

Exploring the Tumor-Suppressing Potential of PSCA in Pancreatic Ductal Adenocarcinoma

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of CM
2.3. Human Peripheral Blood Mononuclear Cells (PBMCs)
2.4. MTT Assay
2.5. Scratch Assay
2.6. Transwell Invasion Assay
2.7. Live-Cell Imaging
2.8. Western Blot Analysis and ELISA
2.9. siRNA Transfection
2.10. Membrane Protein Extraction
2.11. Molecular Docking Analysis
2.12. Immunoprecipitation
2.13. Ex Vivo Tissue Assay
2.14. Evaluation of Tumor-Suppressing Protein Candidates
2.15. Statistical Analysis
3. Results
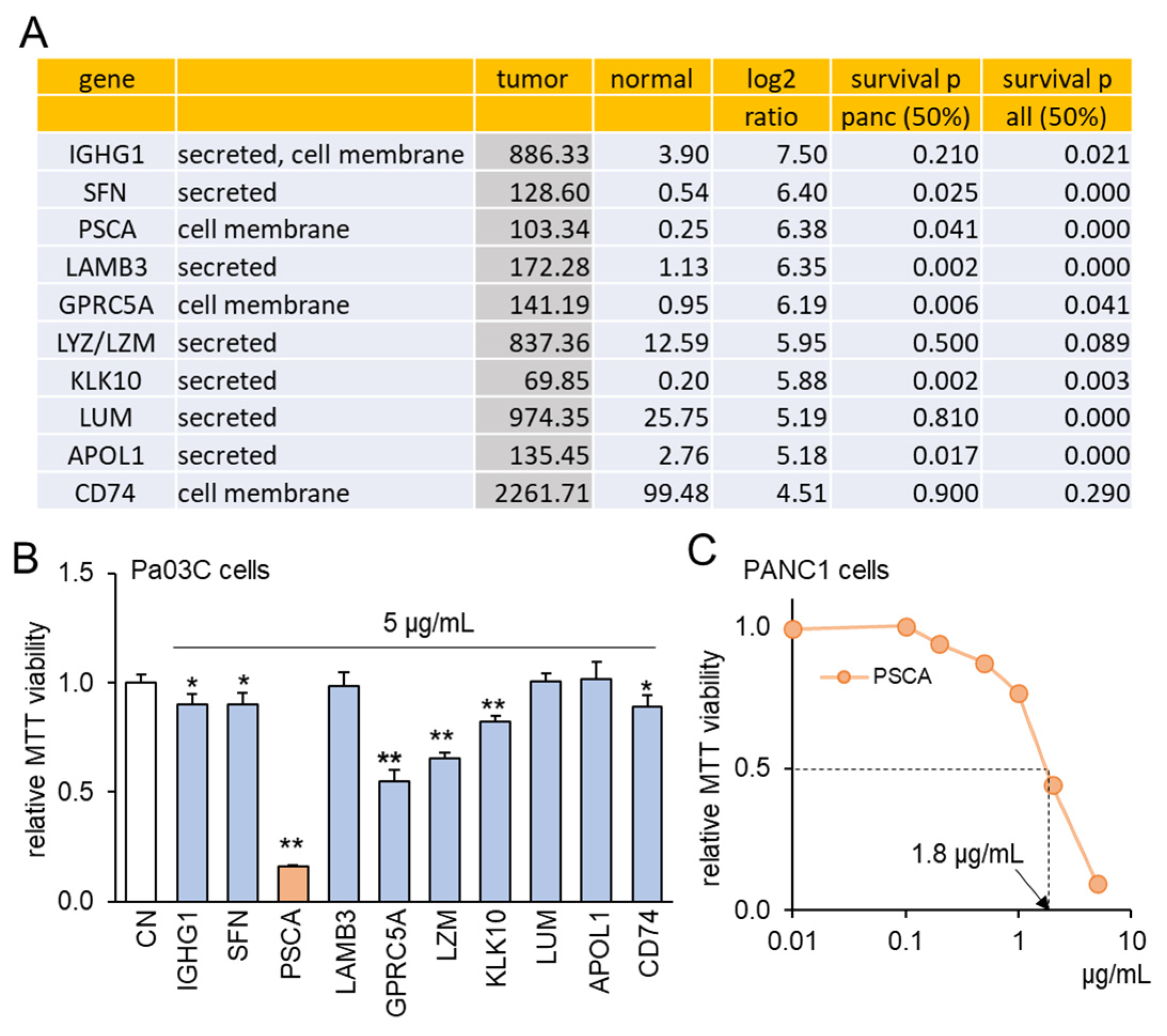
3.1. TCGA Database-Based Prediction of PSCA as a Tumor Suppressor
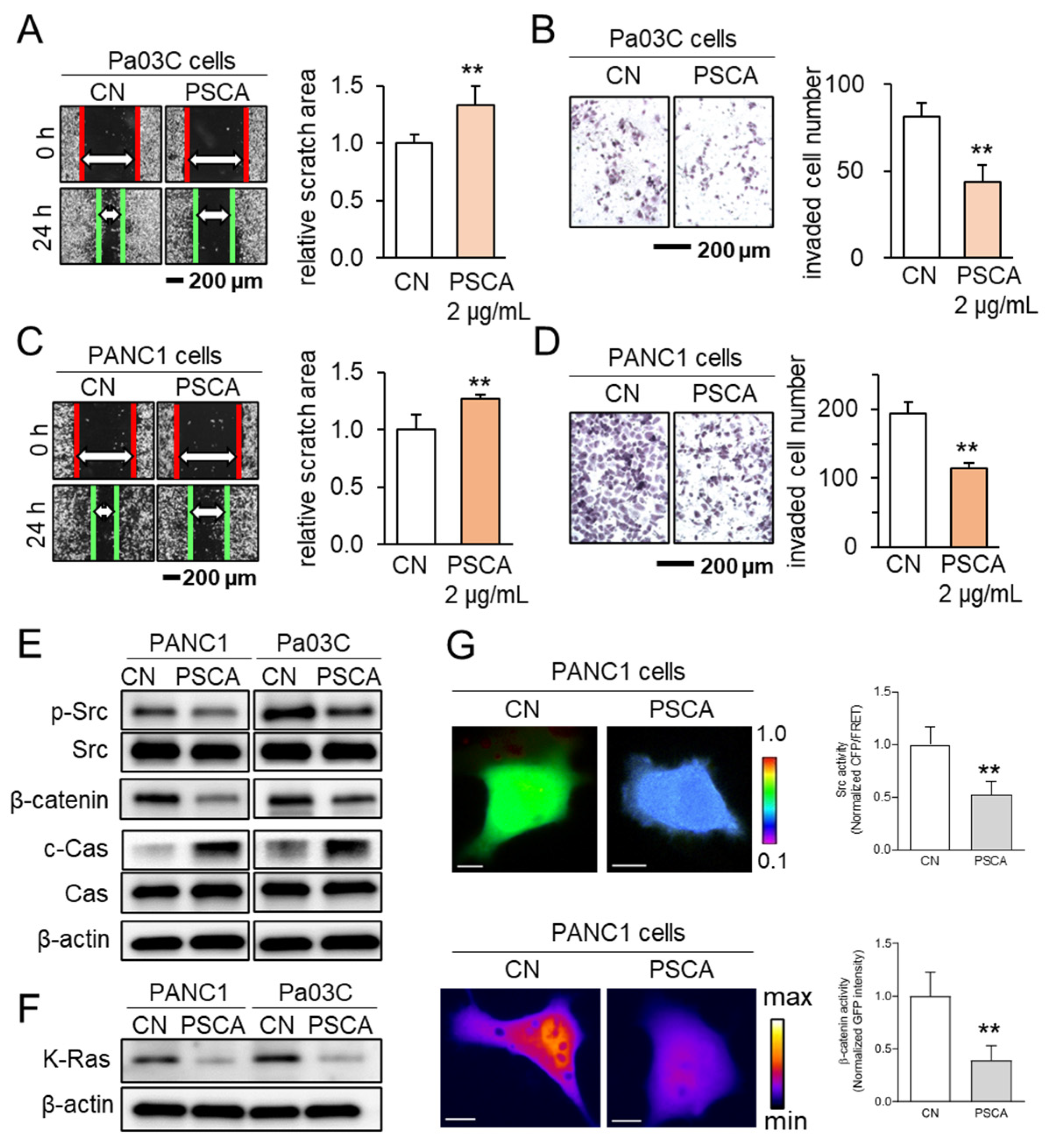
3.2. The Anti-Tumor Effect of PSCA Recombinant Protein
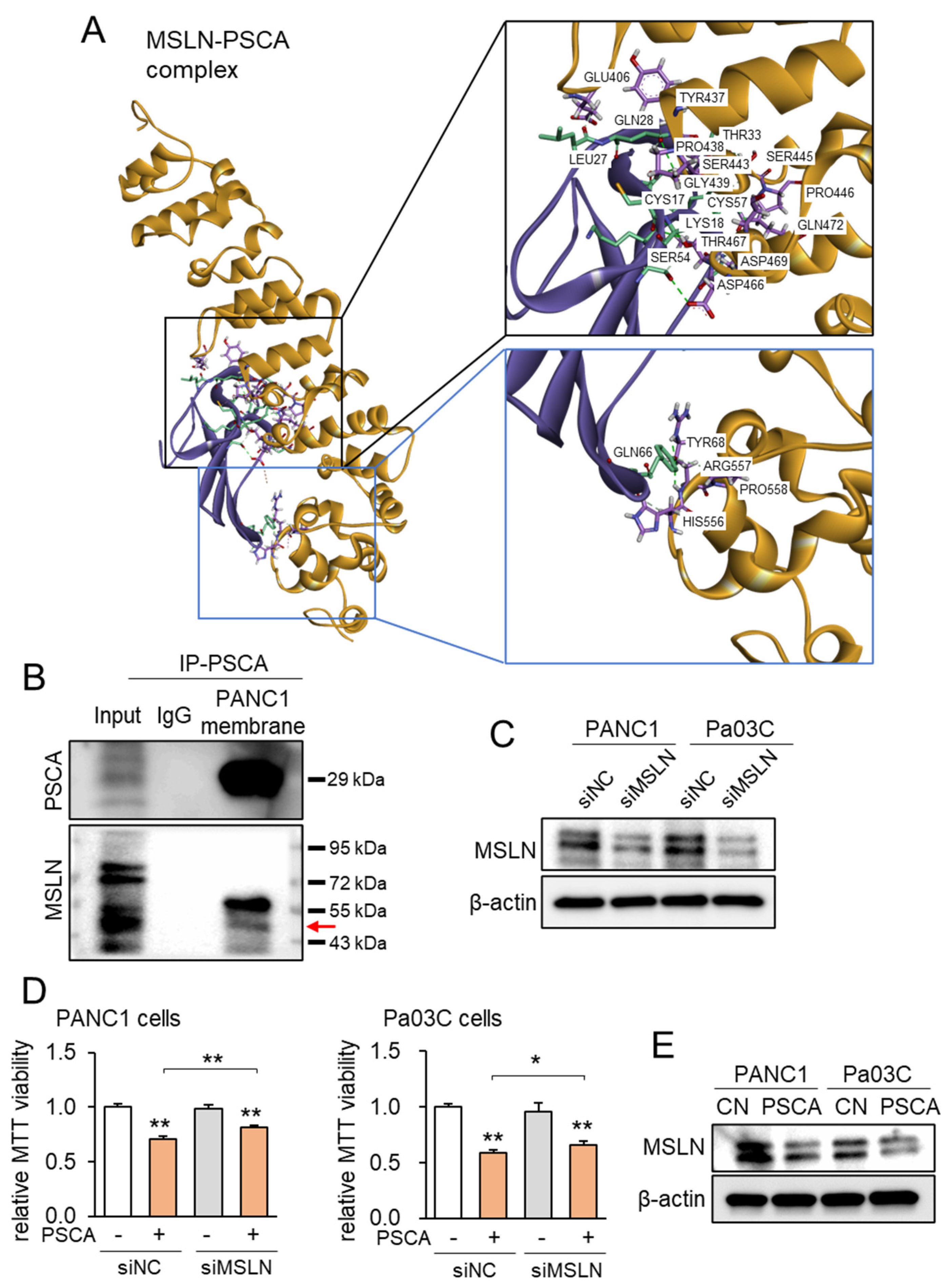
3.3. Linkage of PSCA with Mesothelin (MSLN)
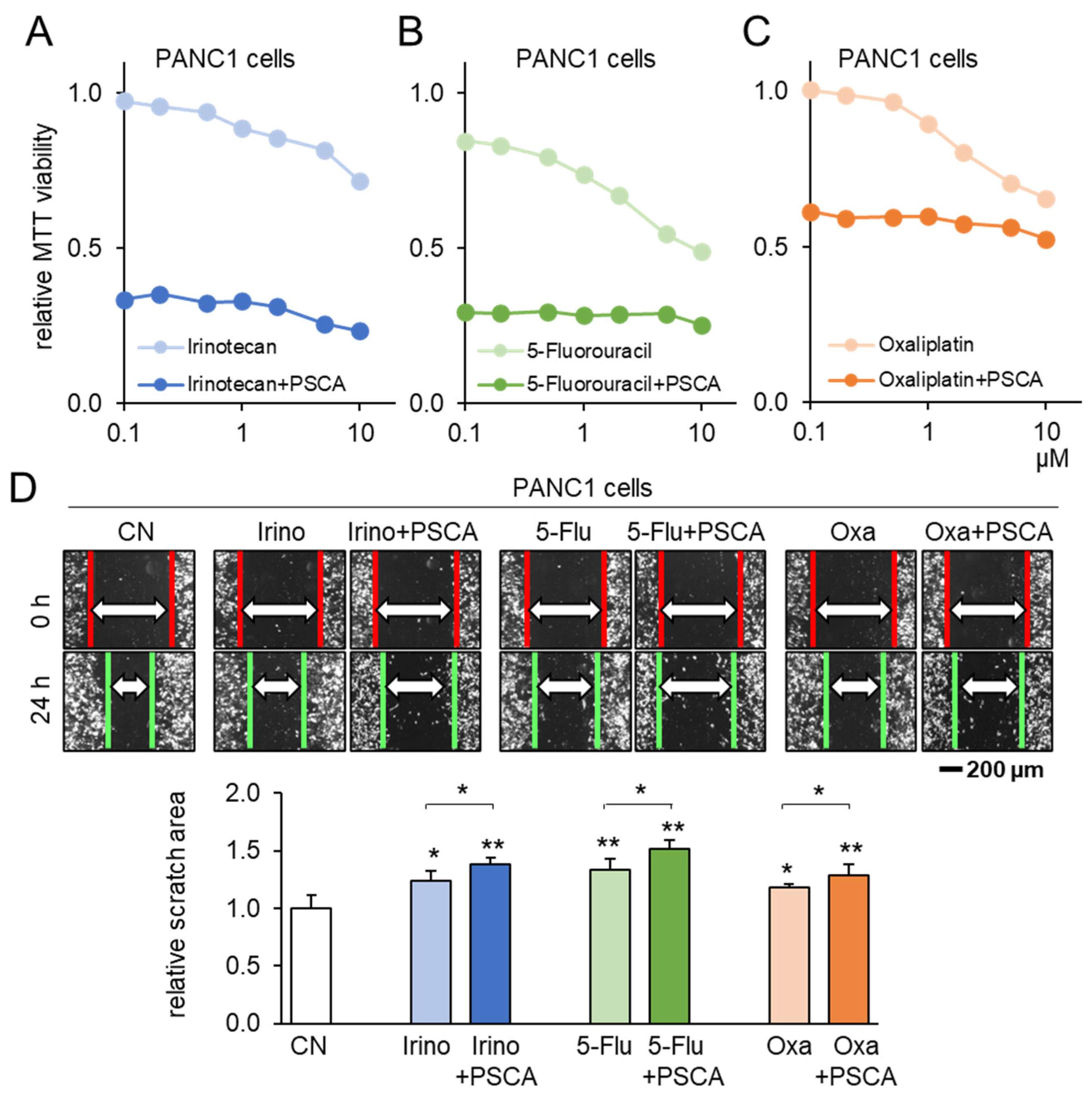
3.4. Additive Anti-Tumor Effects of PSCA with Standard-of-Care Chemotherapy Drugs
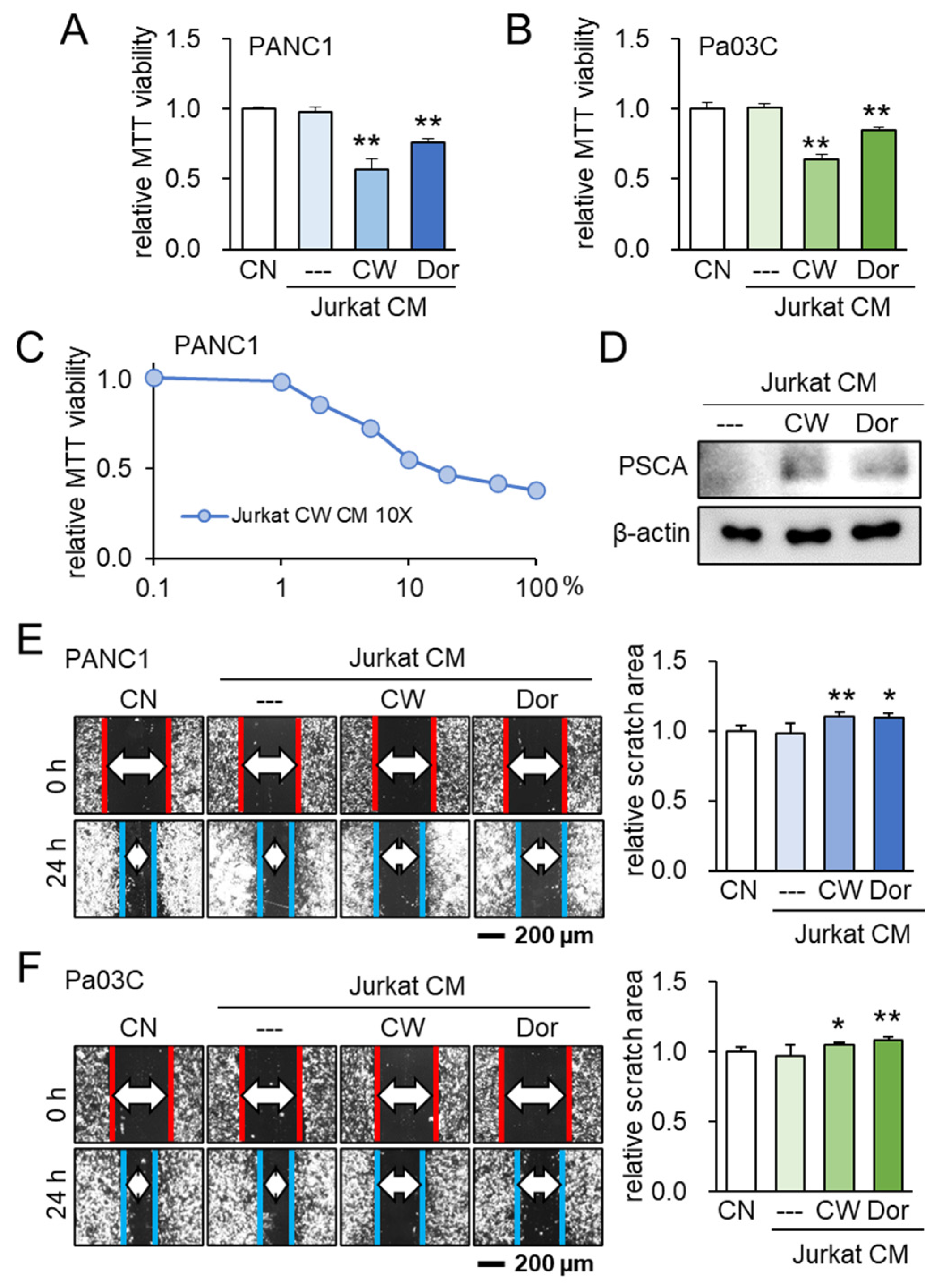
3.5. Lymphocyte-Derived CM and PSCA
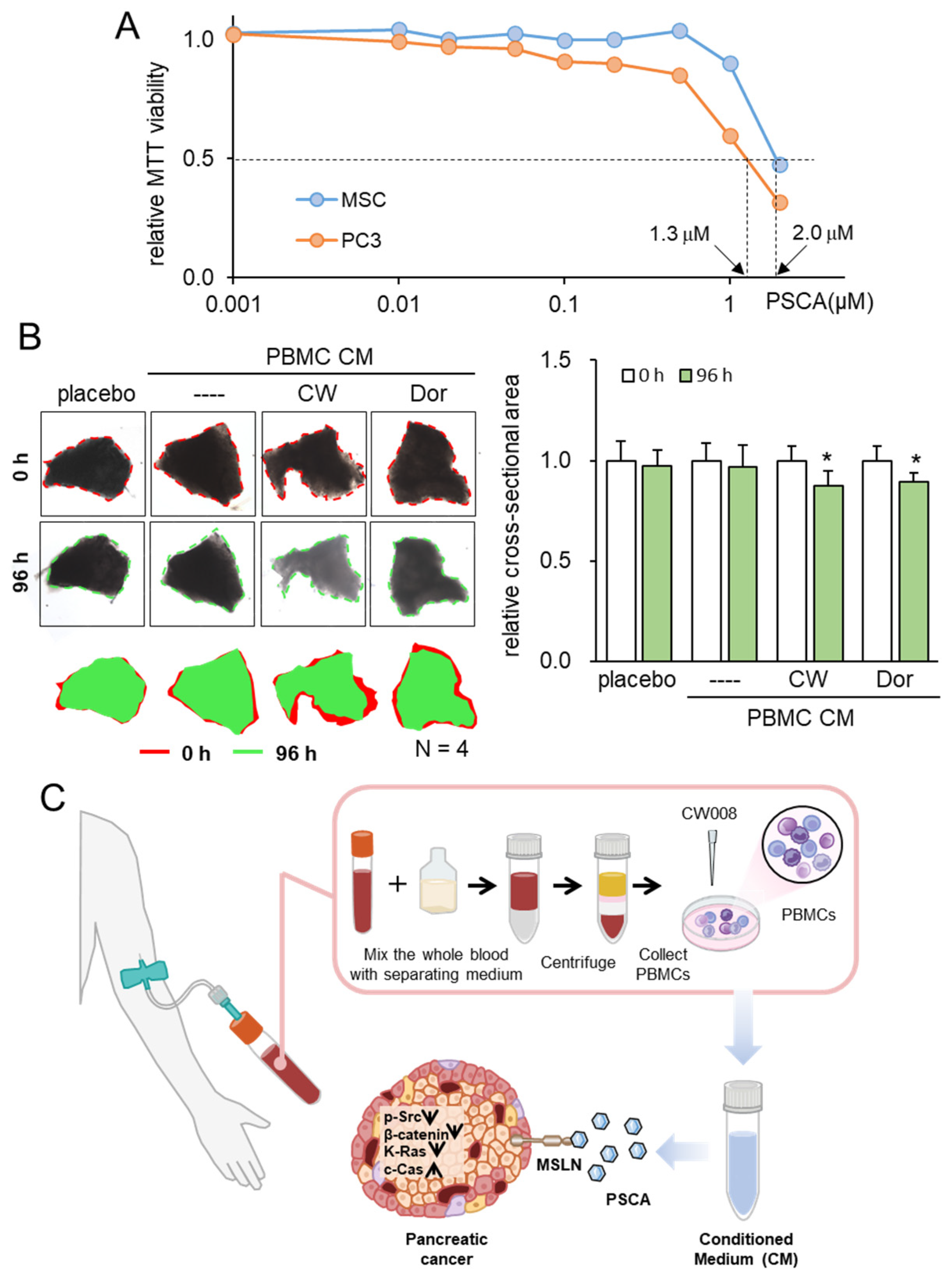
3.6. Generation of Tumor-Suppressive CM from PBMCs
3.7. Tumor Selectivity and Ex Vivo Assay with PDAC Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, S.; Zheng, Y.; Yang, F.; Zhu, L.; Zhu, X.Q.; Wang, Z.F.; Wu, X.L.; Zhou, C.H.; Yan, J.Y.; Hu, B.Y.; et al. The molecular biology of pancreatic adenocarcinoma: Translational challenges and clinical perspectives. Signal Transduct. Target. Ther. 2021, 6, 249. [Google Scholar] [CrossRef]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef]
- Ansari, D.; Gustafsson, A.; Andersson, R. Update on the management of pancreatic cancer: Surgery is not enough. World J. Gastroenterol. 2015, 21, 3157–3165. [Google Scholar] [CrossRef]
- Cho, I.R.; Kang, H.; Jo, J.H.; Lee, H.S.; Chung, M.J.; Park, J.Y.; Park, S.W.; Song, S.Y.; An, C.; Park, M.S.; et al. FOLFIRINOX vs. gemcitabine/nab-paclitaxel for treatment of metastatic pancreatic cancer: Single-center cohort study. World J. Gastrointest. Oncol. 2020, 12, 182–194. [Google Scholar] [CrossRef]
- Nitipir, C.; Vrabie, R.; Parosanu, A.I.; Tulin, R.; Cretu, B.; Cursaru, A.; Slavu, I.; Miron, A.; Calu, V.; Orlov Slavu, M.C. Clinical Impact of the Administration of FOLFIRINOX Beyond Six Months in Advanced Pancreatic Adenocarcinoma: A Cohort Study. Cureus 2021, 13, e19361. [Google Scholar] [CrossRef]
- Sinn, M.; Bahra, M.; Denecke, T.; Travis, S.; Pelzer, U.; Riess, H. Perioperative treatment options in resectable pancreatic cancer—How to improve long-term survival. World J. Gastrointest. Oncol. 2016, 8, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef]
- Liu, X.L.; Ding, J.; Meng, L.H. Oncogene-induced senescence: A double edged sword in cancer. Acta Pharmacol. Sin. 2018, 39, 1553–1558. [Google Scholar] [CrossRef]
- Papapetrou, E.P.; Tomishima, M.J.; Chambers, S.M.; Mica, Y.; Reed, E.; Menon, J.; Tabar, V.; Mo, Q.; Studer, L.; Sadelain, M. Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 12759–12764. [Google Scholar] [CrossRef] [PubMed]
- van Schaijik, B.; Davis, P.F.; Wickremesekera, A.C.; Tan, S.T.; Itinteang, T. Subcellular localisation of the stem cell markers OCT4, SOX2, NANOG, KLF4 and c-MYC in cancer: A review. J. Clin. Pathol. 2018, 71, 88–91. [Google Scholar] [CrossRef]
- Li, K.; Huo, Q.; Li, B.Y.; Yokota, H. The Double-Edged Proteins in Cancer Proteomes and the Generation of Induced Tumor-Suppressing Cells (iTSCs). Proteomes 2023, 11, 5. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Sun, X.; Zha, R.; Liu, S.; Feng, Y.; Sano, T.; Aryal, U.K.; Sudo, A.; Li, B.Y.; Yokota, H. Counterintuitive production of tumor-suppressive secretomes from Oct4- and c-Myc-overexpressing tumor cells and MSCs. Theranostics 2022, 12, 3084–3103. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, K.; Zha, R.; Liu, S.; Fan, Y.; Wu, D.; Hase, M.; Aryal, U.K.; Lin, C.C.; Li, B.Y.; et al. Preventing tumor progression to the bone by induced tumor-suppressing MSCs. Theranostics 2021, 11, 5143–5159. [Google Scholar] [CrossRef]
- Li, K.; Sun, X.; Li, H.; Ma, H.; Zhou, M.; Minami, K.; Tamari, K.; Ogawa, K.; Pandya, P.; Saadatzadeh, M.; et al. Suppression of osteosarcoma progression by engineered lymphocyte-derived proteomes. Genes. Diseases 2022, 10, 1641–1656. [Google Scholar] [CrossRef]
- Liu, S.; Sun, X.; Li, K.; Zha, R.; Feng, Y.; Sano, T.; Dong, C.; Liu, Y.; Aryal, U.K.; Sudo, A.; et al. Generation of the tumor-suppressive secretome from tumor cells. Theranostics 2021, 11, 8517–8534. [Google Scholar] [CrossRef]
- Li, K.X.; Sun, X.; Li, B.Y.; Yokota, H. Conversion of Osteoclasts into Bone-Protective, Tumor-Suppressing Cells. Cancers 2021, 13, 5593. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.C.; Colaprico, A.; Olsen, C.; D’Angelo, F.; Bontempi, G.; Ceccarelli, M.; Noushmehr, H. TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Research 2016, 5, 1542. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Saeki, N.; Gu, J.; Yoshida, T.; Wu, X. Prostate stem cell antigen: A Jekyll and Hyde molecule? Clin. Cancer Res. 2010, 16, 3533–3538. [Google Scholar] [CrossRef] [PubMed]
- Link, T.; Kuithan, F.; Ehninger, A.; Kuhlmann, J.D.; Kramer, M.; Werner, A.; Gatzweiler, A.; Richter, B.; Ehninger, G.; Baretton, G.; et al. Exploratory investigation of PSCA-protein expression in primary breast cancer patients reveals a link to HER2/neu overexpression. Oncotarget 2017, 8, 54592–54603. [Google Scholar] [CrossRef]
- Ali, A.I.; Oliver, A.J.; Samiei, T.; Chan, J.D.; Kershaw, M.H.; Slaney, C.Y. Genetic Redirection of T Cells for the Treatment of Pancreatic Cancer. Front. Oncol. 2019, 9, 56. [Google Scholar] [CrossRef]
- Akce, M.; Zaidi, M.Y.; Waller, E.K.; El-Rayes, B.F.; Lesinski, G.B. The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front. Immunol. 2018, 9, 2166. [Google Scholar] [CrossRef]
- Han, Y.; Li, X.; Zhang, Y.; Han, Y.; Chang, F.; Ding, J. Mesenchymal Stem Cells for Regenerative Medicine. Cells 2019, 8, 886. [Google Scholar] [CrossRef]
- Kim, J.M.; Choi, J.S.; Kim, Y.H.; Jin, S.H.; Lim, S.; Jang, H.J.; Kim, K.T.; Ryu, S.H.; Suh, P.G. An activator of the cAMP/PKA/CREB pathway promotes osteogenesis from human mesenchymal stem cells. J. Cell. Physiol. 2013, 228, 617–626. [Google Scholar] [CrossRef]
- Liu, X.; Chhipa, R.R.; Nakano, I.; Dasgupta, B. The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol. Cancer Ther. 2014, 13, 596–605. [Google Scholar] [CrossRef]
- Na, S.; Collin, O.; Chowdhury, F.; Tay, B.; Ouyang, M.; Wang, Y.; Wang, N. Rapid signal transduction in living cells is a unique feature of mechanotransduction. Proc. Natl. Acad. Sci. USA 2008, 105, 6626–6631. [Google Scholar] [CrossRef] [PubMed]
- Murase, S.; Mosser, E.; Schuman, E.M. Depolarization drives beta-Catenin into neuronal spines promoting changes in synaptic structure and function. Neuron 2002, 35, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Takigawa, S.; Frondorf, B.; Liu, S.; Liu, Y.; Li, B.; Sudo, A.; Wallace, J.M.; Yokota, H.; Hamamura, K. Salubrinal improves mechanical properties of the femur in osteogenesis imperfecta mice. J. Pharmacol. Sci. 2016, 132, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [CrossRef]
- Ma, J.; Tang, W.K.; Esser, L.; Pastan, I.; Xia, D. Recognition of mesothelin by the therapeutic antibody MORAb-009: Structural and mechanistic insights. J. Biol. Chem. 2012, 287, 33123–33131. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Gu, Z.; Thomas, G.; Yamashiro, J.; Shintaku, I.P.; Dorey, F.; Raitano, A.; Witte, O.N.; Said, J.W.; Loda, M.; Reiter, R.E. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene 2000, 19, 1288–1296. [Google Scholar] [CrossRef]
- Amara, N.; Palapattu, G.S.; Schrage, M.; Gu, Z.; Thomas, G.V.; Dorey, F.; Said, J.; Reiter, R.E. Prostate stem cell antigen is overexpressed in human transitional cell carcinoma. Cancer Res. 2001, 61, 4660–4665. [Google Scholar]
- Elsamman, E.M.; Fukumori, T.; Tanimoto, S.; Nakanishi, R.; Takahashi, M.; Toida, K.; Kanayama, H.O. The expression of prostate stem cell antigen in human clear cell renal cell carcinoma: A quantitative reverse transcriptase-polymerase chain reaction analysis. BJU Int. 2006, 98, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Rosty, C.; Reiter, R.E.; Wilentz, R.E.; Murugesan, S.R.; Leach, S.D.; Ryu, B.; Skinner, H.G.; Goggins, M.; Jaffee, E.M.; et al. Discovery of new markers of cancer through serial analysis of gene expression: Prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 2001, 61, 4320–4324. [Google Scholar] [PubMed]
- Cao, D.; Ji, H.; Ronnett, B.M. Expression of mesothelin, fascin, and prostate stem cell antigen in primary ovarian mucinous tumors and their utility in differentiating primary ovarian mucinous tumors from metastatic pancreatic mucinous carcinomas in the ovary. Int. J. Gynecol. Pathol. 2005, 24, 67–72. [Google Scholar] [PubMed]
- Patel, U.; Abernathy, J.; Savani, B.N.; Oluwole, O.; Sengsayadeth, S.; Dholaria, B. CAR T cell therapy in solid tumors: A review of current clinical trials. EJHaem 2022, 3, 24–31. [Google Scholar] [CrossRef]
- Abate-Daga, D.; Lagisetty, K.H.; Tran, E.; Zheng, Z.; Gattinoni, L.; Yu, Z.; Burns, W.R.; Miermont, A.M.; Teper, Y.; Rudloff, U.; et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum. Gene Ther. 2014, 25, 1003–1012. [Google Scholar] [CrossRef]
- Kabacaoglu, D.; Ciecielski, K.J.; Ruess, D.A.; Algül, H. Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options. Front. Immunol. 2018, 9, 1878. [Google Scholar] [CrossRef]
- Bandi, D.S.R.; Sarvesh, S.; Farran, B.; Nagaraju, G.P.; El-Rayes, B.F. Targeting the metabolism and immune system in pancreatic ductal adenocarcinoma: Insights and future directions. Cytokine Growth Factor Rev. 2023, 71–72, 26–39. [Google Scholar] [CrossRef]
- Li, K.; Huo, Q.; Dimmitt, N.H.; Qu, G.; Bao, J.; Pandya, P.H.; Saadatzadeh, M.R.; Bijangi-Vishehsaraei, K.; Kacena, M.A.; Pollok, K.E.; et al. Osteosarcoma-enriched transcripts paradoxically generate osteosarcoma-suppressing extracellular proteins. eLife 2023, 12, e83768. [Google Scholar] [CrossRef]
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef]
- Arifka, M.; Wilar, G.; Elamin, K.M.; Wathoni, N. Polymeric Hydrogels as Mesenchymal Stem Cell Secretome Delivery System in Biomedical Applications. Polymers 2022, 14, 1218. [Google Scholar] [CrossRef] [PubMed]
- Naeimi, R.; Bahmani, A.; Afshar, S. Investigating the role of peptides in effective therapies against cancer. Cancer Cell Int. 2022, 22, 139. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Qian, M.; Ho, M. The role of mesothelin in tumor progression and targeted therapy. Anticancer Agents Med. Chem. 2013, 13, 276–280. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.; Huo, Q.; Minami, K.; Tamari, K.; Ogawa, K.; Na, S.; Fishel, M.L.; Li, B.-Y.; Yokota, H. Exploring the Tumor-Suppressing Potential of PSCA in Pancreatic Ductal Adenocarcinoma. Cancers 2023, 15, 4917. https://doi.org/10.3390/cancers15204917
Li K, Huo Q, Minami K, Tamari K, Ogawa K, Na S, Fishel ML, Li B-Y, Yokota H. Exploring the Tumor-Suppressing Potential of PSCA in Pancreatic Ductal Adenocarcinoma. Cancers. 2023; 15(20):4917. https://doi.org/10.3390/cancers15204917
Chicago/Turabian StyleLi, Kexin, Qingji Huo, Kazumasa Minami, Keisuke Tamari, Kazuhiko Ogawa, Sungsoo Na, Melissa L. Fishel, Bai-Yan Li, and Hiroki Yokota. 2023. "Exploring the Tumor-Suppressing Potential of PSCA in Pancreatic Ductal Adenocarcinoma" Cancers 15, no. 20: 4917. https://doi.org/10.3390/cancers15204917
APA StyleLi, K., Huo, Q., Minami, K., Tamari, K., Ogawa, K., Na, S., Fishel, M. L., Li, B.-Y., & Yokota, H. (2023). Exploring the Tumor-Suppressing Potential of PSCA in Pancreatic Ductal Adenocarcinoma. Cancers, 15(20), 4917. https://doi.org/10.3390/cancers15204917