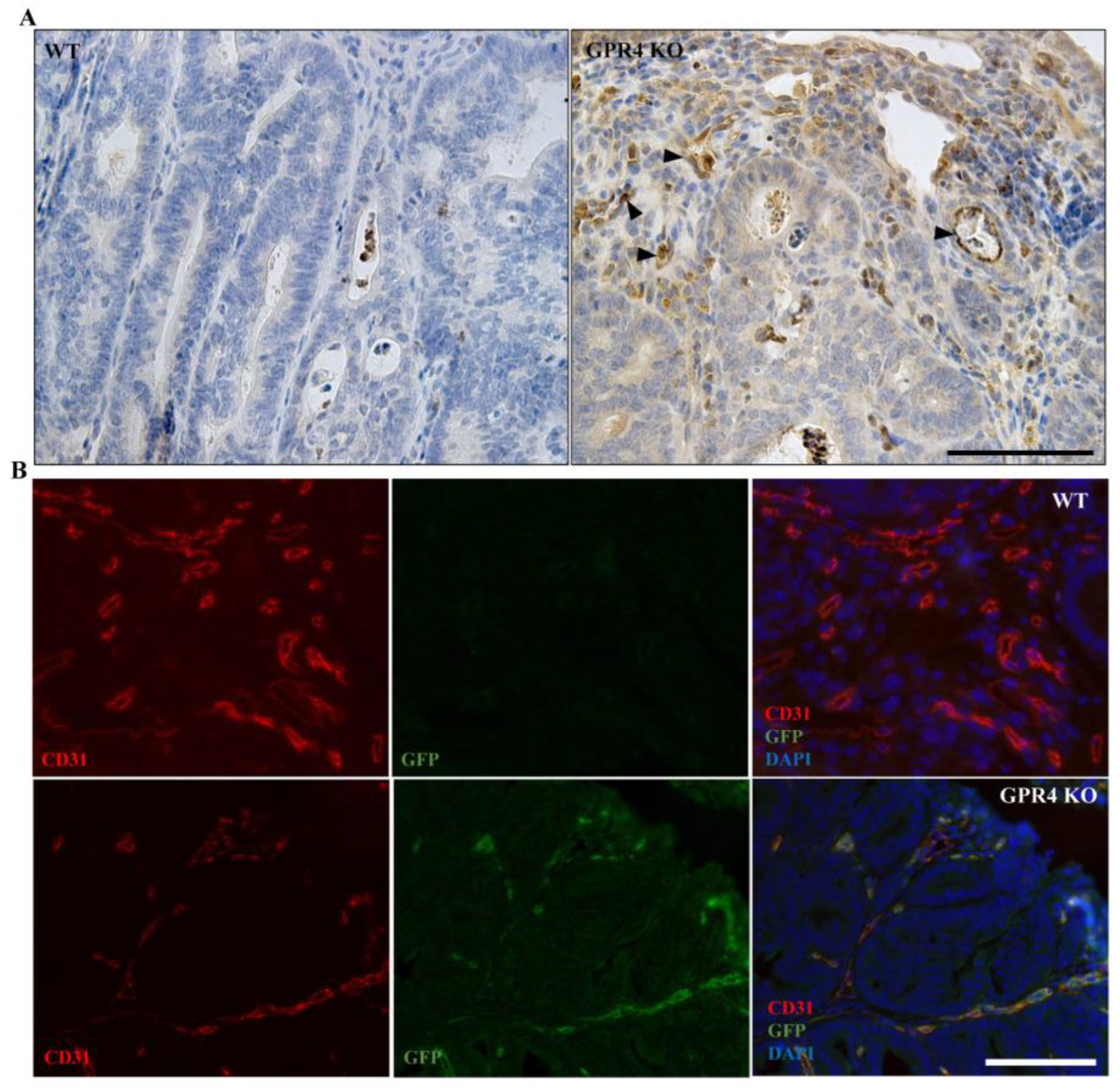
3.6. GPR4 Is Highly Expressed in the Tumor Blood Vessels of AOM/DSS Mice
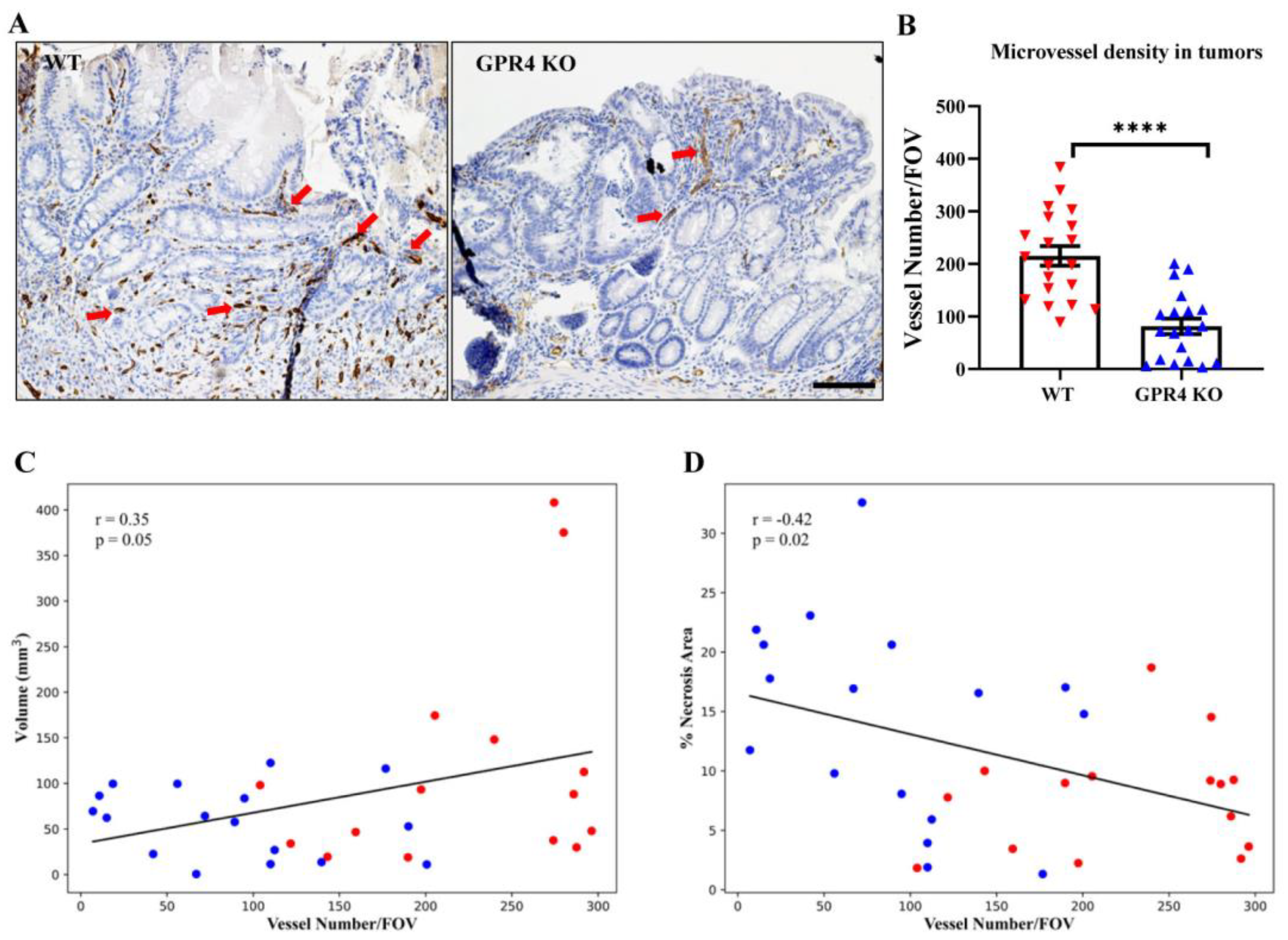
3.7. GPR4 Deletion Decreases Angiogenic Blood Vessel Formation in the Tumors of AOM/DSS Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Porter, R.J.; Arends, M.J.; Churchhouse, A.M.D.; Din, S. Inflammatory Bowel Disease-Associated Colorectal Cancer: Translational Risks from Mechanisms to Medicines. J. Crohn’s Colitis 2021, 15, 2131–2141. [Google Scholar] [CrossRef]
- Maryńczak, K.; Włodarczyk, J.; Sabatowska, Z.; Dziki, A.; Dziki, Ł.; Włodarczyk, M. Colitis-Associated Colorectal Cancer in Patients with Inflammatory Bowel Diseases in a Tertiary Referral Center: A Propensity Score Matching Analysis. J. Clin. Med. 2022, 11, 866. [Google Scholar] [CrossRef] [PubMed]
- Fallingborg, J.; Christensen, L.A.; Jacobsen, B.A.; Rasmussen, S.N. Very low intraluminal colonic pH in patients with active ulcerative colitis. Dig. Dis. Sci. 1993, 38, 1989–1993. [Google Scholar] [CrossRef] [PubMed]
- Nugent, S.G.; Kumar, D.; Rampton, D.S.; Evans, D.F. Intestinal luminal pH in inflammatory bowel disease: Possible determinants and implications for therapy with aminosalicylates and other drugs. Gut 2001, 48, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Colgan, S.P.; Campbell, E.L.; Kominsky, D.J. Hypoxia and Mucosal Inflammation. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 77–100. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef]
- Sanderlin, E.J.; Justus, C.R.; Krewson, E.A.; Yang, L.V. Emerging roles for the pH-sensing G protein-coupled receptors in response to acidotic stress. Cell Health Cytoskelet. 2015, 7, 99–109. [Google Scholar] [CrossRef]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef]
- Jiménez-Vargas, N.N.; Yu, Y.; Jensen, D.D.; Bok, D.D.; Wisdom, M.; Latorre, R.; Lopez, C.; Jaramillo-Polanco, J.O.; Degro, C.; Guzman-Rodriguez, M.; et al. Agonist that activates the µ-opioid receptor in acidified microenvironments inhibits colitis pain without side effects. Gut 2022, 71, 695–704. [Google Scholar] [CrossRef]
- Ludwig, M.G.; Vanek, M.; Guerini, D.; Gasser, J.A.; Jones, C.E.; Junker, U.; Hofstetter, H.; Wolf, R.M.; Seuwen, K. Proton-sensing G-protein-coupled receptors. Nature 2003, 425, 93–98. [Google Scholar] [CrossRef]
- Wang, J.Q.; Kon, J.; Mogi, C.; Tobo, M.; Damirin, A.; Sato, K.; Komachi, M.; Malchinkhuu, E.; Murata, N.; Kimura, T.; et al. TDAG8 is a proton-sensing and psychosine-sensitive G-protein-coupled receptor. J. Biol. Chem. 2004, 279, 45626–45633. [Google Scholar] [CrossRef]
- Ishii, S.; Kihara, Y.; Shimizu, T. Identification of T cell death-associated gene 8 (TDAG8) as a novel acid sensing G-protein-coupled receptor. J. Biol. Chem. 2005, 280, 9083–9087. [Google Scholar] [CrossRef]
- Radu, C.G.; Nijagal, A.; McLaughlin, J.; Wang, L.; Witte, O.N. Differential proton sensitivity of related G protein-coupled receptors T cell death-associated gene 8 and G2A expressed in immune cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1632–1637. [Google Scholar] [CrossRef]
- Nayak, A.P.; Penn, R.B. The proton-sensing receptor ovarian cancer G-protein coupled receptor 1 (OGR1) in airway physiology and disease. Curr. Opin. Pharmacol. 2020, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Saxena, H.; Deshpande, D.A.; Tiegs, B.C.; Yan, H.; Battafarano, R.J.; Burrows, W.M.; Damera, G.; Panettieri, R.A.; Dubose, T.D., Jr.; An, S.S.; et al. The GPCR OGR1 (GPR68) mediates diverse signalling and contraction of airway smooth muscle in response to small reductions in extracellular pH. Br. J. Pharmacol. 2012, 166, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Sanderlin, E.J.; Leffler, N.R.; Lertpiriyapong, K.; Cai, Q.; Hong, H.; Bakthavatchalu, V.; Fox, J.G.; Oswald, J.Z.; Justus, C.R.; Krewson, E.A.; et al. GPR4 deficiency alleviates intestinal inflammation in a mouse model of acute experimental colitis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 569–584. [Google Scholar] [CrossRef]
- Dong, L.; Li, Z.; Leffler, N.R.; Asch, A.S.; Chi, J.T.; Yang, L.V. Acidosis Activation of the Proton-Sensing GPR4 Receptor Stimulates Vascular Endothelial Cell Inflammatory Responses Revealed by Transcriptome Analysis. PLoS ONE 2013, 8, e61991. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; de Valliere, C.; Imenez Silva, P.H.; Leonardi, I.; Gruber, S.; Gerstgrasser, A.; Melhem, H.; Weber, A.; Leucht, K.; Wolfram, L.; et al. The Proton-activated Receptor GPR4 Modulates Intestinal Inflammation. J. Crohn’s Colitis 2018, 12, 355–368. [Google Scholar] [CrossRef]
- Yang, L.V.; Radu, C.G.; Roy, M.; Lee, S.; McLaughlin, J.; Teitell, M.A.; Iruela-Arispe, M.L.; Witte, O.N. Vascular abnormalities in mice deficient for the G protein-coupled receptor GPR4 that functions as a pH sensor. Mol. Cell. Biol. 2007, 27, 1334–1347. [Google Scholar] [CrossRef]
- Weder, B.; Schefer, F.; van Haaften, W.T.; Patsenker, E.; Stickel, F.; Mueller, S.; Hutter, S.; Schuler, C.; Baebler, K.; Wang, Y.; et al. New Therapeutic Approach for Intestinal Fibrosis Through Inhibition of pH-Sensing Receptor GPR4. Inflamm. Bowel Dis. 2022, 28, 109–125. [Google Scholar] [CrossRef]
- Kumar, N.N.; Velic, A.; Soliz, J.; Shi, Y.; Li, K.; Wang, S.; Weaver, J.L.; Sen, J.; Abbott, S.B.G.; Lazarenko, R.M.; et al. Regulation of breathing by CO2 requires the proton-activated receptor GPR4 in retrotrapezoid nucleus neurons. Science 2015, 348, 1255–1260. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Y.; Chen, B. Antagonism of GPR4 with NE 52-QQ57 and the Suppression of AGE-Induced Degradation of Type II Collagen in Human Chondrocytes. Chem. Res. Toxicol. 2020, 33, 1915–1921. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Dong, L.; Leffler, N.R.; Asch, A.S.; Witte, O.N.; Yang, L.V. Activation of GPR4 by acidosis increases endothelial cell adhesion through the cAMP/Epac pathway. PLoS ONE 2011, 6, e27586. [Google Scholar] [CrossRef]
- Gordon, I.O.; Agrawal, N.; Willis, E.; Goldblum, J.R.; Lopez, R.; Allende, D.; Liu, X.; Patil, D.Y.; Yerian, L.; El-Khider, F.; et al. Fibrosis in ulcerative colitis is directly linked to severity and chronicity of mucosal inflammation. Aliment. Pharmacol. Ther. 2018, 47, 922–939. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Fiocchi, C. Intestinal fibrosis in inflammatory bowel disease—Current knowledge and future perspectives. J. Crohn’s Colitis 2008, 2, 279–290. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, Y.; Cai, H.; Ma, H.; Zhao, D.; Zhang, X.; Li, Z.; Wang, S.; Wang, J.; Liu, R.; et al. Human GPR4 and the Notch signaling pathway in endothelial cell tube formation. Mol. Med. Rep. 2016, 14, 1235–1240. [Google Scholar] [CrossRef]
- Jing, Z.; Xu, H.; Chen, X.; Zhong, Q.; Huang, J.; Zhang, Y.; Guo, W.; Yang, Z.; Ding, S.; Chen, P.; et al. The Proton-Sensing G-Protein Coupled Receptor GPR4 Promotes Angiogenesis in Head and Neck Cancer. PLoS ONE 2016, 11, e0152789. [Google Scholar] [CrossRef] [PubMed]
- Wyder, L.; Suply, T.; Ricoux, B.; Billy, E.; Schnell, C.; Baumgarten, B.U.; Maira, S.M.; Koelbing, C.; Ferretti, M.; Kinzel, B.; et al. Reduced pathological angiogenesis and tumor growth in mice lacking GPR4, a proton sensing receptor. Angiogenesis 2011, 14, 533–544. [Google Scholar] [CrossRef]
- Xue, C.; Shao, S.; Yan, Y.; Yang, S.; Bai, S.; Wu, Y.; Zhang, J.; Liu, R.; Ma, H.; Chai, L.; et al. Association between G-protein coupled receptor 4 expression and microvessel density, clinicopathological characteristics and survival in hepatocellular carcinoma. Oncol. Lett. 2020, 19, 2609–2620. [Google Scholar] [CrossRef]
- Yu, M.; Cui, R.; Huang, Y.; Luo, Y.; Qin, S.; Zhong, M. Increased proton-sensing receptor GPR4 signalling promotes colorectal cancer progression by activating the hippo pathway. EBioMedicine 2019, 48, 264–276. [Google Scholar] [CrossRef]
- Krause, P.; Zahner, S.P.; Kim, G.; Shaikh, R.B.; Steinberg, M.W.; Kronenberg, M. The tumor necrosis factor family member TNFSF14 (LIGHT) is required for resolution of intestinal inflammation in mice. Gastroenterology 2014, 146, 1752–1762.e4. [Google Scholar] [CrossRef] [PubMed]
- Sanderlin, E.J.; Marie, M.; Velcicky, J.; Loetscher, P.; Yang, L.V. Pharmacological inhibition of GPR4 remediates intestinal inflammation in a mouse colitis model. Eur. J. Pharmacol. 2019, 852, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Marie, M.A.; Sanderlin, E.J.; Satturwar, S.; Hong, H.; Lertpiriyapong, K.; Donthi, D.; Yang, L.V. GPR65 (TDAG8) inhibits intestinal inflammation and colitis-associated colorectal cancer development in experimental mouse models. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2022, 1868, 166288. [Google Scholar] [CrossRef] [PubMed]
- Clapper, M.L.; Cooper, H.S.; Chang, W.C. Dextran sulfate sodium-induced colitis-associated neoplasia: A promising model for the development of chemopreventive interventions. Acta Pharmacol. Sin. 2007, 28, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Thaker, A.I.; Shaker, A.; Rao, M.S.; Ciorba, M.A. Modeling colitis-associated cancer with azoxymethane (AOM) and dextran sulfate sodium (DSS). J. Vis. Exp. 2012, 67, 4100. [Google Scholar] [CrossRef]
- Ding, S.; Walton, K.L.; Blue, R.E.; McNaughton, K.; Magness, S.T.; Lund, P.K. Mucosal healing and fibrosis after acute or chronic inflammation in wild type FVB-N mice and C57BL6 procollagen alpha1(I)-promoter-GFP reporter mice. PLoS ONE 2012, 7, e42568. [Google Scholar] [CrossRef]
- Crowe, A.R.; Yue, W. Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio Protoc. 2019, 9, e3465. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.; Malleshappa, M.; Vijay-Kumar, M. Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef]
- Kim, J.J.; Shajib, M.S.; Manocha, M.M.; Khan, W.I. Investigating intestinal inflammation in DSS-induced model of IBD. J. Vis. Exp. 2012, 60, 3678. [Google Scholar] [CrossRef]
- Rosenstiel, P.; Fantini, M.; Bräutigam, K.; Kühbacher, T.; Waetzig, G.H.; Seegert, D.; Schreiber, S. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology 2003, 124, 1001–1009. [Google Scholar] [CrossRef]
- Parang, B.; Barrett, C.W.; Williams, C.S. AOM/DSS Model of Colitis-Associated Cancer. In Gastrointestinal Physiology and Diseases. Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; Volume 1422, pp. 297–307. [Google Scholar] [CrossRef]
- De Robertis, M.; Massi, E.; Poeta, M.L.; Carotti, S.; Morini, S.; Cecchetelli, L.; Signori, E.; Fazio, V.M. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J. Carcinog. 2011, 10, 9. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Al-Ostoot, F.H.; Salah, S.; Khamees, H.A.; Khanum, S.A. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat. Res. Commun. 2021, 28, 100422. [Google Scholar] [CrossRef] [PubMed]
- Krewson, E.A.; Sanderlin, E.J.; Marie, M.A.; Akhtar, S.N.; Velcicky, J.; Loetscher, P.; Yang, L.V. The Proton-Sensing GPR4 Receptor Regulates Paracellular Gap Formation and Permeability of Vascular Endothelial Cells. iScience 2020, 23, 100848. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.E.; Akther, M.; Azam, S.; Choi, D.K.; Kim, I.S. GPR4 Knockout Improves the Neurotoxin-Induced, Caspase-Dependent Mitochondrial Apoptosis of the Dopaminergic Neuronal Cell. Int. J. Mol. Sci. 2020, 21, 7517. [Google Scholar] [CrossRef]
- Addante, A.; Wunder, F.; Schroeder, S.; Dietz, L.; Brechmann, M.; Koch, M.; Borissoff, J.; Lizé, M. Preclinical efficacy of GPR4 antagonist in a short-term mouse emphysema-exacerbation model. ERJ Open Res. 2019, 5, PP221. [Google Scholar] [CrossRef]
- Yang, L.V.; Oppelt, K.A.; Thomassen, M.J.; Marie, M.A.; Nik Akhtar, S.; McCallen, J.D. Can GPR4 Be a Potential Therapeutic Target for COVID-19? Front. Med. 2021, 7, 626796. [Google Scholar] [CrossRef]
- Ouyang, S.; Li, Y.; Wu, X.; Wang, Y.; Liu, F.; Zhang, J.; Qiu, Y.; Zhou, Z.; Wang, Z.; Xia, W.; et al. GPR4 signaling is essential for the promotion of acid-mediated angiogenic capacity of endothelial progenitor cells by activating STAT3/VEGFA pathway in patients with coronary artery disease. Stem Cell Res. Ther. 2021, 12, 149. [Google Scholar] [CrossRef]
- Li, R.; Guan, Z.; Bi, S.; Wang, F.; He, L.; Niu, X.; You, Y.; Liu, Y.; Ding, Y.; Siwko, S.; et al. The proton-activated G protein-coupled receptor GPR4 regulates the development of osteoarthritis via modulating CXCL12/CXCR7 signaling. Cell Death Dis. 2022, 13, 152. [Google Scholar] [CrossRef]
- Dong, B.; Zhang, X.; Fan, Y.; Cao, S. GPR4 knockout improves renal ischemia-reperfusion injury and inhibits apoptosis via suppressing the expression of CHOP. Biochem. J. 2017, 474, 4065–4074. [Google Scholar] [CrossRef]
- van Zwieten, R.; Wever, R.; Hamers, M.N.; Weening, R.S.; Roos, D. Extracellular proton release by stimulated neutrophils. J. Clin. Investig. 1981, 68, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.P.; Nakakura, T.; Tomura, H.; Tobo, M.; Mogi, C.; Wang, J.Q.; He, X.D.; Takano, M.; Damirin, A.; Komachi, M.; et al. Each one of certain histidine residues in G-protein-coupled receptor GPR4 is critical for extracellular proton-induced stimulation of multiple G-protein-signaling pathways. Pharmacol. Res. 2010, 61, 499–505. [Google Scholar] [CrossRef]
- Tobo, A.; Tobo, M.; Nakakura, T.; Ebara, M.; Tomura, H.; Mogi, C.; Im, D.S.; Murata, N.; Kuwabara, A.; Ito, S.; et al. Characterization of Imidazopyridine Compounds as Negative Allosteric Modulators of Proton-Sensing GPR4 in Extracellular Acidification-Induced Responses. PLoS ONE 2015, 10, e0129334. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ma, L.; Dai, L.; Zuo, D.; Li, X.; Zhu, H.; Xu, F. TNF-α promotes the malignant transformation of intestinal stem cells through the NF-κB and Wnt/β-catenin signaling pathways. Oncol. Rep. 2020, 44, 577–588. [Google Scholar] [CrossRef]
- Li, C.-W.; Xia, W.; Huo, L.; Lim, S.-O.; Wu, Y.; Hsu, J.L.; Chao, C.-H.; Yamaguchi, H.; Yang, N.-K.; Ding, Q.; et al. Epithelial–Mesenchymal Transition Induced by TNF-α Requires NF-κB–Mediated Transcriptional Upregulation of Twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Hong, K.S.; Chung, J.W.; Kim, J.H.; Hahm, K.B. Prevention of Colitis-Associated Carcinogenesis with Infliximab. Cancer Prev. Res. 2010, 3, 1314–1333. [Google Scholar] [CrossRef]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar] [CrossRef]
- Fukuda, H.; Ito, S.; Watari, K.; Mogi, C.; Arisawa, M.; Okajima, F.; Kurose, H.; Shuto, S. Identification of a Potent and Selective GPR4 Antagonist as a Drug Lead for the Treatment of Myocardial Infarction. ACS Med. Chem. Lett. 2016, 7, 493–497. [Google Scholar] [CrossRef]
- Spisni, E.; Valerii, M.C.; De Fazio, L.; Cavazza, E.; Borsetti, F.; Sgromo, A.; Candela, M.; Centanni, M.; Rizello, F.; Strillacci, A. Cyclooxygenase-2 silencing for the treatment of colitis: A combined in vivo strategy based on RNA interference and engineered Escherichia coli. Mol. Ther. 2015, 23, 278–289. [Google Scholar] [CrossRef]
- Ishioka, T.; Kuwabara, N.; Oohashi, Y.; Wakabayashi, K. Induction of colorectal tumors in rats by sulfated polysaccharides. Crit. Rev. Toxicol. 1987, 17, 215–244. [Google Scholar] [CrossRef]
- Ward, J.M.; Yamamoto, R.S.; Brown, C.A. Pathology of intestinal neoplasms and other lesions in rats exposed to azoxymethane. J. Natl. Cancer Inst. 1973, 51, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Miltz, W.; Velcicky, J.; Dawson, J.; Littlewood-Evans, A.; Ludwig, M.G.; Seuwen, K.; Feifel, R.; Oberhauser, B.; Meyer, A.; Gabriel, D.; et al. Design and synthesis of potent and orally active GPR4 antagonists with modulatory effects on nociception, inflammation, and angiogenesis. Bioorg. Med. Chem. 2017, 25, 4512–4525. [Google Scholar] [CrossRef] [PubMed]
- Velcicky, J.; Miltz, W.; Oberhauser, B.; Orain, D.; Vaupel, A.; Weigand, K.; Dawson King, J.; Littlewood-Evans, A.; Nash, M.; Feifel, R.; et al. Development of Selective, Orally Active GPR4 Antagonists with Modulatory Effects on Nociception, Inflammation, and Angiogenesis. J. Med. Chem. 2017, 60, 3672–3683. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marie, M.A.; Sanderlin, E.J.; Hoffman, A.P.; Cashwell, K.D.; Satturwar, S.; Hong, H.; Sun, Y.; Yang, L.V. GPR4 Knockout Attenuates Intestinal Inflammation and Forestalls the Development of Colitis-Associated Colorectal Cancer in Murine Models. Cancers 2023, 15, 4974. https://doi.org/10.3390/cancers15204974
Marie MA, Sanderlin EJ, Hoffman AP, Cashwell KD, Satturwar S, Hong H, Sun Y, Yang LV. GPR4 Knockout Attenuates Intestinal Inflammation and Forestalls the Development of Colitis-Associated Colorectal Cancer in Murine Models. Cancers. 2023; 15(20):4974. https://doi.org/10.3390/cancers15204974
Chicago/Turabian StyleMarie, Mona A., Edward J. Sanderlin, Alexander P. Hoffman, Kylie D. Cashwell, Swati Satturwar, Heng Hong, Ying Sun, and Li V. Yang. 2023. "GPR4 Knockout Attenuates Intestinal Inflammation and Forestalls the Development of Colitis-Associated Colorectal Cancer in Murine Models" Cancers 15, no. 20: 4974. https://doi.org/10.3390/cancers15204974
APA StyleMarie, M. A., Sanderlin, E. J., Hoffman, A. P., Cashwell, K. D., Satturwar, S., Hong, H., Sun, Y., & Yang, L. V. (2023). GPR4 Knockout Attenuates Intestinal Inflammation and Forestalls the Development of Colitis-Associated Colorectal Cancer in Murine Models. Cancers, 15(20), 4974. https://doi.org/10.3390/cancers15204974