
TGFβ1-Induced EMT in the MCF10A Mammary Epithelial Cell Line Model Is Executed Independently of SNAIL1 and ZEB1 but Relies on JUNB-Coordinated Transcriptional Regulation

 , and
, and 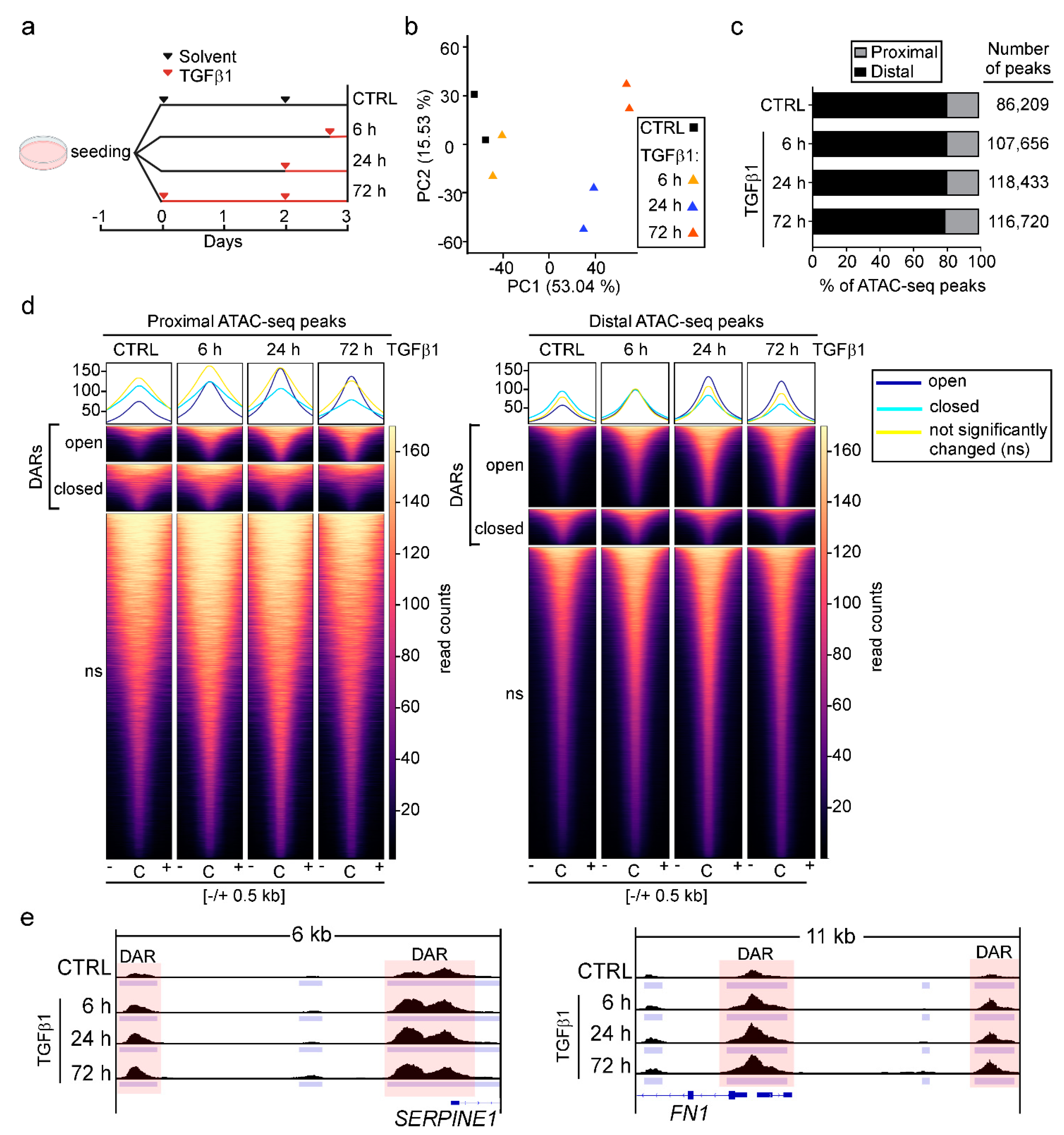{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Oligonucleotides, and Antibodies
2.2. CRISPR/Cas9-Mediated Genome Editing
2.3. Generation of Cell Lines Stably Expressing JUNB-ER Fusion Proteins
2.4. TGFβ1 Treatment of Cells
2.5. Protein Isolation and Western Blotting
2.6. Phase Contrast and Fluorescence Microscopy
2.7. RNA Isolation and Targeted Gene Expression Analysis by qRT-PCR
2.8. Population Dynamics and Cell Cycle Analysis
2.9. Transwell Migration Assays
2.10. Spheroid Invasion Assays
2.11. ChIP-qPCR
2.12. ATAC-seq
2.13. ATAC-seq Data Processing and Analysis
2.14. RNA Sequencing (RNA-seq) Data Processing and Analysis
2.15. Combined Analysis of Time-Resolved RNA-seq and ATAC-seq Data
2.16. Survival Analysis
3. Results
3.1. TGFβ Pathway Activation Increases Chromatin Accessibility Predominantly at Promoter-Distal Candidate DNA Regulatory Elements
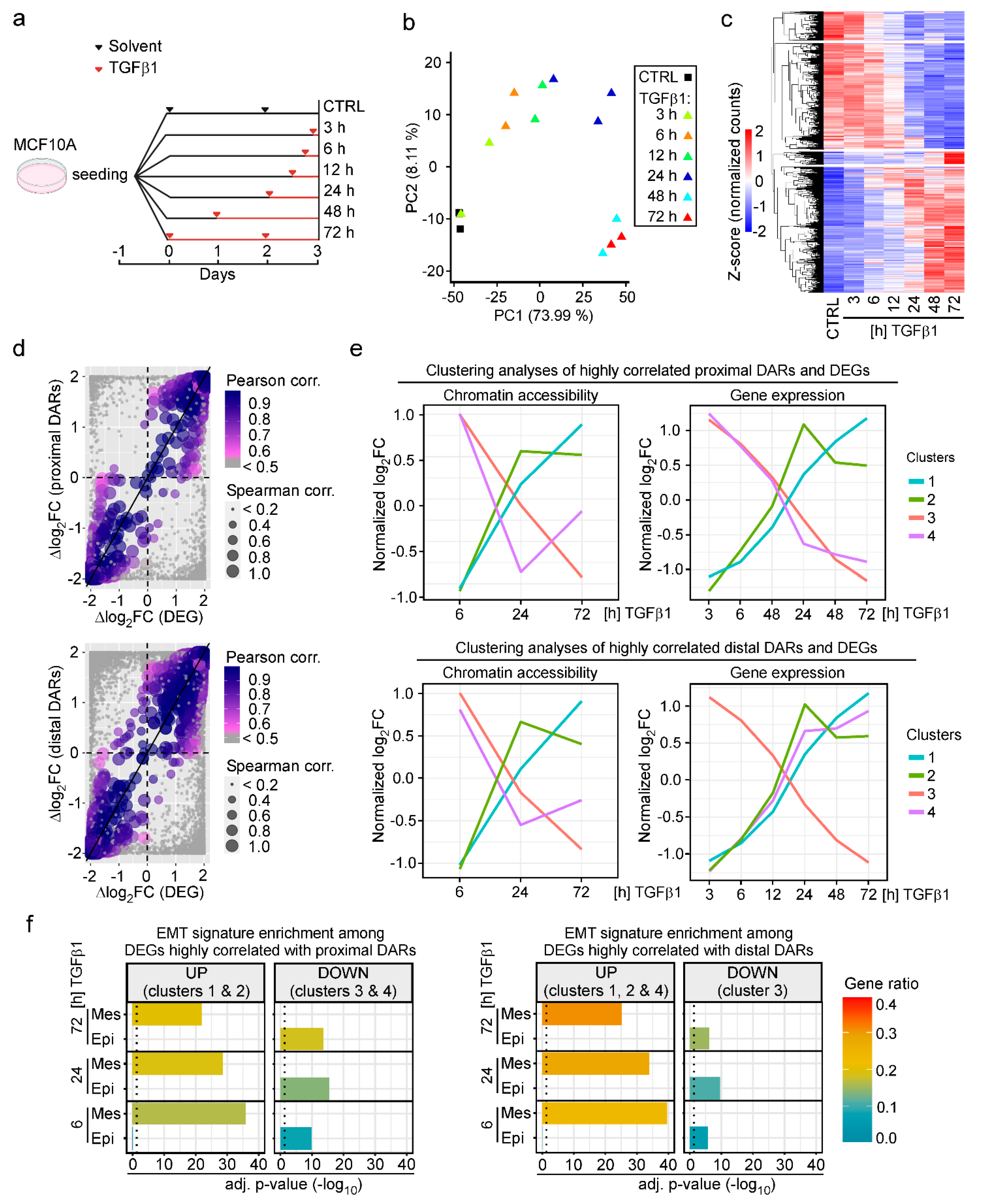
3.2. Changes in Chromatin States Parallel Transcriptional Reprogramming Indicative of TGFβ1-Induced EMT
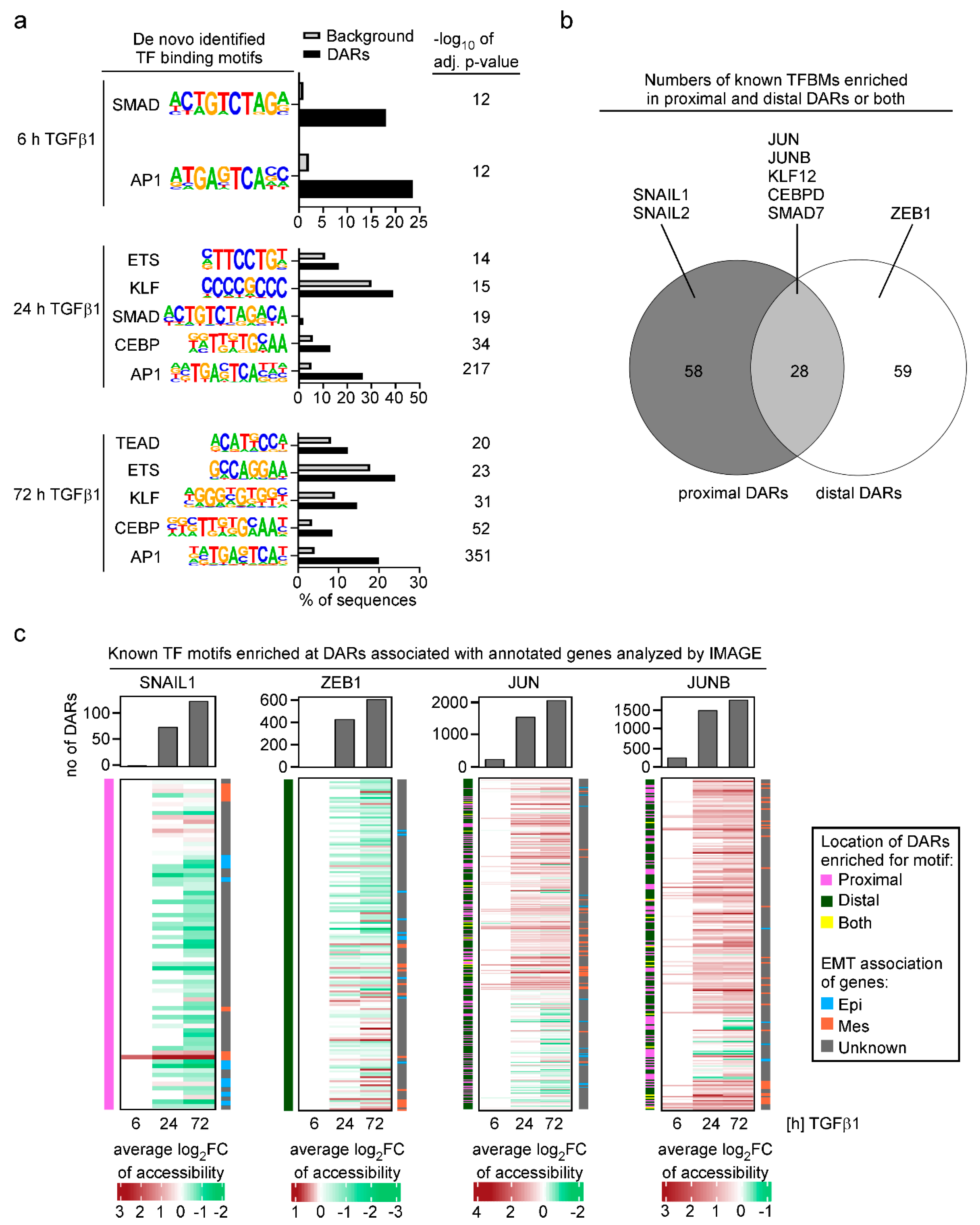
3.3. Analysis of Transcription Factor Motif Activity Predicts AP-1 Subunits JUN and JUNB as Key Regulators of TGFβ1-Induced EMT
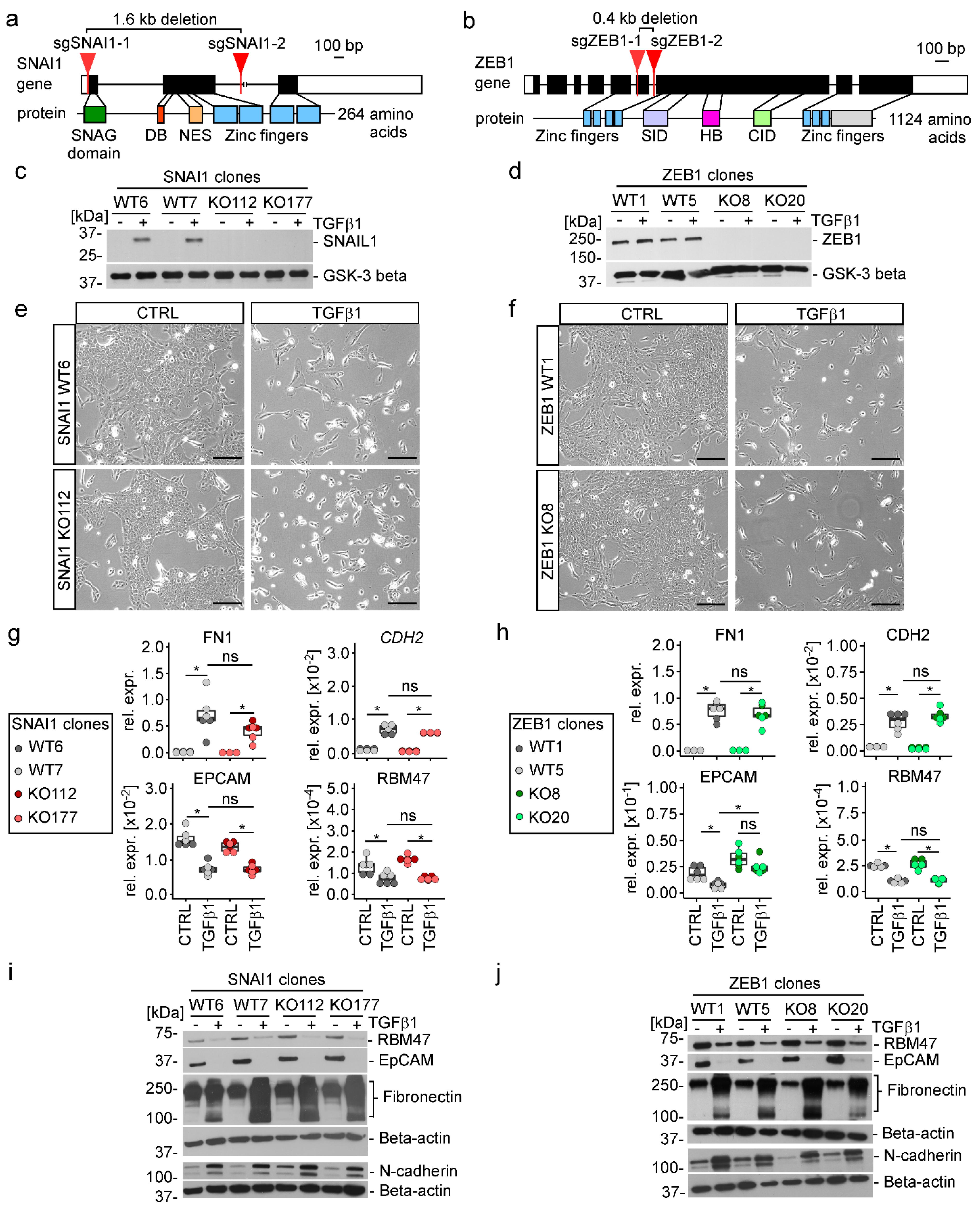
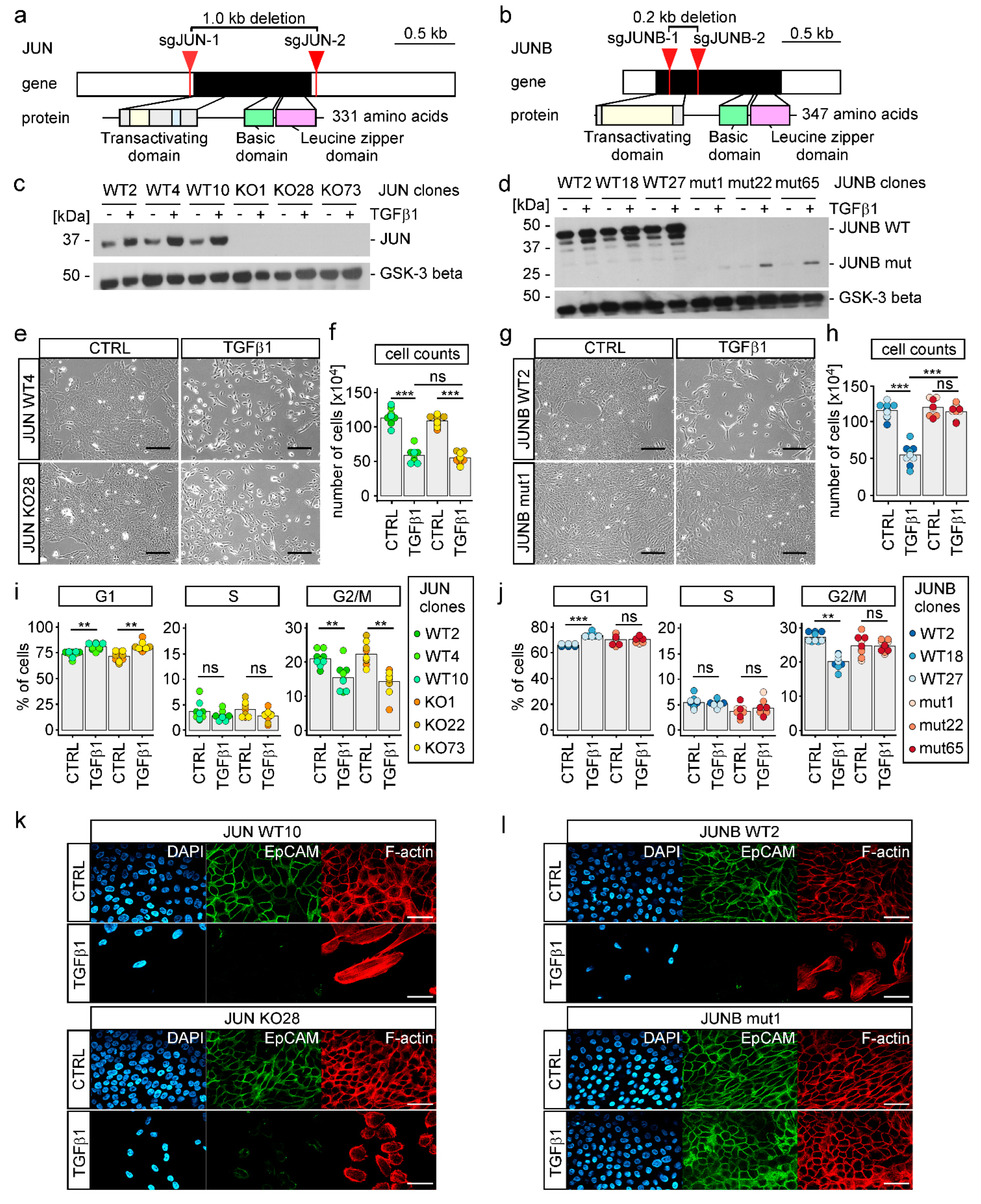
3.4. TGFβ1-Induced EMT Proceeds in the Absence of SNAIL1, SNAIL2, and ZEB1
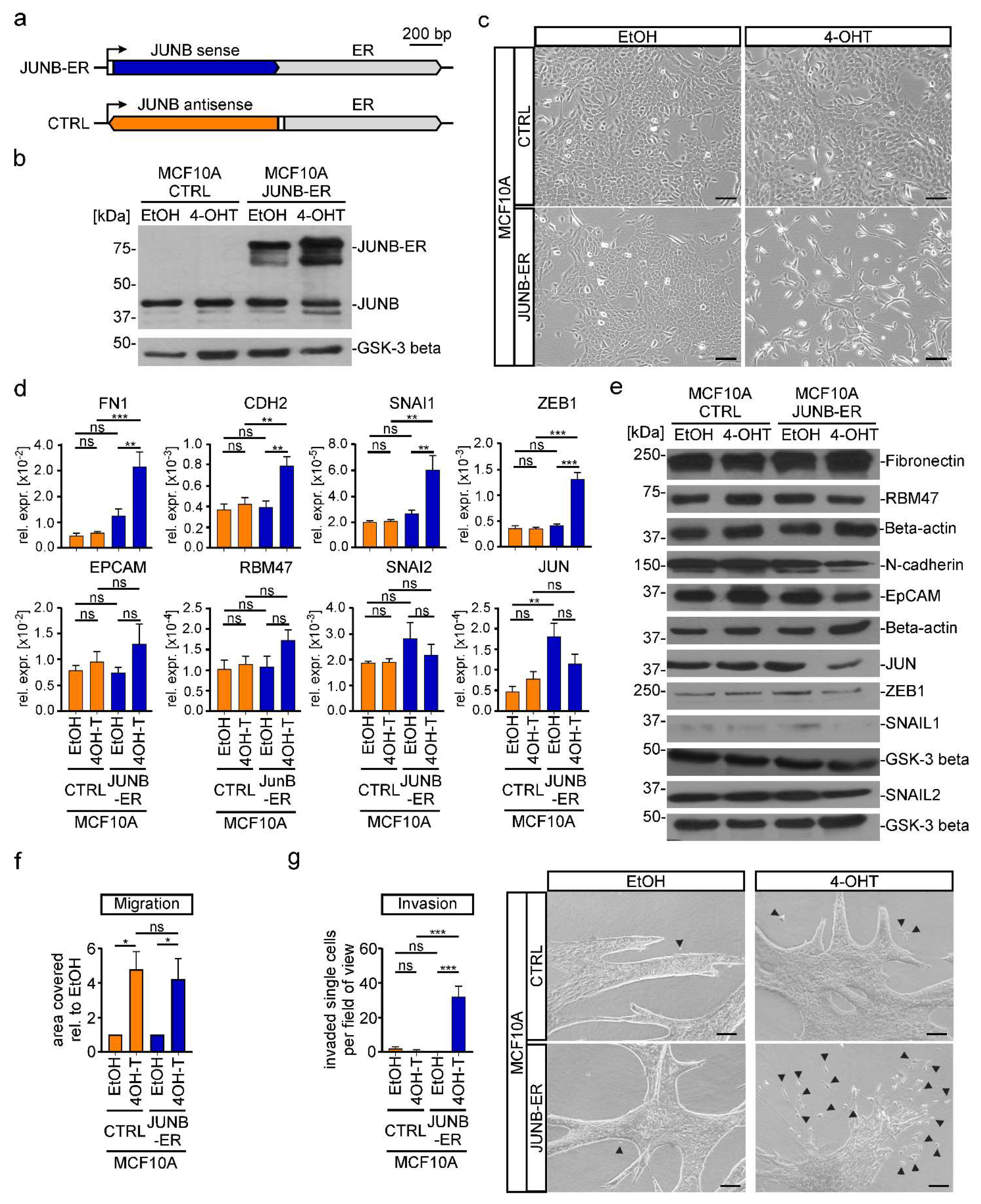
3.5. TGFβ1-Induced EMT Depends on JUNB in MCF10A Cells
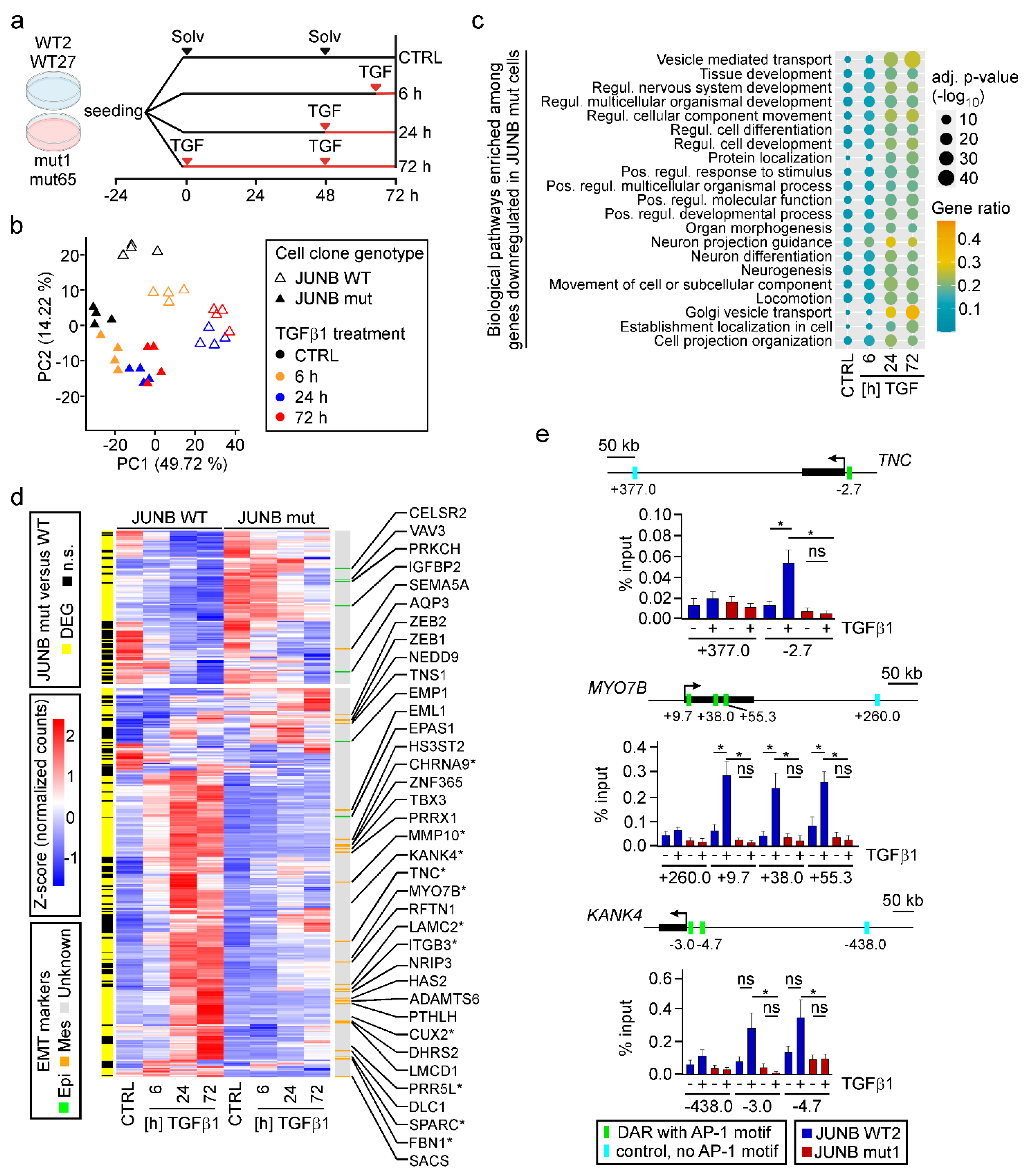
3.6. JUNB Loss-Of-Function Impairs TGFβ1-Mediated Transcriptional Reprogramming
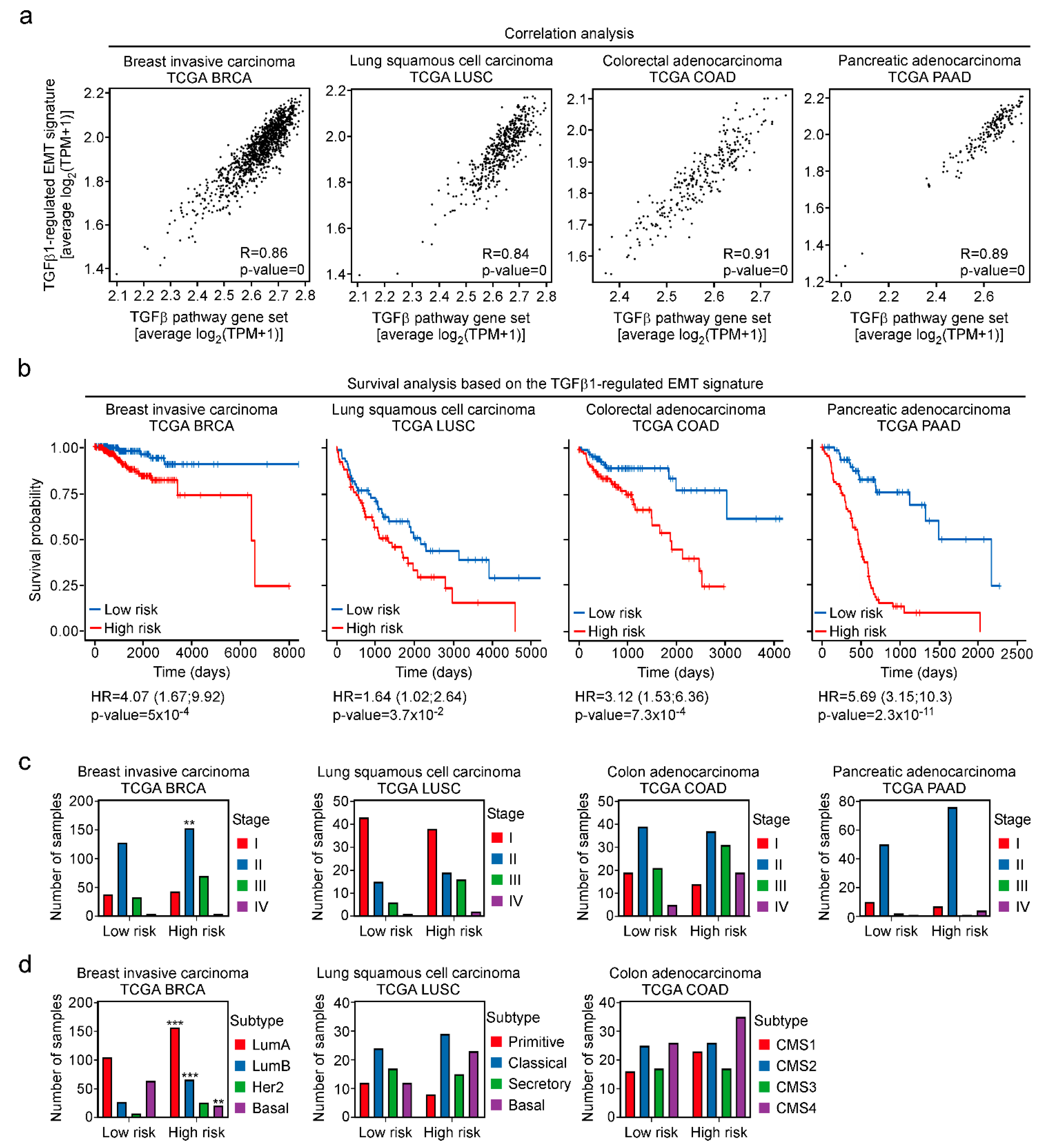
3.7. Definition of a TGFβ-Regulated EMT Signature Predictive of Patient Survival across Cancer Types
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esposito, M.; Ganesan, S.; Kang, Y. Emerging strategies for treating metastasis. Nat. Cancer 2021, 2, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.-J.; Zhang, H.; Teng, Y.; Li, R.; Bai, F.; Elankumaran, S.; Xing, J. TGF-β-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci. Signal. 2014, 7, ra91. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- McFaline-Figueroa, J.L.; Hill, A.J.; Qiu, X.; Jackson, D.; Shendure, J.; Trapnell, C. A pooled single-cell genetic screen identifies regulatory checkpoints in the continuum of the epithelial-to-mesenchymal transition. Nat. Genet. 2019, 51, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.P.; Vanderhyden, B.C. Transcriptional census of epithelial-mesenchymal plasticity in cancer. Sci. Adv. 2022, 8, eabi7640. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef]
- Meyer-Schaller, N.; Cardner, M.; Diepenbruck, M.; Saxena, M.; Tiede, S.; Lüönd, F.; Ivanek, R.; Beerenwinkel, N.; Christofori, G. A Hierarchical Regulatory Landscape during the Multiple Stages of EMT. Dev. Cell 2019, 48, 539–553.e6. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Bolós, V.; Peinado, H.; Pérez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci. 2003, 116, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005, 24, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.P.; Vanderhyden, B.C. Context specificity of the EMT transcriptional response. Nat. Commun. 2020, 11, 2142. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Flum, M.; Dicks, S.; Teng, Y.-H.; Schrempp, M.; Nyström, A.; Boerries, M.; Hecht, A. Canonical TGFβ signaling induces collective invasion in colorectal carcinogenesis through a Snail1- and Zeb1-independent partial EMT. Oncogene 2022, 41, 1492–1506. [Google Scholar] [CrossRef] [PubMed]
- Kröger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-K.; Shiekhattar, R. Architectural and Functional Commonalities between Enhancers and Promoters. Cell 2015, 162, 948–959. [Google Scholar] [CrossRef]
- Grandi, F.C.; Modi, H.; Kampman, L.; Corces, M.R. Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 2022; online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Madsen, J.G.S.; Rauch, A.; van Hauwaert, E.L.; Schmidt, S.F.; Winnefeld, M.; Mandrup, S. Integrated analysis of motif activity and gene expression changes of transcription factors. Genome Res. 2018, 28, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.; Danzer, C.; Rechsteiner, M.; Lehmann, H.; Brandt, L.P.; Hejhal, T.; Catalano, A.; Busenhart, P.; Gonçalves, A.F.; Brandt, S.; et al. A versatile modular vector system for rapid combinatorial mammalian genetics. J. Clin. Investig. 2015, 125, 1603–1619. [Google Scholar] [CrossRef] [PubMed]
- Freihen, V.; Rönsch, K.; Mastroianni, J.; Frey, P.; Rose, K.; Boerries, M.; Zeiser, R.; Busch, H.; Hecht, A. SNAIL1 employs β-Catenin-LEF1 complexes to control colorectal cancer cell invasion and proliferation. Int. J. Cancer 2020, 146, 2229–2242. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Littlewood, T.D.; Hancock, D.C.; Danielian, P.S.; Parker, M.G.; Evan, G.I. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995, 23, 1686–1690. [Google Scholar] [CrossRef]
- Rönsch, K.; Jägle, S.; Rose, K.; Seidl, M.; Baumgartner, F.; Freihen, V.; Yousaf, A.; Metzger, E.; Lassmann, S.; Schüle, R.; et al. SNAIL1 combines competitive displacement of ASCL2 and epigenetic mechanisms to rapidly silence the EPHB3 tumor suppressor in colorectal cancer. Mol. Oncol. 2015, 9, 335–354. [Google Scholar] [CrossRef]
- Jägle, S.; Rönsch, K.; Timme, S.; Andrlová, H.; Bertrand, M.; Jäger, M.; Proske, A.; Schrempp, M.; Yousaf, A.; Michoel, T.; et al. Silencing of the EPHB3 tumor-suppressor gene in human colorectal cancer through decommissioning of a transcriptional enhancer. Proc. Natl. Acad. Sci. USA 2014, 111, 4886–4891. [Google Scholar] [CrossRef]
- Jägle, S.; Busch, H.; Freihen, V.; Beyes, S.; Schrempp, M.; Boerries, M.; Hecht, A. SNAIL1-mediated downregulation of FOXA proteins facilitates the inactivation of transcriptional enhancer elements at key epithelial genes in colorectal cancer cells. PLoS Genet. 2017, 13, e1007109. [Google Scholar] [CrossRef]
- Frey, P.; Devisme, A.; Rose, K.; Schrempp, M.; Freihen, V.; Andrieux, G.; Boerries, M.; Hecht, A. SMAD4 mutations do not preclude epithelial-mesenchymal transition in colorectal cancer. Oncogene 2022, 41, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, L.; Richter, A.S.; Betancourt, E.; Bruder, K.; Diehl, S.; Manke, T.; Bönisch, U. Standardizing chromatin research: A simple and universal method for ChIP-seq. Nucleic Acids Res. 2016, 44, e67. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2019, 35, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute. Picard Tools. Available online: http://broadinstitute.github.io/picard/ (accessed on 20 November 2020).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160-5. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Gaspar, J.M. Genrich. Available online: https://github.com/jsh58/Genrich (accessed on 20 November 2020).
- Yu, G.; Wang, L.-G.; He, Q.-Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef]
- Reske, J.J.; Wilson, M.R.; Chandler, R.L. ATAC-seq normalization method can significantly affect differential accessibility analysis and interpretation. Epigenetics Chromatin 2020, 13, 22. [Google Scholar] [CrossRef]
- Lun, A.T.L.; Smyth, G.K. csaw: A Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 2016, 44, e45. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.L.; Lee, T. Fuzzy competitive learning. Neural Netw. 1994, 7, 539–551. [Google Scholar] [CrossRef]
- Pal, N.R.; Bezdek, J.C.; Hathaway, R.J. Sequential Competitive Learning and the Fuzzy c-Means Clustering Algorithms. Neural Netw. 1996, 9, 787–796. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Therneau, T. A Package for Survival Analysis in R. Available online: https://CRAN.R-project.org/package=survival (accessed on 22 June 2022).
- Wickham, H.; Sievert, C. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer: Cham, Switzerland, 2016; ISBN 331924275X. [Google Scholar]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef]
- Foroutan, M.; Cursons, J.; Hediyeh-Zadeh, S.; Thompson, E.W.; Davis, M.J. A transcriptional program for detecting TGF\beta-induced EMT in cancer. Mol. Cancer Res. 2017, 15, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef] [PubMed]
- Gröger, C.J.; Grubinger, M.; Waldhör, T.; Vierlinger, K.; Mikulits, W. Meta-Analysis of Gene Expression Signatures Defining the Epithelial to Mesenchymal Transition during Cancer Progression. PLoS ONE 2012, 7, e51136. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Liu, Y.; Xue, M.; Liu, H.; Du, S.; Zhang, L.; Wang, P. Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nucleic Acids Res. 2016, 44, 2514–2527. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, N.; Meyer-Schaller, N.; Arnold, P.; Antoniadis, H.; Pachkov, M.; van Nimwegen, E.; Christofori, G. Klf4 is a transcriptional regulator of genes critical for EMT, including Jnk1 (Mapk8). PLoS ONE 2013, 8, e57329. [Google Scholar] [CrossRef]
- Miyashita, N.; Enokido, T.; Horie, M.; Fukuda, K.; Urushiyama, H.; Strell, C.; Brunnström, H.; Micke, P.; Saito, A.; Nagase, T. TGF-β-mediated epithelial-mesenchymal transition and tumor-promoting effects in CMT64 cells are reflected in the transcriptomic signature of human lung adenocarcinoma. Sci. Rep. 2021, 11, 22380. [Google Scholar] [CrossRef]
- Vincent, T.; Neve, E.P.A.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef]
- Phillips, S.; Kuperwasser, C. SLUG: Critical regulator of epithelial cell identity in breast development and cancer. Cell Adh. Migr. 2014, 8, 578–587. [Google Scholar] [CrossRef]
- Jägle, S.; Dertmann, A.; Schrempp, M.; Hecht, A. ZEB1 is neither sufficient nor required for epithelial-mesenchymal transition in LS174T colorectal cancer cells. Biochem. Biophys. Res. Commun. 2017, 482, 1226–1232. [Google Scholar] [CrossRef]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Glass, C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Martínez, J.A.; Ceballos-Chávez, M.; Koehler, F.; Peiró, S.; Reyes, J.C. TGFβ promotes widespread enhancer chromatin opening and operates on genomic regulatory domains. Nat. Commun. 2020, 11, 6196. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.S.; Hussein, S.; Chakraborty, P.; Muruganantham, A.; Mikhail, S.; Gonzalez, G.; Song, S.; Jolly, M.K.; Toneff, M.J.; Benton, M.L.; et al. CTCF Expression and Dynamic Motif Accessibility Modulates Epithelial-Mesenchymal Gene Expression. Cancers 2022, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- Arase, M.; Tamura, Y.; Kawasaki, N.; Isogaya, K.; Nakaki, R.; Mizutani, A.; Tsutsumi, S.; Aburatani, H.; Miyazono, K.; Koinuma, D. Dynamics of chromatin accessibility during TGF-β-induced EMT of Ras-transformed mammary gland epithelial cells. Sci. Rep. 2017, 7, 1166. [Google Scholar] [CrossRef]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K.; et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef]
- Barnett, K.R.; Decato, B.E.; Scott, T.J.; Hansen, T.J.; Chen, B.; Attalla, J.; Smith, A.D.; Hodges, E. ATAC-Me Captures Prolonged DNA Methylation of Dynamic Chromatin Accessibility Loci during Cell Fate Transitions. Mol. Cell 2020, 77, 1350–1364.e6. [Google Scholar] [CrossRef]
- Sanghi, A.; Gruber, J.J.; Metwally, A.; Jiang, L.; Reynolds, W.; Sunwoo, J.; Orloff, L.; Chang, H.Y.; Kasowski, M.; Snyder, M.P. Chromatin accessibility associates with protein-RNA correlation in human cancer. Nat. Commun. 2021, 12, 5732. [Google Scholar] [CrossRef]
- Segert, J.A.; Gisselbrecht, S.S.; Bulyk, M.L. Transcriptional Silencers: Driving Gene Expression with the Brakes On. Trends Genet. 2021, 37, 514–527. [Google Scholar] [CrossRef]
- Pang, B.; Snyder, M.P. Systematic identification of silencers in human cells. Nat. Genet. 2020, 52, 254–263. [Google Scholar] [CrossRef]
- Feldker, N.; Ferrazzi, F.; Schuhwerk, H.; Widholz, S.A.; Guenther, K.; Frisch, I.; Jakob, K.; Kleemann, J.; Riegel, D.; Bönisch, U.; et al. Genome-wide cooperation of EMT transcription factor ZEB1 with YAP and AP-1 in breast cancer. EMBO J. 2020, 39, e103209. [Google Scholar] [CrossRef]
- Beyes, S.; Andrieux, G.; Schrempp, M.; Aicher, D.; Wenzel, J.; Antón-García, P.; Boerries, M.; Hecht, A. Genome-wide mapping of DNA-binding sites identifies stemness-related genes as directly repressed targets of SNAIL1 in colorectal cancer cells. Oncogene 2019, 38, 6647–6661. [Google Scholar] [CrossRef] [PubMed]
- Rosmaninho, P.; Mükusch, S.; Piscopo, V.; Teixeira, V.; Raposo, A.A.; Warta, R.; Bennewitz, R.; Tang, Y.; Herold-Mende, C.; Stifani, S.; et al. Zeb1 potentiates genome-wide gene transcription with Lef1 to promote glioblastoma cell invasion. EMBO J. 2018, 37, e97115. [Google Scholar] [CrossRef] [PubMed]
- Smita, S.; Ahad, A.; Ghosh, A.; Biswas, V.K.; Koga, M.M.; Gupta, B.; Acha-Orbea, H.; Raghav, S.K. Importance of EMT Factor ZEB1 in cDC1 “MutuDC Line” Mediated Induction of Th1 Immune Response. Front. Immunol. 2018, 9, 2604. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.D.; Corsa, C.A.S.; Biswas, H.; Aft, R.L.; Longmore, G.D. Temporal and Spatial Cooperation of Snail1 and Twist1 during Epithelial-Mesenchymal Transition Predicts for Human Breast Cancer Recurrence. Mol. Cancer Res. 2011, 9, 1644–1657. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Betel, D.; Miller, M.L.; Sander, C.; Leslie, C.S.; Marks, D.S. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat. Biotechnol. 2009, 27, 549–555. [Google Scholar] [CrossRef]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Expósito-Villén, A.; Aránega, A.E.; Franco, D. Functional Role of Non-Coding RNAs during Epithelial-To-Mesenchymal Transition. Noncoding RNA 2018, 4, 14. [Google Scholar] [CrossRef]
- Díaz-López, A.; Moreno-Bueno, G.; Cano, A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag. Res. 2014, 6, 205–216. [Google Scholar] [CrossRef]
- Hu, Y.; Tang, H. MicroRNAs regulate the epithelial to mesenchymal transition (EMT) in cancer progression. Microrna 2014, 3, 108–117. [Google Scholar] [CrossRef]
- Frey, P.; Devisme, A.; Schrempp, M.; Andrieux, G.; Boerries, M.; Hecht, A. Canonical BMP Signaling Executes Epithelial-Mesenchymal Transition Downstream of SNAIL1. Cancers 2020, 12, 1019. [Google Scholar] [CrossRef]
- Javaid, S.; Zhang, J.; Anderssen, E.; Black, J.C.; Wittner, B.S.; Tajima, K.; Ting, D.T.; Smolen, G.A.; Zubrowski, M.; Desai, R.; et al. Dynamic Chromatin Modification Sustains Epithelial-Mesenchymal Transition following Inducible Expression of Snail-1. Cell Rep. 2013, 5, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Preca, B.-T.; Bajdak, K.; Mock, K.; Sundararajan, V.; Pfannstiel, J.; Maurer, J.; Wellner, U.; Hopt, U.T.; Brummer, T.; Brabletz, S.; et al. A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int. J. Cancer 2015, 137, 2566–2577. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Peinado, H.; Molina, P.; Olmeda, D.; Cubillo, E.; Santos, V.; Palacios, J.; Portillo, F.; Cano, A. The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat. Protoc. 2009, 4, 1591–1613. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Sundqvist, A.; Zieba, A.; Vasilaki, E.; Herrera Hidalgo, C.; Söderberg, O.; Koinuma, D.; Miyazono, K.; Heldin, C.-H.; Landegren, U.; Dijke, P.T.; et al. Specific interactions between Smad proteins and AP-1 components determine TGFβ-induced breast cancer cell invasion. Oncogene 2013, 32, 3606–3615. [Google Scholar] [CrossRef]
- Sundqvist, A.; Morikawa, M.; Ren, J.; Vasilaki, E.; Kawasaki, N.; Kobayashi, M.; Koinuma, D.; Aburatani, H.; Miyazono, K.; Heldin, C.-H.; et al. JUNB governs a feed-forward network of TGFβ signaling that aggravates breast cancer invasion. Nucleic Acids Res. 2018, 46, 1180–1195. [Google Scholar] [CrossRef]
- Biddle, A.; Liang, X.; Gammon, L.; Fazil, B.; Harper, L.J.; Emich, H.; Costea, D.E.; Mackenzie, I.C. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. 2011, 71, 5317–5326. [Google Scholar] [CrossRef]
- Vega, S.; Morales, A.V.; Ocaña, O.H.; Valdés, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef]
- Bejjani, F.; Evanno, E.; Zibara, K.; Piechaczyk, M.; Jariel-Encontre, I. The AP-1 transcriptional complex: Local switch or remote command? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 11–23. [Google Scholar] [CrossRef]
- Seo, J.; Koçak, D.D.; Bartelt, L.C.; Williams, C.A.; Barrera, A.; Gersbach, C.A.; Reddy, T.E. AP-1 subunits converge promiscuously at enhancers to potentiate transcription. Genome Res. 2021, 31, 538–550. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antón-García, P.; Haghighi, E.B.; Rose, K.; Vladimirov, G.; Boerries, M.; Hecht, A. TGFβ1-Induced EMT in the MCF10A Mammary Epithelial Cell Line Model Is Executed Independently of SNAIL1 and ZEB1 but Relies on JUNB-Coordinated Transcriptional Regulation. Cancers 2023, 15, 558. https://doi.org/10.3390/cancers15020558
Antón-García P, Haghighi EB, Rose K, Vladimirov G, Boerries M, Hecht A. TGFβ1-Induced EMT in the MCF10A Mammary Epithelial Cell Line Model Is Executed Independently of SNAIL1 and ZEB1 but Relies on JUNB-Coordinated Transcriptional Regulation. Cancers. 2023; 15(2):558. https://doi.org/10.3390/cancers15020558
Chicago/Turabian StyleAntón-García, Pablo, Elham Bavafaye Haghighi, Katja Rose, Georg Vladimirov, Melanie Boerries, and Andreas Hecht. 2023. "TGFβ1-Induced EMT in the MCF10A Mammary Epithelial Cell Line Model Is Executed Independently of SNAIL1 and ZEB1 but Relies on JUNB-Coordinated Transcriptional Regulation" Cancers 15, no. 2: 558. https://doi.org/10.3390/cancers15020558
APA StyleAntón-García, P., Haghighi, E. B., Rose, K., Vladimirov, G., Boerries, M., & Hecht, A. (2023). TGFβ1-Induced EMT in the MCF10A Mammary Epithelial Cell Line Model Is Executed Independently of SNAIL1 and ZEB1 but Relies on JUNB-Coordinated Transcriptional Regulation. Cancers, 15(2), 558. https://doi.org/10.3390/cancers15020558