
Shallow Whole-Genome Sequencing of Cell-Free DNA (cfDNA) Detects Epithelial Ovarian Cancer and Predicts Patient Prognosis
 , ,
, ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Preparation of cfDNA and NGS Data Preparation
2.3. Shallow WGS of cfDNA
2.4. Data Processing for CNV Detection and Z-Score Grouping
2.5. Genome-Wide Instability Score
2.6. Identification of OC-Specific Genes and CNV Validation Using the TCGA Dataset
2.7. Statistical Analysis
3. Results
3.1. Clinical and Pathology Data of Subjects
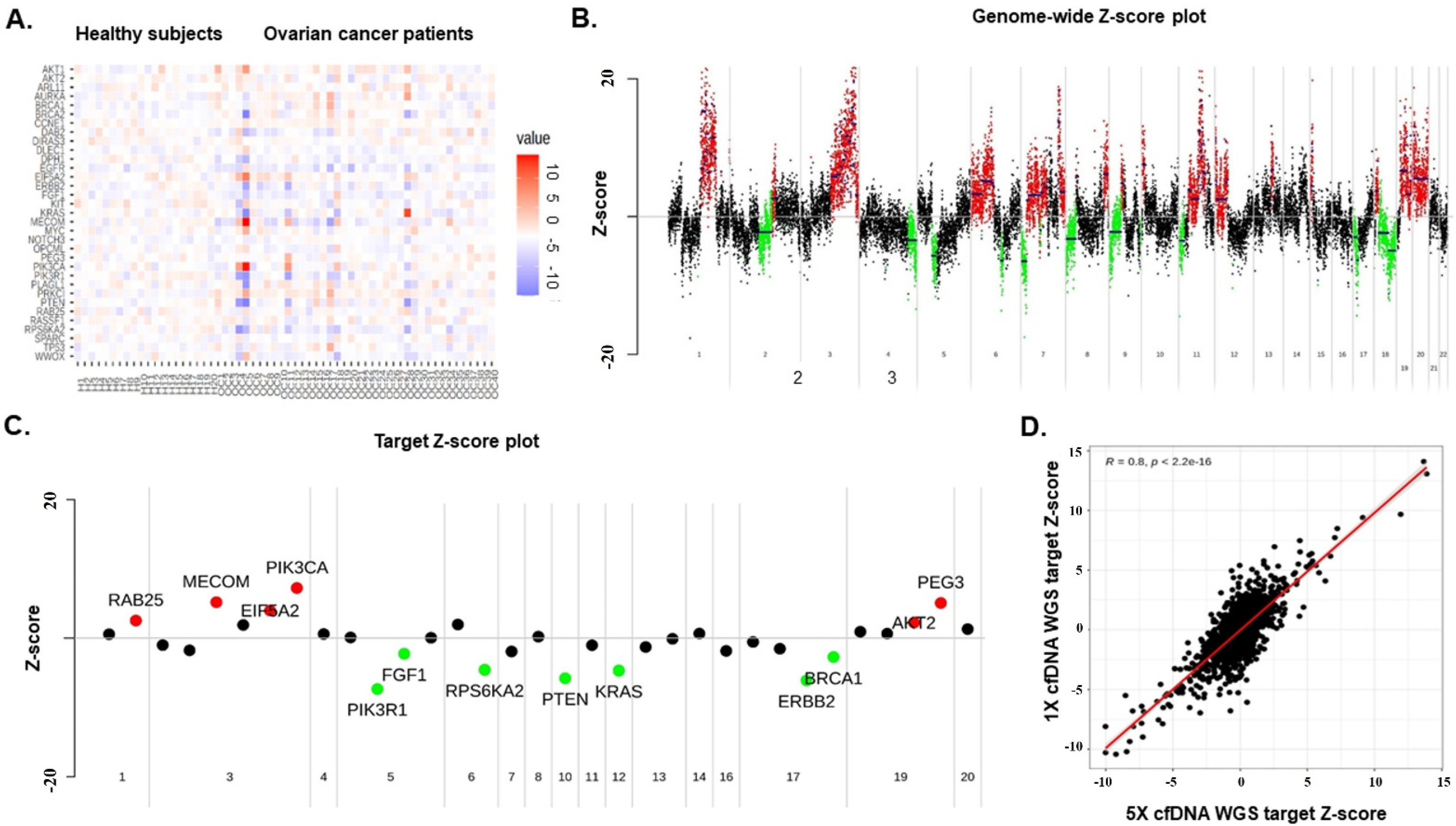
3.2. Genome-Wide Z-Scores from Shallow WGS Detect Chromosomal Instability
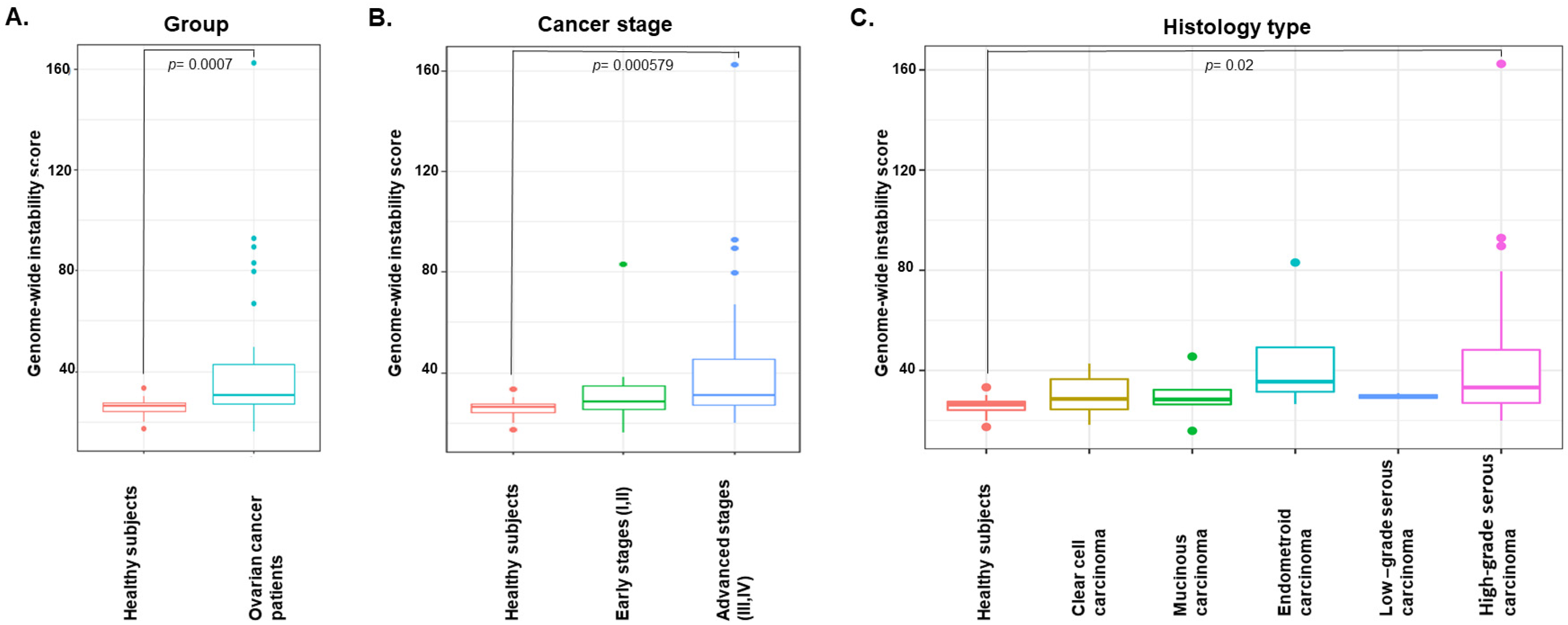
3.3. Genome-Wide Instability by Shallow Whole-Genome Sequencing Characterizes EOC
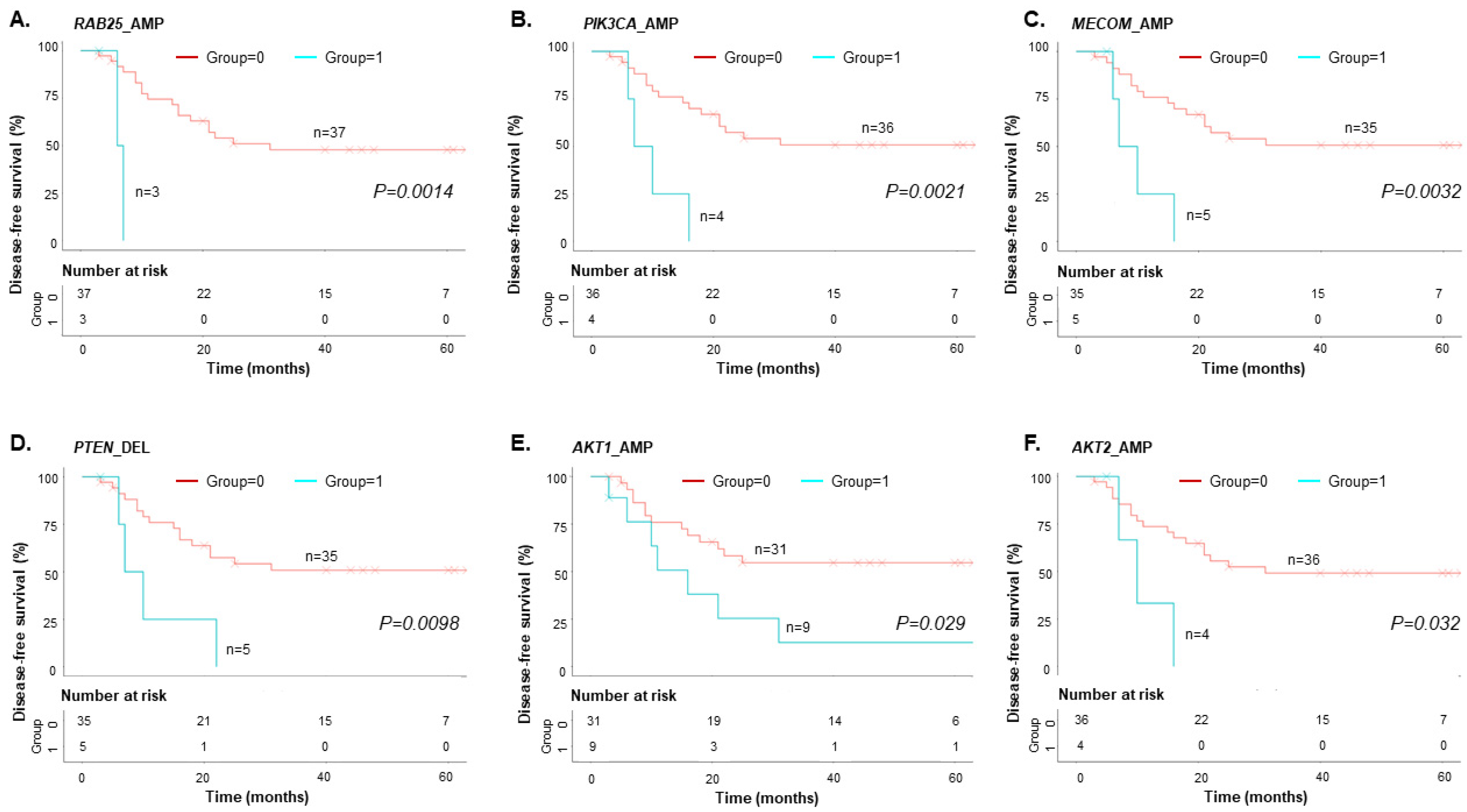
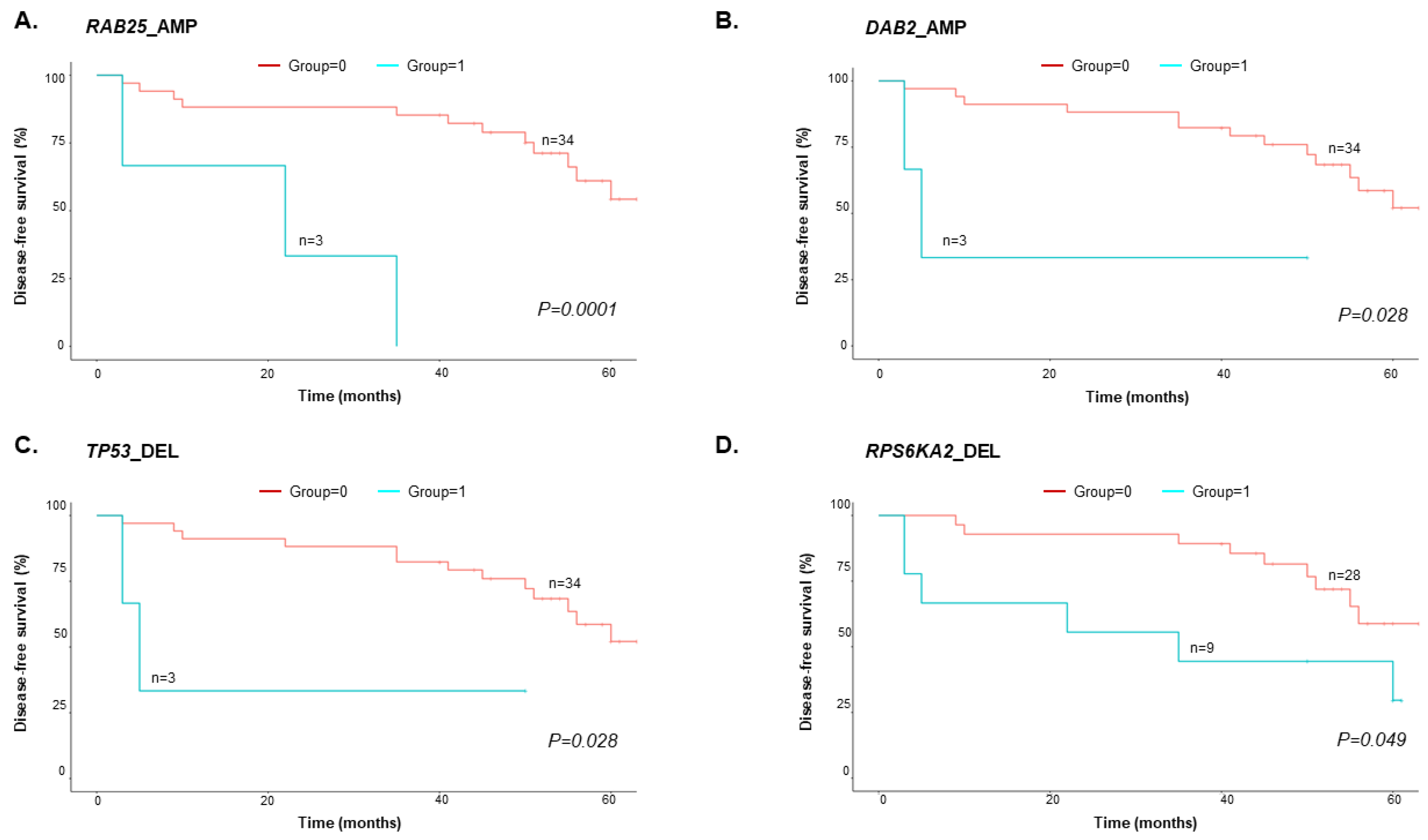
3.4. Copy-Number Variations in cfDNA predict EOC Patient Survival
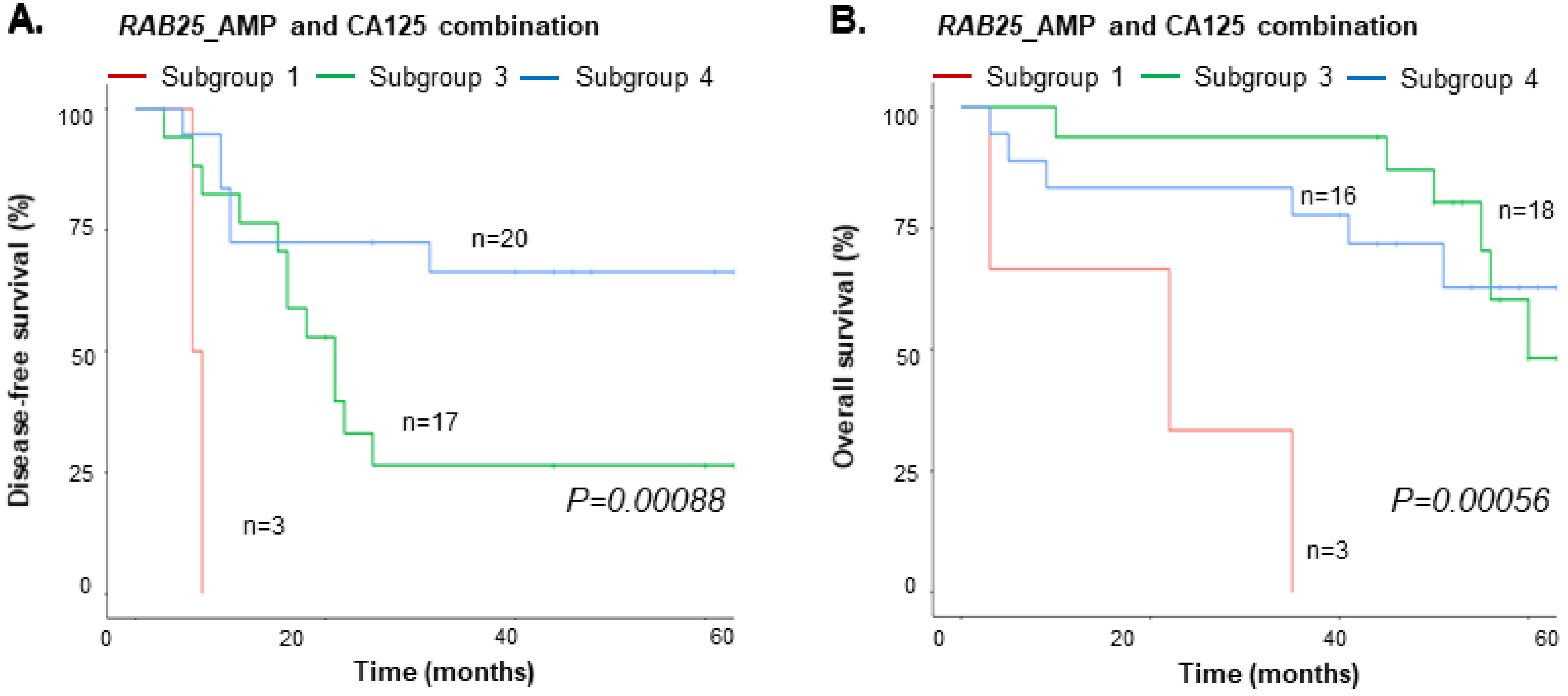
3.5. Integrated Value of cfDNA and CA125
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Hess, L.M.; Rong, N.; Monahan, P.O.; Gupta, P.; Thomaskutty, C.; Matei, D. Continued chemotherapy after complete response to primary therapy among women with advanced ovarian cancer: A meta-analysis. Cancer 2010, 116, 5251–5260. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, K. Treatment for recurrent ovarian cancer-at first relapse. J. Oncol. 2010, 2010, 497429. [Google Scholar] [CrossRef]
- Takaya, H.; Nakai, H.; Takamatsu, S.; Mandai, M.; Matsumura, N. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci. Rep. 2020, 10, 2757. [Google Scholar] [CrossRef] [PubMed]
- Stover, E.H.; Fuh, K.; Konstantinopoulos, P.A.; Matulonis, U.A.; Liu, J.F. Clinical assays for assessment of homologous recombination DNA repair deficiency. Gynecol. Oncol. 2020, 159, 887–898. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef]
- Tamura, N.; Shaikh, N.; Muliaditan, D.; Soliman, T.N.; McGuinness, J.R.; Maniati, E.; Moralli, D.; Durin, M.A.; Green, C.M.; Balkwill, F.R.; et al. Specific Mechanisms of Chromosomal Instability Indicate Therapeutic Sensitivities in High-Grade Serous Ovarian Carcinoma. Cancer Res. 2020, 80, 4946–4959. [Google Scholar] [CrossRef]
- Castellarin, M.; Milne, K.; Zeng, T.; Tse, K.; Mayo, M.; Zhao, Y.; Webb, J.R.; Watson, P.H.; Nelson, B.H.; Holt, R.A. Clonal evolution of high-grade serous ovarian carcinoma from primary to recurrent disease. J. Pathol. 2013, 229, 515–524. [Google Scholar] [CrossRef]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef]
- Reid, B.M.; Permuth, J.B.; Chen, Y.A.; Fridley, B.L.; Iversen, E.S.; Chen, Z.; Jim, H.; Vierkant, R.A.; Cunningham, J.M.; Barnholtz-Sloan, J.S.; et al. Genome-wide Analysis of Common Copy Number Variation and Epithelial Ovarian Cancer Risk. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1117–1126. [Google Scholar] [CrossRef]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J.; Bockmayr, M.; Denkert, C.; Klauschen, F.; Gröschel, S.; Darb-Esfahani, S.; Pfarr, N.; Leichsenring, J.; Onozato, M.L.; Lennerz, J.K.; et al. Pan-cancer analysis of copy number changes in programmed death-ligand 1 (PD-L1, CD274)—Associations with gene expression, mutational load, and survival. Genes Chromosomes Cancer 2016, 55, 626–639. [Google Scholar] [CrossRef]
- Stranger, B.E.; Forrest, M.S.; Dunning, M.; Ingle, C.E.; Beazley, C.; Thorne, N.; Redon, R.; Bird, C.P.; de Grassi, A.; Lee, C.; et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007, 315, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.C.; Birkbak, N.J.; Culhane, A.C.; Drapkin, R.; Fatima, A.; Tian, R.; Schwede, M.; Alsop, K.; Daniels, K.E.; Piao, H.; et al. Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clin. Cancer Res. 2012, 18, 5806–5815. [Google Scholar] [CrossRef] [PubMed]
- Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef] [PubMed]
- Macintyre, G.; Goranova, T.E.; De Silva, D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.A.; Hanif, A.; Wilson, C.; et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Park, R.W.; Kim, T.M.; Kasif, S.; Park, P.J. Identification of rare germline copy number variations over-represented in five human cancer types. Mol. Cancer 2015, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, M.; Schmidt, B. Circulating nucleic acids (CNAs) and cancer—A survey. Biochim. Biophys. Acta 2007, 1775, 181–232. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Venesio, T.; Marsoni, S.; Seoane, J.; Dive, C.; Papadopoulos, N.; Kopetz, S.; Corcoran, R.B.; Siu, L.L.; et al. How liquid biopsies can change clinical practice in oncology. Ann. Oncol. 2019, 30, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [CrossRef] [PubMed]
- Heitzer, E.; Ulz, P.; Belic, J.; Gutschi, S.; Quehenberger, F.; Fischereder, K.; Benezeder, T.; Auer, M.; Pischler, C.; Mannweiler, S.; et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Thusgaard, C.F.; Korsholm, M.; Koldby, K.M.; Kruse, T.A.; Thomassen, M.; Jochumsen, K.M. Epithelial ovarian cancer and the use of circulating tumor DNA: A systematic review. Gynecol. Oncol. 2021, 161, 884–895. [Google Scholar] [CrossRef]
- Steffensen, K.D.; Madsen, C.V.; Andersen, R.F.; Waldstrøm, M.; Adimi, P.; Jakobsen, A. Prognostic importance of cell-free DNA in chemotherapy resistant ovarian cancer treated with bevacizumab. Eur. J. Cancer 2014, 50, 2611–2618. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, Z.H.; Wang, S.; Lang, J.H. Circulating Cell-Free DNA or Circulating Tumor DNA in the Management of Ovarian and Endometrial Cancer. Onco. Targets Ther. 2019, 12, 11517–11530. [Google Scholar] [CrossRef]
- Didion, J.P.; Martin, M.; Collins, F.S. Atropos: Specific, sensitive, and speedy trimming of sequencing reads. PeerJ 2017, 5, e3720. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 2018, 34, 867–868. [Google Scholar] [CrossRef]
- Benjamini, Y.; Speed, T.P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012, 40, e72. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Vanderstichele, A.; Busschaert, P.; Smeets, D.; Landolfo, C.; Van Nieuwenhuysen, E.; Leunen, K.; Neven, P.; Amant, F.; Mahner, S.; Braicu, E.I.; et al. Chromosomal Instability in Cell-Free DNA as a Highly Specific Biomarker for Detection of Ovarian Cancer in Women with Adnexal Masses. Clin. Cancer Res. 2017, 23, 2223–2231. [Google Scholar] [CrossRef] [PubMed]
- Paracchini, L.; Beltrame, L.; Grassi, T.; Inglesi, A.; Fruscio, R.; Landoni, F.; Ippolito, D.; Delle Marchette, M.; Paderno, M.; Adorni, M.; et al. Genome-wide Copy-number Alterations in Circulating Tumor DNA as a Novel Biomarker for Patients with High-grade Serous Ovarian Cancer. Clin. Cancer Res. 2021, 27, 2549–2559. [Google Scholar] [CrossRef] [PubMed]
- Braicu, E.I.; du Bois, A.; Sehouli, J.; Beck, J.; Prader, S.; Kulbe, H.; Eiben, B.; Harter, P.; Traut, A.; Pietzner, K.; et al. Cell-Free-DNA-Based Copy Number Index Score in Epithelial Ovarian Cancer-Impact for Diagnosis and Treatment Monitoring. Cancers 2021, 14, 168. [Google Scholar] [CrossRef]
- Raman, L.; Van der Linden, M.; Van der Eecken, K.; Vermaelen, K.; Demedts, I.; Surmont, V.; Himpe, U.; Dedeurwaerdere, F.; Ferdinande, L.; Lievens, Y.; et al. Shallow whole-genome sequencing of plasma cell-free DNA accurately differentiates small from non-small cell lung carcinoma. Genome Med. 2020, 12, 35. [Google Scholar] [CrossRef]
- Bouzidi, A.; Labreche, K.; Baron, M.; Veyri, M.; Denis, J.A.; Touat, M.; Sanson, M.; Davi, F.; Guillerm, E.; Jouannet, S.; et al. Low-Coverage Whole Genome Sequencing of Cell-Free DNA From Immunosuppressed Cancer Patients Enables Tumor Fraction Determination and Reveals Relevant Copy Number Alterations. Front. Cell Dev. Biol. 2021, 9, 661272. [Google Scholar] [CrossRef]
- Sabatier, R.; Garnier, S.; Guille, A.; Carbuccia, N.; Pakradouni, J.; Adelaide, J.; Provansal, M.; Cappiello, M.; Rousseau, F.; Chaffanet, M.; et al. Whole-genome/exome analysis of circulating tumor DNA and comparison to tumor genomics from patients with heavily pre-treated ovarian cancer: Subset analysis of the PERMED-01 trial. Front. Oncol. 2022, 12, 946257. [Google Scholar] [CrossRef]
- Mitra, S.; Cheng, K.W.; Mills, G.B. Rab25 in cancer: A brief update. Biochem. Soc. Trans. 2012, 40, 1404–1408. [Google Scholar] [CrossRef]
- Cho, K.H.; Lee, H.Y. Rab25 and RCP in cancer progression. Arch. Pharm. Res. 2019, 42, 101–112. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Domchek, S.M.; Nathanson, K.L.; Robson, M.E. Population Frequency of Germline BRCA1/2 Mutations. J. Clin. Oncol. 2016, 34, 4183–4185. [Google Scholar] [CrossRef]
- Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br. J. Cancer 2000, 83, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, S.P.; Huang, T.; Rajendran, B.K.; Guo, M.; Luo, J.; Qin, Z.; Zhao, B.; Chian, J.; Li, S.; Wang, S.M. Ethnic-specific BRCA1/2 variation within Asia population: Evidence from over 78,000 cancer and 40,000 non-cancer cases of Indian, Chinese, Korean and Japanese populations. J. Med. Genet. 2021, 58, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Momozawa, Y.; Iwasaki, Y.; Parsons, M.T.; Kamatani, Y.; Takahashi, A.; Tamura, C.; Katagiri, T.; Yoshida, T.; Nakamura, S.; Sugano, K.; et al. Germline pathogenic variants of 11 breast cancer genes in 7051 Japanese patients and 11,241 controls. Nat. Commun. 2018, 9, 4083. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ovarian Cancer Patients (n = 40) | |
|---|---|
| Age (median) [quartile 1; quartile 3] | 54 yr [47.5; 61.5] |
| FIGO stage | |
| I | 9 |
| II | 2 |
| III | 26 |
| IV | 3 |
| BRCA mutation | |
| Yes | 6 |
| No | 34 |
| Pathologic types | |
| High-grade serous carcinoma | 23 |
| Low-grade serous carcinoma | 3 |
| Mucinous carcinoma | 5 |
| Endometrioid carcinoma | 4 |
| Clear cell carcinoma | 5 |
| Recurrence/ progression | |
| Yes | 20 |
| No | 20 |
| Disease-free time (median) [quartile 1; quartile 3] | 11 mo [7; 19.5] |
| Follow- up time (median) [quartile 1; quartile 3] | 50 mo [37.5; 58] |
| CA125 (U/mL) (median) [quartile 1; quartile 3] | 575.5 [91.5; 1175.8] |
| Chr | Start (hg19) | End (hg19) | Gene | Strand | CNV | Occuring Events_Frequency | TCGA_Frequency | Oncogenic Information |
|---|---|---|---|---|---|---|---|---|
| chr1 | 156,030,965 | 156,040,295 | RAB25 | + | AMP | 7.50% | 7.20% | Oncogene |
| chr3 | 168,801,286 | 169,381,563 | MECOM | − | AMP | 12.50% | 24.70% | Oncogene |
| chr3 | 169,940,219 | 170,023,770 | PRKCI | + | AMP | 15.00% | 22.10% | Oncogene |
| chr3 | 170,606,203 | 170,626,426 | EIF5A2 | − | AMP | 22.50% | 20.70% | Oncogene |
| chr3 | 178,866,310 | 178,952,497 | PIK3CA | + | AMP | 10.00% | 18.00% | Oncogene |
| chr4 | 55,524,094 | 55,606,881 | KIT | + | AMP | 2.50% | 1.20% | Oncogene |
| chr5 | 67,511,583 | 67,597,649 | PIK3R1 | + | DEL | 20.00% | 2.00% | Oncogene |
| chr5 | 141,971,742 | 142,077,635 | FGF1 | − | AMP | 5.00% | 1.00% | Oncogene |
| chr7 | 55,086,724 | 55,275,031 | EGFR | + | AMP | 2.50% | 0.40% | Oncogene |
| chr8 | 128,748,314 | 128,753,680 | MYC | + | AMP | 7.50% | 31.50% | Oncogene |
| chr12 | 25,358,179 | 25,403,854 | KRAS | − | AMP | 2.50% | 9.80% | Oncogene |
| chr14 | 105,235,686 | 105,262,080 | AKT1 | − | AMP | 22.50% | 2.90% | Oncogene |
| chr17 | 37,844,392 | 37,884,915 | ERBB2 | + | AMP | 0.00% | 2.20% | Oncogene |
| chr19 | 15,270,443 | 15,311,792 | NOTCH3 | − | AMP | 0.00% | 11.50% | Oncogene |
| chr19 | 30,302,900 | 30,315,215 | CCNE1 | + | AMP | 2.50% | 21.70% | Oncogene |
| chr19 | 40,736,223 | 40,791,302 | AKT2 | − | AMP | 10.00% | 7.00% | Oncogene |
| chr20 | 54,944,444 | 54,967,351 | AURKA | − | AMP | 10.00% | 3.90% | Oncogene |
| chr1 | 68,511,644 | 68,516,460 | DIRAS3 | − | AMP | 2.50% | 1.00% | Tumor suppressor gene |
| chr3 | 38,080,695 | 38,164,228 | DLEC1 | + | AMP | 2.50% | 0.60% | Tumor suppressor gene |
| chr3 | 50,367,216 | 50,378,367 | RASSF1 | − | AMP | 7.50% | 1.00% | Tumor suppressor gene |
| chr5 | 39,371,775 | 39,425,335 | DAB2 | − | AMP | 10.00% | 3.10% | Tumor suppressor gene |
| chr5 | 151,040,656 | 151,066,615 | SPARC | − | AMP | 2.50% | 1.20% | Tumor suppressor gene |
| chr6 | 144,261,436 | 144,385,735 | PLAGL1 | − | DEL | 12.50% | 0.60% | Tumor suppressor gene |
| chr6 | 166,822,853 | 167,275,771 | RPS6KA2 | − | DEL | 22.50% | 1.40% | Tumor suppressor gene |
| chr10 | 89,623,194 | 89,728,532 | PTEN | + | DEL | 12.50% | 6.10% | Tumor suppressor gene |
| chr11 | 132,284,874 | 133,402,403 | OPCML | − | AMP | 7.50% | 2.50% | Tumor suppressor gene |
| chr13 | 32,889,616 | 32,973,809 | BRCA2 | + | DEL | 10.00% | 0.80% | Tumor suppressor gene |
| chr13 | 50,202,434 | 50,208,008 | ARL11 | + | DEL | 2.50% | 2.20% | Tumor suppressor gene |
| chr16 | 78,133,326 | 79,246,564 | WWOX | + | DEL | 10.00% | 5.70% | Tumor suppressor gene |
| chr17 | 1,933,430 | 1,946,725 | DPH1 | + | AMP | 0.00% | 1.00% | Tumor suppressor gene |
| chr17 | 7,571,719 | 7,590,868 | TP53 | − | DEL | 10.00% | 0.60% | Tumor suppressor gene |
| chr17 | 41,196,311 | 41,277,500 | BRCA1 | − | DEL | 7.50% | 0.60% | Tumor suppressor gene |
| chr19 | 57,321,444 | 57,352,094 | PEG3 | − | AMP | 5.00% | 1.80% | Tumor suppressor gene |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bak, S.E.; Kim, H.; Ho, J.Y.; Cho, E.-H.; Lee, J.; Youn, S.M.; Park, S.-W.; Han, M.-R.; Hur, S.Y.; Lee, S.J.; et al. Shallow Whole-Genome Sequencing of Cell-Free DNA (cfDNA) Detects Epithelial Ovarian Cancer and Predicts Patient Prognosis. Cancers 2023, 15, 530. https://doi.org/10.3390/cancers15020530
Bak SE, Kim H, Ho JY, Cho E-H, Lee J, Youn SM, Park S-W, Han M-R, Hur SY, Lee SJ, et al. Shallow Whole-Genome Sequencing of Cell-Free DNA (cfDNA) Detects Epithelial Ovarian Cancer and Predicts Patient Prognosis. Cancers. 2023; 15(2):530. https://doi.org/10.3390/cancers15020530
Chicago/Turabian StyleBak, Seong Eun, Hanwool Kim, Jung Yoon Ho, Eun-Hae Cho, Junnam Lee, Sung Min Youn, Seong-Woo Park, Mi-Ryung Han, Soo Young Hur, Sung Jong Lee, and et al. 2023. "Shallow Whole-Genome Sequencing of Cell-Free DNA (cfDNA) Detects Epithelial Ovarian Cancer and Predicts Patient Prognosis" Cancers 15, no. 2: 530. https://doi.org/10.3390/cancers15020530
APA StyleBak, S. E., Kim, H., Ho, J. Y., Cho, E.-H., Lee, J., Youn, S. M., Park, S.-W., Han, M.-R., Hur, S. Y., Lee, S. J., & Choi, Y. J. (2023). Shallow Whole-Genome Sequencing of Cell-Free DNA (cfDNA) Detects Epithelial Ovarian Cancer and Predicts Patient Prognosis. Cancers, 15(2), 530. https://doi.org/10.3390/cancers15020530