

Mass Spectrometry-Based Proteomics Workflows in Cancer Research: The Relevance of Choosing the Right Steps


Abstract
Simple Summary
Abstract

1. Introduction
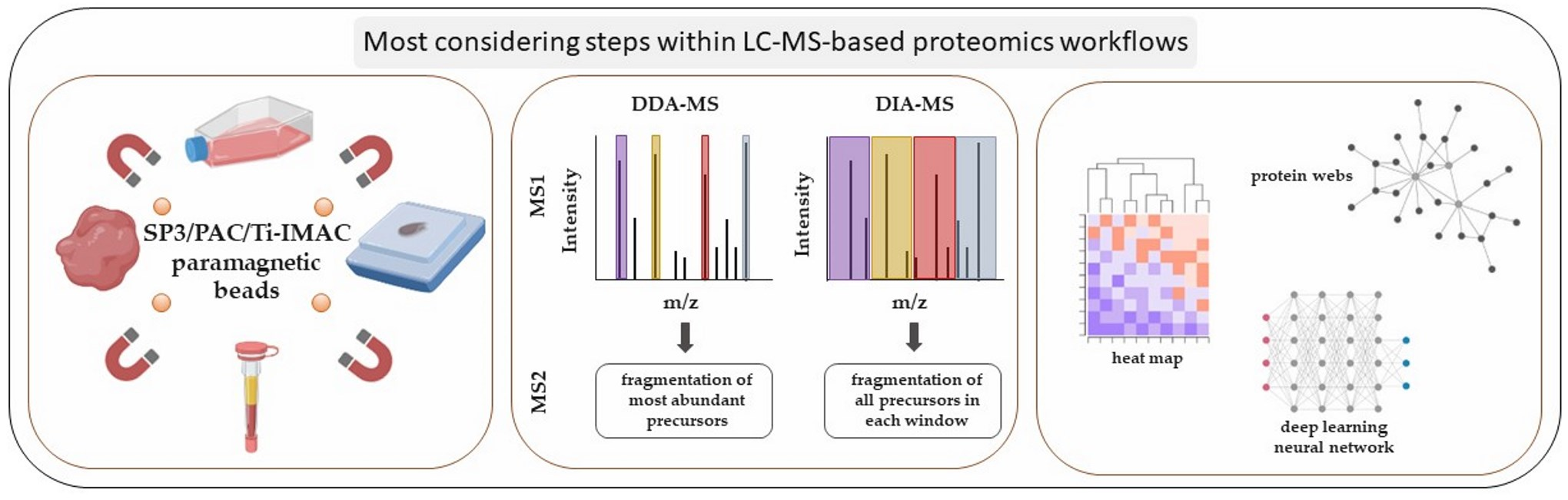
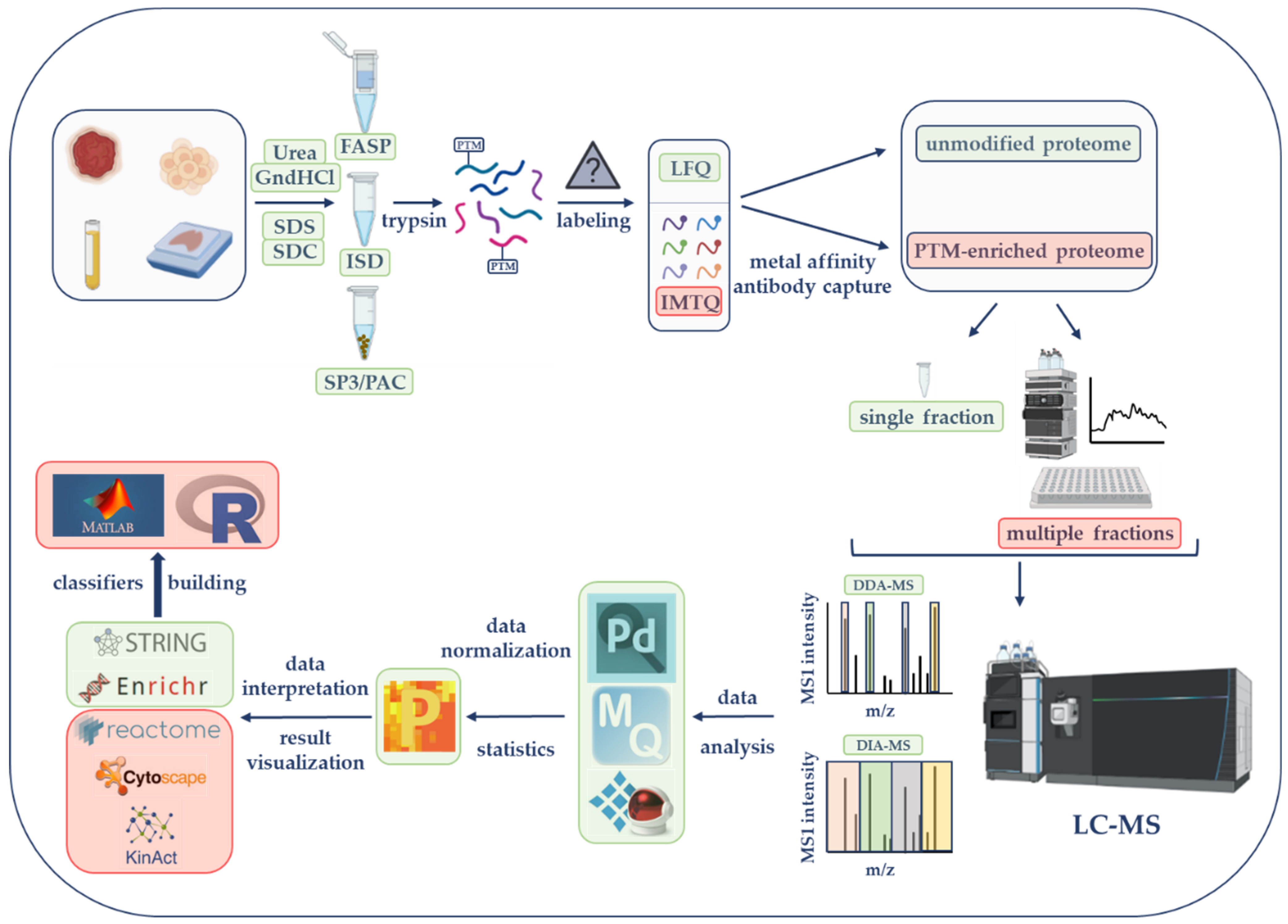
2. LC-MS-Based Proteomics Strategies from Sample Selection to Data Acquisition in Cancer Research: Steps and Main Considerations
2.1. Sample Type’s Selection and Cohort Size
2.2. Sample Preparation Strategies
2.3. Quantification Strategies
2.4. PTM Enrichments
2.5. Peptide Fractionation to Increase Proteome Coverage
2.6. MS Methods for Data Acquisition
3. LC-MS-Based Proteomics Data Analysis and Bioinformatics

{kind=link}
{kind=link}
| LC-MS Data | Data Processing | Gene Ontology (GO)/Pathway Analysis | Protein Networks | Visualization Tools |
|---|---|---|---|---|
| MaxQuant [90,91] | Perseus [94,95] | Enrichr [97] | STRING [98] | Cytoscape [99] |
| MSFragger [92] | Prostar [108] | A GO tool [109] | Omnipath [107] | OpenPIP [110] |
| DIA-NN [93] | Proteome Discoverer 1 | Reactome [111] | PINA [112,113] | Perseus |
| Proteome Discoverer (F) 1 | Qlucore 1 | DAVID [114] | Perseus | |
| Mascot Distiller/Server 1 | InnateDB [115] | |||
| Spectronaut (F) 1 | FunRich [116] | |||
| PEAKS Xpro (F) 1 MSStats [117] Progenesis QI for Proteomics 1 | QIAGEN IPA 1 |
4. Artificial Intelligence Strategies on Proteomics Data
5. Fever of Single-Cell LC-MS-Based Proteomics
6. Conclusions and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martinez-Val, A.; Guzman, U.H.; Olsen, J.V. Obtaining Complete Human Proteomes. Annu. Rev. Genom. Hum. Genet. 2022, 23, 99–121. [Google Scholar] [CrossRef] [PubMed]
- Toby, T.K.; Fornelli, L.; Srzentic, K.; DeHart, C.J.; Levitsky, J.; Friedewald, J.; Kelleher, N.L. A comprehensive pipeline for translational top-down proteomics from a single blood draw. Nat. Protoc. 2019, 14, 119–152. [Google Scholar] [CrossRef] [PubMed]
- van Bentum, M.; Selbach, M. An Introduction to Advanced Targeted Acquisition Methods. Mol. Cell. Proteom. 2021, 20, 100165. [Google Scholar] [CrossRef]
- Lee, H.; Kim, S.I. Review of Liquid Chromatography-Mass Spectrometry-Based Proteomic Analyses of Body Fluids to Diagnose Infectious Diseases. Int. J. Mol. Sci. 2022, 23, 2187. [Google Scholar] [CrossRef]
- Martelli, C.; Iavarone, F.; D’Angelo, L.; Arba, M.; Vincenzoni, F.; Inserra, I.; Delfino, D.; Rossetti, D.V.; Caretto, M.; Massimi, L.; et al. Integrated proteomic platforms for the comparative characterization of medulloblastoma and pilocytic astrocytoma pediatric brain tumors: A preliminary study. Mol. Biosyst. 2015, 11, 1668–1683. [Google Scholar] [CrossRef] [PubMed]
- Borras, E.; Sabido, E. What is targeted proteomics? A concise revision of targeted acquisition and targeted data analysis in mass spectrometry. Proteomics 2017, 17, 17–18. [Google Scholar] [CrossRef]
- Ntai, I.; Fornelli, L.; DeHart, C.J.; Hutton, J.E.; Doubleday, P.F.; LeDuc, R.D.; van Nispen, A.J.; Fellers, R.T.; Whiteley, G.; Boja, E.S.; et al. Precise characterization of KRAS4b proteoforms in human colorectal cells and tumors reveals mutation/modification cross-talk. Proc. Natl. Acad. Sci. USA 2018, 115, 4140–4145. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Martinez-Val, A.; Steigerwald, S.; Ruther, P.; Fort, K.L.; Arrey, T.N.; Harder, A.; Makarov, A.; Olsen, J.V. A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients. Mol. Cell. Proteom. 2020, 19, 716–729. [Google Scholar] [CrossRef]
- Ni, M.; Zhou, J.; Zhu, Z.; Yuan, J.; Gong, W.; Zhu, J.; Zheng, Z.; Zhao, H. A Novel Classifier Based on Urinary Proteomics for Distinguishing Between Benign and Malignant Ovarian Tumors. Front. Cell Dev. Biol. 2021, 9, 712196. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, J.; Wei, J.; Zhao, Y.; Gao, Y. Urinary biomarker discovery in gliomas using mass spectrometry-based clinical proteomics. Chin. Neurosurg. J. 2020, 6, 11. [Google Scholar] [CrossRef]
- Nakayasu, E.S.; Gritsenko, M.; Piehowski, P.D.; Gao, Y.; Orton, D.J.; Schepmoes, A.A.; Fillmore, T.L.; Frohnert, B.I.; Rewers, M.; Krischer, J.P.; et al. Tutorial: Best practices and considerations for mass-spectrometry-based protein biomarker discovery and validation. Nat. Protoc. 2021, 16, 3737–3760. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.Z.L.; Capra, M.; Cassi, C.; Taliento, G.; Ivanova, M.; Guerini-Rocco, E.; Fumagali, M.; Monturano, M.; Albini, A.; Viale, G.; et al. Standard operating procedures for biobank in oncology. Front. Mol. Biosci. 2022, 9. [Google Scholar] [CrossRef]
- Greco, V.; Piras, C.; Pieroni, L.; Urbani, A. Direct Assessment of Plasma/Serum Sample Quality for Proteomics Biomarker Investigation. Methods Mol. Biol. 2017, 1619, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Valladares, M.; Aasebo, E.; Mjaavatten, O.; Vaudel, M.; Bruserud, O.; Berven, F.; Selheim, F. Reliable FASP-based procedures for optimal quantitative proteomic and phosphoproteomic analysis on samples from acute myeloid leukemia patients. Biol. Proced. Online 2016, 18, 13. [Google Scholar] [CrossRef]
- Aasebo, E.; Brenner, A.K.; Hernandez-Valladares, M.; Birkeland, E.; Berven, F.S.; Selheim, F.; Bruserud, O. Proteomic Comparison of Bone Marrow Derived Osteoblasts and Mesenchymal Stem Cells. Int. J. Mol. Sci. 2021, 22, 5665. [Google Scholar] [CrossRef] [PubMed]
- Aasebo, E.; Brenner, A.K.; Hernandez-Valladares, M.; Birkeland, E.; Mjaavatten, O.; Reikvam, H.; Selheim, F.; Berven, F.S.; Bruserud, O. Patient Heterogeneity in Acute Myeloid Leukemia: Leukemic Cell Communication by Release of Soluble Mediators and Its Effects on Mesenchymal Stem Cells. Diseases 2021, 9, 74. [Google Scholar] [CrossRef]
- Aasebo, E.; Mjaavatten, O.; Vaudel, M.; Farag, Y.; Selheim, F.; Berven, F.; Bruserud, O.; Hernandez-Valladares, M. Freezing effects on the acute myeloid leukemia cell proteome and phosphoproteome revealed using optimal quantitative workflows. J. Proteom. 2016, 145, 214–225. [Google Scholar] [CrossRef]
- Dapic, I.; Uwugiaren, N.; Kers, J.; Mohammed, Y.; Goodlett, D.R.; Corthals, G. Evaluation of Fast and Sensitive Proteome Profiling of FF and FFPE Kidney Patient Tissues. Molecules 2022, 27, 1137. [Google Scholar] [CrossRef]
- Dressler, F.F.; Schoenfeld, J.; Revyakina, O.; Vogele, D.; Kiefer, S.; Kirfel, J.; Gemoll, T.; Perner, S. Systematic evaluation and optimization of protein extraction parameters in diagnostic FFPE specimens. Clin. Proteom. 2022, 19, 10. [Google Scholar] [CrossRef]
- Neset, L.; Takayidza, G.; Berven, F.S.; Hernandez-Valladares, M. Comparing Efficiency of Lysis Buffer Solutions and Sample Preparation Methods for Liquid Chromatography-Mass Spectrometry Analysis of Human Cells and Plasma. Molecules 2022, 27, 3390. [Google Scholar] [CrossRef]
- Buczak, K.; Kirkpatrick, J.M.; Truckenmueller, F.; Santinha, D.; Ferreira, L.; Roessler, S.; Singer, S.; Beck, M.; Ori, A. Spatially resolved analysis of FFPE tissue proteomes by quantitative mass spectrometry. Nat. Protoc. 2020, 15, 2956–2979. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Sandberg, A.; Araujo, J.E.; Cvetkovski, F.; Berglund, E.; Eriksson, L.E.; Pernemalm, M. Evaluation of Spin Columns for Human Plasma Depletion to Facilitate MS-Based Proteomics Analysis of Plasma. J. Proteome Res. 2021, 20, 4610–4620. [Google Scholar] [CrossRef] [PubMed]
- Keshishian, H.; Burgess, M.W.; Specht, H.; Wallace, L.; Clauser, K.R.; Gillette, M.A.; Carr, S.A. Quantitative, multiplexed workflow for deep analysis of human blood plasma and biomarker discovery by mass spectrometry. Nat. Protoc. 2017, 12, 1683–1701. [Google Scholar] [CrossRef] [PubMed]
- Kverneland, A.H.; Ostergaard, O.; Emdal, K.B.; Svane, I.M.; Olsen, J.V. Differential ultracentrifugation enables deep plasma proteomics through enrichment of extracellular vesicles. Proteomics 2022, e2200039. [Google Scholar] [CrossRef] [PubMed]
- Blume, J.E.; Manning, W.C.; Troiano, G.; Hornburg, D.; Figa, M.; Hesterberg, L.; Platt, T.L.; Zhao, X.; Cuaresma, R.A.; Everley, P.A.; et al. Rapid, deep and precise profiling of the plasma proteome with multi-nanoparticle protein corona. Nat. Commun. 2020, 11, 3662. [Google Scholar] [CrossRef]
- Ferdosi, S.; Stukalov, A.; Hasan, M.; Tangeysh, B.; Brown, T.R.; Wang, T.; Elgierari, E.M.; Zhao, X.; Huang, Y.; Alavi, A.; et al. Enhanced Competition at the Nano-Bio Interface Enables Comprehensive Characterization of Protein Corona Dynamics and Deep Coverage of Proteomes. Adv. Mater. 2022, 34, e2206008. [Google Scholar] [CrossRef]
- Levin, Y. The role of statistical power analysis in quantitative proteomics. Proteomics 2011, 11, 2565–2567. [Google Scholar] [CrossRef]
- Alexovic, M.; Sabo, J.; Longuespee, R. Microproteomic sample preparation. Proteomics 2021, 21, e2000318. [Google Scholar] [CrossRef]
- Varnavides, G.; Madern, M.; Anrather, D.; Hartl, N.; Reiter, W.; Hartl, M. In Search of a Universal Method: A Comparative Survey of Bottom-Up Proteomics Sample Preparation Methods. J. Proteome Res. 2022, 21, 2397–2411. [Google Scholar] [CrossRef]
- Foster, L.J.; De Hoog, C.L.; Mann, M. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc. Natl. Acad. Sci. USA 2003, 100, 5813–5818. [Google Scholar] [CrossRef]
- Kelstrup, C.D.; Jersie-Christensen, R.R.; Batth, T.S.; Arrey, T.N.; Kuehn, A.; Kellmann, M.; Olsen, J.V. Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra-high-field Orbitrap mass spectrometer. J. Proteome Res. 2014, 13, 6187–6195. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.A.; Rhoads, A.; Cocco, A.R.; Peckner, R.; Haber, A.L.; Schweitzer, L.D.; Krug, K.; Mani, D.R.; Clauser, K.R.; Rozenblatt-Rosen, O.; et al. Streamlined Protocol for Deep Proteomic Profiling of FAC-sorted Cells and Its Application to Freshly Isolated Murine Immune Cells. Mol. Cell. Proteom. 2019, 18, 995–1009. [Google Scholar] [CrossRef] [PubMed]
- Sialana, F.J.; Roumeliotis, T.I.; Bouguenina, H.; Chan Wah Hak, L.; Wang, H.; Caldwell, J.; Collins, I.; Chopra, R.; Choudhary, J.S. SimPLIT: Simplified Sample Preparation for Large-Scale Isobaric Tagging Proteomics. J. Proteome Res. 2022, 21, 1842–1856. [Google Scholar] [CrossRef] [PubMed]
- Manza, L.L.; Stamer, S.L.; Ham, A.J.; Codreanu, S.G.; Liebler, D.C. Sample preparation and digestion for proteomic analyses using spin filters. Proteomics 2005, 5, 1742–1745. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Mann, M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 2012, 84, 2631–2637. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Rakus, D. Multi-enzyme digestion FASP and the ’Total Protein Approach’-based absolute quantification of the Escherichia coli proteome. J. Proteom. 2014, 109, 322–331. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Mann, M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 2009, 8, 5674–5678. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Muller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Hayoun, K.; Gouveia, D.; Grenga, L.; Pible, O.; Armengaud, J.; Alpha-Bazin, B. Evaluation of Sample Preparation Methods for Fast Proteotyping of Microorganisms by Tandem Mass Spectrometry. Front. Microbiol. 2019, 10, 1985. [Google Scholar] [CrossRef]
- Sielaff, M.; Kuharev, J.; Bohn, T.; Hahlbrock, J.; Bopp, T.; Tenzer, S.; Distler, U. Evaluation of FASP, SP3, and iST Protocols for Proteomic Sample Preparation in the Low Microgram Range. J. Proteome Res. 2017, 16, 4060–4072. [Google Scholar] [CrossRef] [PubMed]
- Waas, M.; Pereckas, M.; Jones Lipinski, R.A.; Ashwood, C.; Gundry, R.L. SP2: Rapid and Automatable Contaminant Removal from Peptide Samples for Proteomic Analyses. J. Proteome Res. 2019, 18, 1644–1656. [Google Scholar] [CrossRef] [PubMed]
- Batth, T.S.; Tollenaere, M.X.; Ruther, P.; Gonzalez-Franquesa, A.; Prabhakar, B.S.; Bekker-Jensen, S.; Deshmukh, A.S.; Olsen, J.V. Protein Aggregation Capture on Microparticles Enables Multipurpose Proteomics Sample Preparation. Mol. Cell. Proteom. 2019, 18, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Franciosa, G.; Smits, J.G.A.; Minuzzo, S.; Martinez-Val, A.; Indraccolo, S.; Olsen, J.V. Proteomics of resistance to Notch1 inhibition in acute lymphoblastic leukemia reveals targetable kinase signatures. Nat. Commun. 2021, 12, 2507. [Google Scholar] [CrossRef]
- Ruther, P.L.; Husic, I.M.; Bangsgaard, P.; Gregersen, K.M.; Pantmann, P.; Carvalho, M.; Godinho, R.M.; Friedl, L.; Cascalheira, J.; Taurozzi, A.J.; et al. SPIN enables high throughput species identification of archaeological bone by proteomics. Nat. Commun. 2022, 13, 2458. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Kalxdorf, M.; Longuespee, R.; Kazdal, D.N.; Stenzinger, A.; Krijgsveld, J. Automated sample preparation with SP3 for low-input clinical proteomics. Mol. Syst. Biol. 2020, 16, e9111. [Google Scholar] [CrossRef]
- Moggridge, S.; Sorensen, P.H.; Morin, G.B.; Hughes, C.S. Extending the Compatibility of the SP3 Paramagnetic Bead Processing Approach for Proteomics. J. Proteome Res. 2018, 17, 1730–1740. [Google Scholar] [CrossRef]
- Dagley, L.F.; Infusini, G.; Larsen, R.H.; Sandow, J.J.; Webb, A.I. Universal Solid-Phase Protein Preparation (USP(3)) for Bottom-up and Top-down Proteomics. J. Proteome Res. 2019, 18, 2915–2924. [Google Scholar] [CrossRef]
- van der Pan, K.; Kassem, S.; Khatri, I.; de Ru, A.H.; Janssen, G.M.C.; Tjokrodirijo, R.T.N.; Al Makindji, F.; Stavrakaki, E.; de Jager, A.L.; Naber, B.A.E.; et al. Quantitative proteomics of small numbers of closely-related cells: Selection of the optimal method for a clinical setting. Front. Med. 2022, 9, 997305. [Google Scholar] [CrossRef]
- Tape, C.J.; Worboys, J.D.; Sinclair, J.; Gourlay, R.; Vogt, J.; McMahon, K.M.; Trost, M.; Lauffenburger, D.A.; Lamont, D.J.; Jorgensen, C. Reproducible automated phosphopeptide enrichment using magnetic TiO2 and Ti-IMAC. Anal. Chem. 2014, 86, 10296–10302. [Google Scholar] [CrossRef]
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031. [Google Scholar] [CrossRef]
- Geiger, T.; Wisniewski, J.R.; Cox, J.; Zanivan, S.; Kruger, M.; Ishihama, Y.; Mann, M. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat. Protoc. 2011, 6, 147–157. [Google Scholar] [CrossRef]
- Rigbolt, K.T.; Blagoev, B. Proteome-wide quantitation by SILAC. Methods Mol. Biol. 2010, 658, 187–204. [Google Scholar] [CrossRef]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef]
- Hsu, J.L.; Huang, S.Y.; Chow, N.H.; Chen, S.H. Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 2003, 75, 6843–6852. [Google Scholar] [CrossRef]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Yao, X.; Freas, A.; Ramirez, J.; Demirev, P.A.; Fenselau, C. Proteolytic 18O labeling for comparative proteomics: Model studies with two serotypes of adenovirus. Anal. Chem. 2001, 73, 2836–2842. [Google Scholar] [CrossRef]
- Unwin, R.D. Quantification of proteins by iTRAQ. Methods Mol. Biol. 2010, 658, 205–215. [Google Scholar] [CrossRef]
- Wong, J.W.; Cagney, G. An overview of label-free quantitation methods in proteomics by mass spectrometry. Methods Mol. Biol. 2010, 604, 273–283. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Bielow, C.; Mastrobuoni, G.; Kempa, S. Proteomics Quality Control: Quality Control Software for MaxQuant Results. J. Proteome Res. 2016, 15, 777–787. [Google Scholar] [CrossRef]
- Yu, S.H.; Kyriakidou, P.; Cox, J. Isobaric Matching between Runs and Novel PSM-Level Normalization in MaxQuant Strongly Improve Reporter Ion-Based Quantification. J. Proteome Res. 2020, 19, 3945–3954. [Google Scholar] [CrossRef]
- Hutchinson-Bunch, C.; Sanford, J.A.; Hansen, J.R.; Gritsenko, M.A.; Rodland, K.D.; Piehowski, P.D.; Qian, W.J.; Adkins, J.N. Assessment of TMT Labeling Efficiency in Large-Scale Quantitative Proteomics: The Critical Effect of Sample pH. ACS Omega 2021, 6, 12660–12666. [Google Scholar] [CrossRef]
- Yu, K.; Wang, Z.; Wu, Z.; Tan, H.; Mishra, A.; Peng, J. High-Throughput Profiling of Proteome and Posttranslational Modifications by 16-Plex TMT Labeling and Mass Spectrometry. Methods Mol. Biol. 2021, 2228, 205–224. [Google Scholar] [CrossRef]
- Zecha, J.; Satpathy, S.; Kanashova, T.; Avanessian, S.C.; Kane, M.H.; Clauser, K.R.; Mertins, P.; Carr, S.A.; Kuster, B. TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Mol. Cell. Proteom. 2019, 18, 1468–1478. [Google Scholar] [CrossRef]
- Pieroni, L.; Iavarone, F.; Olianas, A.; Greco, V.; Desiderio, C.; Martelli, C.; Manconi, B.; Sanna, M.T.; Messana, I.; Castagnola, M.; et al. Enrichments of post-translational modifications in proteomic studies. J. Sep. Sci. 2020, 43, 313–336. [Google Scholar] [CrossRef]
- Hernandez-Valladares, M.; Wangen, R.; Berven, F.S.; Guldbrandsen, A. Protein Post-Translational Modification Crosstalk in Acute Myeloid Leukemia Calls for Action. Curr. Med. Chem. 2019, 26, 5317–5337. [Google Scholar] [CrossRef]
- Thingholm, T.E.; Jensen, O.N.; Robinson, P.J.; Larsen, M.R. SIMAC (sequential elution from IMAC), a phosphoproteomics strategy for the rapid separation of monophosphorylated from multiply phosphorylated peptides. Mol. Cell. Proteom. 2008, 7, 661–671. [Google Scholar] [CrossRef]
- Thingholm, T.E.; Jorgensen, T.J.; Jensen, O.N.; Larsen, M.R. Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat. Protoc. 2006, 1, 1929–1935. [Google Scholar] [CrossRef]
- Thingholm, T.E.; Larsen, M.R. Phosphopeptide Enrichment by Immobilized Metal Affinity Chromatography. Methods Mol. Biol. 2016, 1355, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Aasebo, E.; Berven, F.S.; Bartaula-Brevik, S.; Stokowy, T.; Hovland, R.; Vaudel, M.; Doskeland, S.O.; McCormack, E.; Batth, T.S.; Olsen, J.V.; et al. Proteome and Phosphoproteome Changes Associated with Prognosis in Acute Myeloid Leukemia. Cancers 2020, 12, 709. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020, 11, 787. [Google Scholar] [CrossRef]
- Manadas, B.; Mendes, V.M.; English, J.; Dunn, M.J. Peptide fractionation in proteomics approaches. Expert Rev. Proteom. 2010, 7, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, D.B.; Kelstrup, C.D.; Batth, T.S.; Larsen, S.C.; Haldrup, C.; Bramsen, J.B.; Sorensen, K.D.; Hoyer, S.; Orntoft, T.F.; Andersen, C.L.; et al. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017, 4, 587–599. [Google Scholar] [CrossRef]
- Branca, R.M.; Orre, L.M.; Johansson, H.J.; Granholm, V.; Huss, M.; Perez-Bercoff, A.; Forshed, J.; Kall, L.; Lehtio, J. HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat. Methods 2014, 11, 59–62. [Google Scholar] [CrossRef]
- Lehtio, J.; Arslan, T.; Siavelis, I.; Pan, Y.; Socciarelli, F.; Berkovska, O.; Umer, H.M.; Mermelekas, G.; Pirmoradian, M.; Jonsson, M.; et al. Proteogenomics of non-small cell lung cancer reveals molecular subtypes associated with specific therapeutic targets and immune evasion mechanisms. Nat. Cancer 2021, 2, 1224–1242. [Google Scholar] [CrossRef]
- Panizza, E.; Branca, R.M.M.; Oliviusson, P.; Orre, L.M.; Lehtio, J. Isoelectric point-based fractionation by HiRIEF coupled to LC-MS allows for in-depth quantitative analysis of the phosphoproteome. Sci. Rep. 2017, 7, 4513. [Google Scholar] [CrossRef]
- Hodge, K.; Have, S.T.; Hutton, L.; Lamond, A.I. Cleaning up the masses: Exclusion lists to reduce contamination with HPLC-MS/MS. J. Proteom. 2013, 88, 92–103. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, Y.; Jia, L.; Zhang, C.; Cao, W.; Alam, N.; Wang, R.; Wang, W.; Bai, L.; Zhao, S.; et al. TMT-based quantitative proteomic profiling of human monocyte-derived macrophages and foam cells. Proteome Sci. 2022, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Ping, L.; Kundinger, S.R.; Duong, D.M.; Yin, L.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Global quantitative analysis of the human brain proteome and phosphoproteome in Alzheimer’s disease. Sci. Data 2020, 7, 315. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, K.; Brombacher, E.; Fahrner, M.; Vogele, D.; Kook, L.; Pinter, N.; Bronsert, P.; Timme-Bronsert, S.; Schmidt, A.; Barenfaller, K.; et al. Benchmarking of analysis strategies for data-independent acquisition proteomics using a large-scale dataset comprising inter-patient heterogeneity. Nat. Commun. 2022, 13, 2622. [Google Scholar] [CrossRef] [PubMed]
- Skowronek, P.; Thielert, M.; Voytik, E.; Tanzer, M.C.; Hansen, F.M.; Willems, S.; Karayel, O.; Brunner, A.D.; Meier, F.; Mann, M. Rapid and In-Depth Coverage of the (Phospho-)Proteome With Deep Libraries and Optimal Window Design for dia-PASEF. Mol. Cell. Proteom. 2022, 21, 100279. [Google Scholar] [CrossRef] [PubMed]
- Erickson, B.K.; Mintseris, J.; Schweppe, D.K.; Navarrete-Perea, J.; Erickson, A.R.; Nusinow, D.P.; Paulo, J.A.; Gygi, S.P. Active Instrument Engagement Combined with a Real-Time Database Search for Improved Performance of Sample Multiplexing Workflows. J. Proteome Res. 2019, 18, 1299–1306. [Google Scholar] [CrossRef]
- McAlister, G.C.; Nusinow, D.P.; Jedrychowski, M.P.; Wuhr, M.; Huttlin, E.L.; Erickson, B.K.; Rad, R.; Haas, W.; Gygi, S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014, 86, 7150–7158. [Google Scholar] [CrossRef]
- Schweppe, D.K.; Eng, J.K.; Yu, Q.; Bailey, D.; Rad, R.; Navarrete-Perea, J.; Huttlin, E.L.; Erickson, B.K.; Paulo, J.A.; Gygi, S.P. Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics. J. Proteome Res. 2020, 19, 2026–2034. [Google Scholar] [CrossRef]
- Gatto, L.; Breckels, L.M.; Naake, T.; Gibb, S. Visualization of proteomics data using R and bioconductor. Proteomics 2015, 15, 1375–1389. [Google Scholar] [CrossRef]
- Schessner, J.P.; Voytik, E.; Bludau, I. A practical guide to interpreting and generating bottom-up proteomics data visualizations. Proteomics 2022, 22, e2100103. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Sinitcyn, P.; Hamzeiy, H.; Salinas Soto, F.; Itzhak, D.; McCarthy, F.; Wichmann, C.; Steger, M.; Ohmayer, U.; Distler, U.; Kaspar-Schoenefeld, S.; et al. MaxDIA enables library-based and library-free data-independent acquisition proteomics. Nat. Biotechnol. 2021, 39, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.T.; Leprevost, F.V.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat. Methods 2017, 14, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Rudolph, J.D.; Cox, J. A Network Module for the Perseus Software for Computational Proteomics Facilitates Proteome Interaction Graph Analysis. J. Proteome Res. 2019, 18, 2052–2064. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.H.; Ferretti, D.; Schessner, J.P.; Rudolph, J.D.; Borner, G.H.H.; Cox, J. Expanding the Perseus Software for Omics Data Analysis With Custom Plugins. Curr. Protoc. Bioinform. 2020, 71, e105. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Rost, H.L.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.C.; Gutenbrunner, P.; Kenar, E.; et al. OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Mendoza, L.; Shteynberg, D.; Slagel, J.; Sun, Z.; Moritz, R.L. Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteom. Clin. Appl. 2015, 9, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Casado, P.; Rodriguez-Prados, J.C.; Cosulich, S.C.; Guichard, S.; Vanhaesebroeck, B.; Joel, S.; Cutillas, P.R. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 2013, 6, rs6. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.H.; Ascher, D.B.; Pires, D.E. Kinact: A computational approach for predicting activating missense mutations in protein kinases. Nucleic Acids Res. 2018, 46, W127–W132. [Google Scholar] [CrossRef]
- Wiredja, D.D.; Koyuturk, M.; Chance, M.R. The KSEA App: A web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics 2017, 33, 3489–3491. [Google Scholar] [CrossRef]
- Turei, D.; Valdeolivas, A.; Gul, L.; Palacio-Escat, N.; Klein, M.; Ivanova, O.; Olbei, M.; Gabor, A.; Theis, F.; Modos, D.; et al. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. Mol. Syst. Biol. 2021, 17, e9923. [Google Scholar] [CrossRef]
- Wieczorek, S.; Combes, F.; Lazar, C.; Giai Gianetto, Q.; Gatto, L.; Dorffer, A.; Hesse, A.M.; Coute, Y.; Ferro, M.; Bruley, C.; et al. DAPAR & ProStaR: Software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics 2017, 33, 135–136. [Google Scholar] [CrossRef]
- Scholz, C.; Lyon, D.; Refsgaard, J.C.; Jensen, L.J.; Choudhary, C.; Weinert, B.T. Avoiding abundance bias in the functional annotation of post-translationally modified proteins. Nat. Methods 2015, 12, 1003–1004. [Google Scholar] [CrossRef]
- Helmy, M.; Mee, M.; Ranjan, A.; Hao, T.; Vidal, M.; Calderwood, M.A.; Luck, K.; Bader, G.D. OpenPIP: An Open-source Platform for Hosting, Visualizing and Analyzing Protein Interaction Data. J. Mol. Biol. 2022, 434, 167603. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Cai, M.; Xing, X.; Ji, J.; Yang, E.; Wu, J. PINA 3.0: Mining cancer interactome. Nucleic Acids Res. 2020, 49, D1351–D1357. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.J.; Pinese, M.; Kassahn, K.S.; Waddell, N.; Pearson, J.V.; Grimmond, S.M.; Biankin, A.V.; Hautaniemi, S.; Wu, J. PINA v2.0: Mining interactome modules. Nucleic Acids Res. 2012, 40, D862–D865. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.; Brinkman, F.S.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [CrossRef]
- Benito-Martin, A.; Peinado, H. FunRich proteomics software analysis, let the fun begin! Proteomics 2015, 15, 2555–2556. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Chang, C.Y.; Clough, T.; Broudy, D.; Killeen, T.; MacLean, B.; Vitek, O. MSstats: An R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 2014, 30, 2524–2526. [Google Scholar] [CrossRef]
- Yin, X.; Liao, H.; Yun, H.; Lin, N.; Li, S.; Xiang, Y.; Ma, X. Artificial intelligence-based prediction of clinical outcome in immunotherapy and targeted therapy of lung cancer. Semin. Cancer Biol. 2022, 86, 146–159. [Google Scholar] [CrossRef]
- Kawahara, R.; Meirelles, G.V.; Heberle, H.; Domingues, R.R.; Granato, D.C.; Yokoo, S.; Canevarolo, R.R.; Winck, F.V.; Ribeiro, A.C.; Brandao, T.B.; et al. Integrative analysis to select cancer candidate biomarkers to targeted validation. Oncotarget 2015, 6, 43635–43652. [Google Scholar] [CrossRef]
- Swan, A.L.; Mobasheri, A.; Allaway, D.; Liddell, S.; Bacardit, J. Application of machine learning to proteomics data: Classification and biomarker identification in postgenomics biology. OMICS 2013, 17, 595–610. [Google Scholar] [CrossRef]
- Li, X.; Zheng, N.R.; Wang, L.H.; Li, Z.W.; Liu, Z.C.; Fan, H.; Wang, Y.; Dai, J.; Ni, X.T.; Wei, X.; et al. Proteomic profiling identifies signatures associated with progression of precancerous gastric lesions and risk of early gastric cancer. EBioMedicine 2021, 74, 103714. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Ishizawa, K.; Tanaka, A.; Kimura, H.; Kitabatake, K.; Sugano, A.; Edamatsu, K.; Ueda, S.; Iino, M. Identification of Salivary Proteomic Biomarkers for Oral Cancer Screening. Vivo 2021, 35, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Toth, R.; Schiffmann, H.; Hube-Magg, C.; Buscheck, F.; Hoflmayer, D.; Weidemann, S.; Lebok, P.; Fraune, C.; Minner, S.; Schlomm, T.; et al. Random forest-based modelling to detect biomarkers for prostate cancer progression. Clin. Epigenetics 2019, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Leng, W.; Sun, C.; Lu, T.; Chen, Z.; Men, X.; Wang, Y.; Wang, G.; Zhen, B.; Qin, J. Urine Proteome Profiling Predicts Lung Cancer from Control Cases and Other Tumors. EBioMedicine 2018, 30, 120–128. [Google Scholar] [CrossRef]
- Song, C.; Li, X. Cost-Sensitive KNN Algorithm for Cancer Prediction Based on Entropy Analysis. Entropy 2022, 24, 253. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Long, Y.; Li, W.; Dai, W.; Xie, S.; Liu, Y.; Zhang, Y.; Liu, M.; Tian, Y.; Li, Q.; et al. Exploratory study on classification of lung cancer subtypes through a combined K-nearest neighbor classifier in breathomics. Sci. Rep. 2020, 10, 5880. [Google Scholar] [CrossRef]
- Tyanova, S.; Albrechtsen, R.; Kronqvist, P.; Cox, J.; Mann, M.; Geiger, T. Proteomic maps of breast cancer subtypes. Nat. Commun. 2016, 7, 10259. [Google Scholar] [CrossRef]
- Wu, J.; Ji, Y.; Zhao, L.; Ji, M.; Ye, Z.; Li, S. A Mass Spectrometric Analysis Method Based on PPCA and SVM for Early Detection of Ovarian Cancer. Comput. Math. Methods Med. 2016, 2016, 6169249. [Google Scholar] [CrossRef]
- Mann, M.; Kumar, C.; Zeng, W.F.; Strauss, M.T. Artificial intelligence for proteomics and biomarker discovery. Cell Syst. 2021, 12, 759–770. [Google Scholar] [CrossRef]
- Meyer, J.G. Deep learning neural network tools for proteomics. Cell Rep. Methods 2021, 1, 100003. [Google Scholar] [CrossRef]
- Kall, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Degroeve, S.; Martens, L. MS2PIP: A tool for MS/MS peak intensity prediction. Bioinformatics 2013, 29, 3199–3203. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Sheng, J.; Bai, M.; Shu, K.; Zhu, Y.; Chang, C. A Comprehensive Evaluation of MS/MS Spectrum Prediction Tools for Shotgun Proteomics. Proteomics 2020, 20, e1900345. [Google Scholar] [CrossRef] [PubMed]
- Desiere, F.; Deutsch, E.W.; King, N.L.; Nesvizhskii, A.I.; Mallick, P.; Eng, J.; Chen, S.; Eddes, J.; Loevenich, S.N.; Aebersold, R. The PeptideAtlas project. Nucleic Acids Res. 2006, 34, D655–D658. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Wang, M.; Wang, J.; Carver, J.; Pullman, B.S.; Cha, S.W.; Bandeira, N. Assembling the Community-Scale Discoverable Human Proteome. Cell Syst. 2018, 7, 412–421. [Google Scholar] [CrossRef]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef]
- Okuda, S.; Watanabe, Y.; Moriya, Y.; Kawano, S.; Yamamoto, T.; Matsumoto, M.; Takami, T.; Kobayashi, D.; Araki, N.; Yoshizawa, A.C.; et al. jPOSTrepo: An international standard data repository for proteomes. Nucleic Acids Res. 2017, 45, D1107–D1111. [Google Scholar] [CrossRef]
- Sharma, V.; Eckels, J.; Schilling, B.; Ludwig, C.; Jaffe, J.D.; MacCoss, M.J.; MacLean, B. Panorama Public: A Public Repository for Quantitative Data Sets Processed in Skyline. Mol. Cell. Proteom. 2018, 17, 1239–1244. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Gessulat, S.; Schmidt, T.; Zolg, D.P.; Samaras, P.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Rechenberger, J.; Delanghe, B.; Huhmer, A.; et al. Prosit: Proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 2019, 16, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Tiwary, S.; Levy, R.; Gutenbrunner, P.; Salinas Soto, F.; Palaniappan, K.K.; Deming, L.; Berndl, M.; Brant, A.; Cimermancic, P.; Cox, J. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nat. Methods 2019, 16, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhu, Z.; Ye, J.; Yang JPei, J.; Xu, S.; Zhou, R.; Yu, C.; Mo, F.; Wen, B.; Liu, S. DeepRT: Deep learning for peptide retention time time prediction in proteomics. arXiv 2017, arXiv:1705.05368. [Google Scholar]
- Ma, C.; Ren, Y.; Yang, J.; Ren, Z.; Yang, H.; Liu, S. Improved Peptide Retention Time Prediction in Liquid Chromatography through Deep Learning. Anal. Chem. 2018, 90, 10881–10888. [Google Scholar] [CrossRef] [PubMed]
- Serrano, G.; Guruceaga, E.; Segura, V. DeepMSPeptide: Peptide detectability prediction using deep learning. Bioinformatics 2019, 36, 1279–1280. [Google Scholar] [CrossRef] [PubMed]
- Zohora, F.T.; Rahman, M.Z.; Tran, N.H.; Xin, L.; Shan, B.; Li, M. DeepIso: A Deep Learning Model for Peptide Feature Detection from LC-MS map. Sci. Rep. 2019, 9, 17168. [Google Scholar] [CrossRef]
- Tran, N.H.; Zhang, X.; Xin, L.; Shan, B.; Li, M. De novo peptide sequencing by deep learning. Proc. Natl. Acad. Sci. USA 2017, 114, 8247–8252. [Google Scholar] [CrossRef]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Karpathy, A.; Fei-Fei, L. Deep visual-semantic alignments for generating image descriptions. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Boston, MA, USA, 7–12 June 2015; pp. 3128–3137. [Google Scholar]
- Vinyals, O.; Toshev, A.; Bengio, S.; Erhan, D. Show and tell: A neural image caption generator. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Boston, MA, USA, 7–12 June 2015; pp. 3156–3164. [Google Scholar]
- Tran, N.H.; Qiao, R.; Xin, L.; Chen, X.; Liu, C.; Zhang, X.; Shan, B.; Ghodsi, A.; Li, M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat. Methods 2019, 16, 63–66. [Google Scholar] [CrossRef]
- Zolg, D.P.; Gessulat, S.; Paschke, C.; Graber, M.; Rathke-Kuhnert, M.; Seefried, F.; Fitzemeier, K.; Berg, F.; Lopez-Ferrer, D.; Horn, D.; et al. INFERYS rescoring: Boosting peptide identifications and scoring confidence of database search results. Rapid Commun. Mass Spectrom. 2021, e9128. [Google Scholar] [CrossRef]
- Kim, M.; Eetemadi, A.; Tagkopoulos, I. DeepPep: Deep proteome inference from peptide profiles. PLoS Comput. Biol. 2017, 13, e1005661. [Google Scholar] [CrossRef] [PubMed]
- Muntel, J.; Gandhi, T.; Verbeke, L.; Bernhardt, O.M.; Treiber, T.; Bruderer, R.; Reiter, L. Surpassing 10 000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Mol. Omics 2019, 15, 348–360. [Google Scholar] [CrossRef]
- Zhu, Y.; Piehowski, P.D.; Zhao, R.; Chen, J.; Shen, Y.; Moore, R.J.; Shukla, A.K.; Petyuk, V.A.; Campbell-Thompson, M.; Mathews, C.E.; et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells. Nat. Commun. 2018, 9, 882. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Huang, M.; Wang, X.K.; Zhu, Y.; Li, J.S.; Wong, C.C.L.; Fang, Q. Nanoliter-Scale Oil-Air-Droplet Chip-Based Single Cell Proteomic Analysis. Anal. Chem. 2018, 90, 5430–5438. [Google Scholar] [CrossRef] [PubMed]
- Ctortecka, C.H.D.; Seth, A.; Mendjan, S.; Tourniaire, G.; Mechtler, K. An automated workflow for multiplexed single-cell proteomics sample preparation at unprecedented sensitivity. bioRxiv 2022. [Google Scholar] [CrossRef]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161. [Google Scholar] [CrossRef]
- Schoof, E.M.; Furtwangler, B.; Uresin, N.; Rapin, N.; Savickas, S.; Gentil, C.; Lechman, E.; Keller, U.A.D.; Dick, J.E.; Porse, B.T. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021, 12, 3341. [Google Scholar] [CrossRef]
- Cheung, T.K.; Lee, C.Y.; Bayer, F.P.; McCoy, A.; Kuster, B.; Rose, C.M. Defining the carrier proteome limit for single-cell proteomics. Nat. Methods 2021, 18, 76–83. [Google Scholar] [CrossRef]
- Brunner, A.D.; Thielert, M.; Vasilopoulou, C.; Ammar, C.; Coscia, F.; Mund, A.; Hoerning, O.B.; Bache, N.; Apalategui, A.; Lubeck, M.; et al. Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol. Syst. Biol. 2022, 18, e10798. [Google Scholar] [CrossRef]
- Mund, A.; Coscia, F.; Kriston, A.; Hollandi, R.; Kovacs, F.; Brunner, A.D.; Migh, E.; Schweizer, L.; Santos, A.; Bzorek, M.; et al. Deep Visual Proteomics defines single-cell identity and heterogeneity. Nat. Biotechnol. 2022, 40, 1231–1240. [Google Scholar] [CrossRef]
- Mund, A.; Brunner, A.D.; Mann, M. Unbiased spatial proteomics with single-cell resolution in tissues. Mol. Cell 2022, 82, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- An, R.; Yu, H.; Wang, Y.; Lu, J.; Gao, Y.; Xie, X.; Zhang, J. Integrative analysis of plasma metabolomics and proteomics reveals the metabolic landscape of breast cancer. Cancer Metab. 2022, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.; Schallenberg, S.; Kirchner, M.; Ziehm, M.; Niquet, S.; Haji, M.; Beier, C.; Neudecker, J.; Klauschen, F.; Mertins, P. Comprehensive micro-scaled proteome and phosphoproteome characterization of archived retrospective cancer repositories. Nat. Commun. 2021, 12, 3576. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrillo-Rodriguez, P.; Selheim, F.; Hernandez-Valladares, M. Mass Spectrometry-Based Proteomics Workflows in Cancer Research: The Relevance of Choosing the Right Steps. Cancers 2023, 15, 555. https://doi.org/10.3390/cancers15020555
Carrillo-Rodriguez P, Selheim F, Hernandez-Valladares M. Mass Spectrometry-Based Proteomics Workflows in Cancer Research: The Relevance of Choosing the Right Steps. Cancers. 2023; 15(2):555. https://doi.org/10.3390/cancers15020555
Chicago/Turabian StyleCarrillo-Rodriguez, Paula, Frode Selheim, and Maria Hernandez-Valladares. 2023. "Mass Spectrometry-Based Proteomics Workflows in Cancer Research: The Relevance of Choosing the Right Steps" Cancers 15, no. 2: 555. https://doi.org/10.3390/cancers15020555
APA StyleCarrillo-Rodriguez, P., Selheim, F., & Hernandez-Valladares, M. (2023). Mass Spectrometry-Based Proteomics Workflows in Cancer Research: The Relevance of Choosing the Right Steps. Cancers, 15(2), 555. https://doi.org/10.3390/cancers15020555