
Mechanisms of Acquired Resistance and Tolerance to EGFR Targeted Therapy in Non-Small Cell Lung Cancer


Abstract
Simple Summary
Abstract
1. Introduction
2. Targeted Therapy
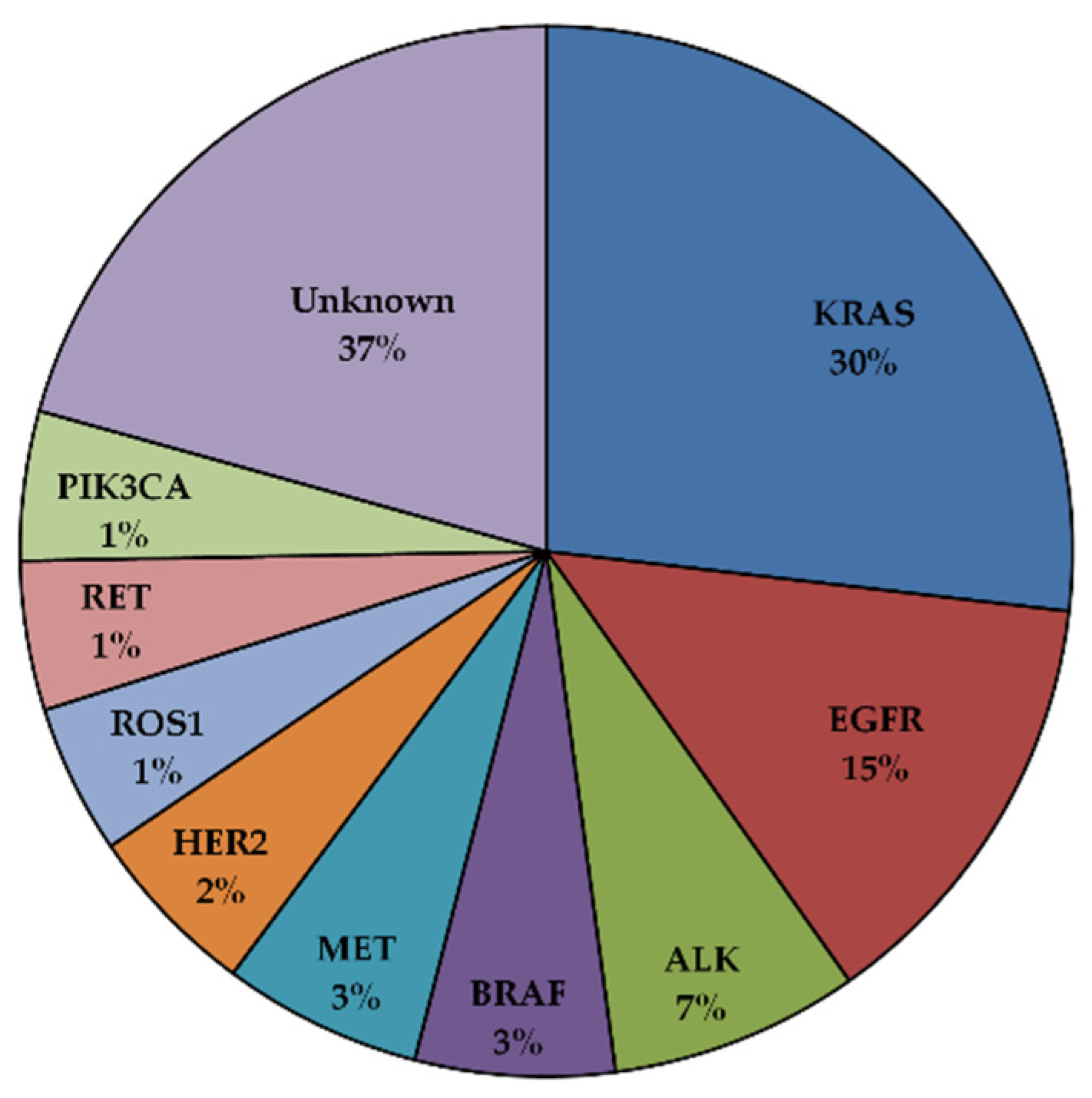
3. EGFR Mutations in NSCLC
4. EGFR Targeted Therapy
4.1. First Generation EGFR-TKIs
4.2. Second Generation EGFR-TKIs
4.3. Third Generation EGFR-TKIs
4.4. Fourth Generation EGFR-TKIs
4.5. EGFR Degraders
5. Drug Tolerant Persister States
5.1. Persisters in NSCLC
5.2. Molecular Characteristics of Drug Tolerance in NSCLC
5.2.1. Epigenetic Modifications
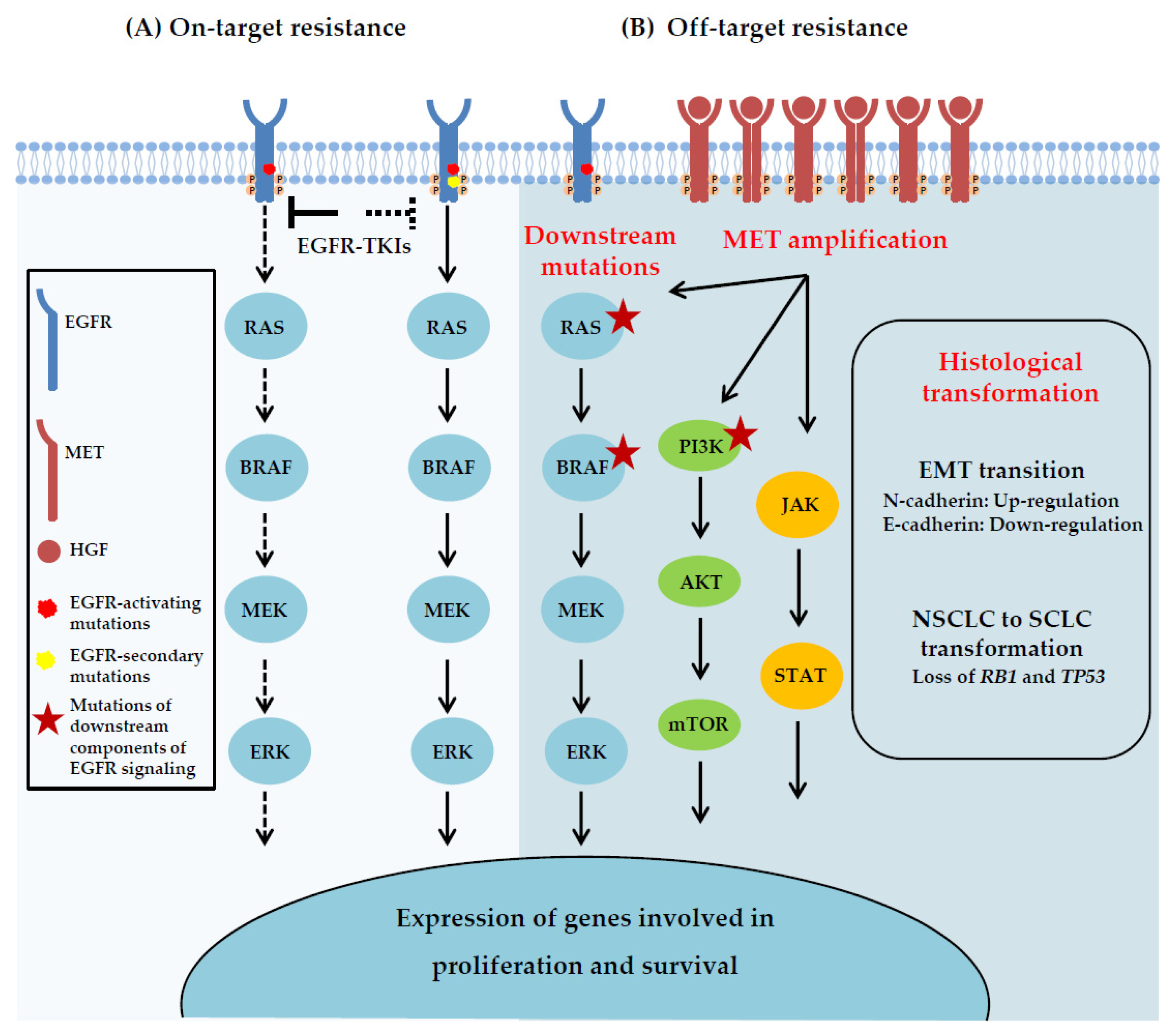
5.2.2. Reactivation of EGFR Signaling and Up-Regulation of Other Pathways
5.2.3. Metabolic Reprogramming
5.3. Origin of DTP Cells: Darwinian Selection or Lamarckian Induction?
6. Cellular Barcoding
6.1. Lentiviral Barcodes to Study Resistance to Anti-Cancer Drugs
6.2. Viral-Barcoding Compatible with Single Cell RNA Sequencing
6.3. CRISPR-Barcoding
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-Cell Lung Cancer: What We Know, What We Need to Know and the Path Forward. Nat. Rev. Cancer 2017, 17, 725–737. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The Biology and Management of Non-Small Cell Lung Cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-Small-Cell Lung Cancers: A Heterogeneous Set of Diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Kenfield, S.A.; Wei, E.K.; Stampfer, M.J.; Rosner, B.A.; Colditz, G.A. Comparison of Aspects of Smoking Among Four Histologic Types of Lung Cancer. Tob. Control 2008, 17, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.R.; Gazdar, A.F.; Clarke, B.E. The Pivotal Role of Pathology in the Management of Lung Cancer. J. Thorac. Dis. 2013, 5 (Suppl. 5), S463–S478. [Google Scholar] [CrossRef] [PubMed]
- Rekhtman, N.; Travis, W.D. Large No More: The Journey of Pulmonary Large Cell Carcinoma from Common to Rare Entity. J. Thorac. Oncol. 2019, 14, 1125–1127. [Google Scholar] [CrossRef]
- Rodak, O.; Peris-Díaz, M.D.; Olbromski, M.; Podhorska-Okołów, M.; Dzięgiel, P. Current Landscape of Non-Small Cell Lung Cancer: Epidemiology, Histological Classification, Targeted Therapies, and Immunotherapy. Cancers 2021, 13, 4705. [Google Scholar] [CrossRef]
- Wang, M.; Herbst, R.S.; Boshoff, C. Toward Personalized Treatment Approaches for Non-Small-Cell Lung Cancer. Nat. Med. 2021, 27, 1345–1356. [Google Scholar] [CrossRef]
- Weinstein, I.B. Addiction to Oncogenes—The Achilles Heal of Cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef]
- Rotow, J.; Bivona, T.G. Understanding and Targeting Resistance Mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Tsao, A.S.; Scagliotti, G.V.; Bunn, P.A.; Carbone, D.P.; Warren, G.W.; Bai, C.; de Koning, H.J.; Yousaf-Khan, A.U.; McWilliams, A.; Tsao, M.S.; et al. Scientific Advances in Lung Cancer 2015. J. Thorac. Oncol. 2016, 11, 613–638. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. The ErbB/HER Family of Protein-Tyrosine Kinases and Cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Au, J.S.-K.; Thongprasert, S.; Srinivasan, S.; Tsai, C.-M.; Khoa, M.T.; Heeroma, K.; Itoh, Y.; Cornelio, G.; Yang, P.-C. A Prospective, Molecular Epidemiology Study of EGFR Mutations in Asian Patients with Advanced Non-Small-Cell Lung Cancer of Adenocarcinoma Histology (PIONEER). J. Thorac. Oncol. 2014, 9, 154–162. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Canepa, H.M.; Bailey, A.S.; Nakayama, S.; Yamaguchi, N.; Goldstein, M.A.; Huberman, M.S.; Costa, D.B. Compound EGFR Mutations and Response to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2013, 8, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Eastwood, M.P.; Zhang, X.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic Mutations Counteract Intrinsic Disorder in the EGFR Kinase and Promote Receptor Dimerization. Cell 2012, 149, 860–870. [Google Scholar] [CrossRef]
- Kate, S.; Chougule, A.; Joshi, A.; Noronha, V.; Patil, V.; Dusane, R.; Solanki, L.; Tiwrekar, P.; Trivedi, V.; Prabhash, K. Outcome of Uncommon EGFR Mutation Positive Newly Diagnosed Advanced Non-Small Cell Lung Cancer Patients: A Single Center Retrospective Analysis. Lung Cancer 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal Growth Factor Receptor Mutations in Lung Cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Lee, C.K.; Wu, Y.-L.; Ding, P.N.; Lord, S.J.; Inoue, A.; Zhou, C.; Mitsudomi, T.; Rosell, R.; Pavlakis, N.; Links, M.; et al. Impact of Specific Epidermal Growth Factor Receptor (EGFR) Mutations and Clinical Characteristics on Outcomes After Treatment With EGFR Tyrosine Kinase Inhibitors Versus Chemotherapy in EGFR-Mutant Lung Cancer: A Meta-Analysis. J. Clin. Oncol. 2015, 33, 1958–1965. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus Cisplatin plus Docetaxel in Patients with Non-Small-Cell Lung Cancer Harbouring Mutations of the Epidermal Growth Factor Receptor (WJTOG3405): An Open Label, Randomised Phase 3 Trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Final Overall Survival Results from a Randomised, Phase III Study of Erlotinib versus Chemotherapy as First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer (OPTIMAL, CTONG-0802). Ann. Oncol. 2015, 26, 1877–1883. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus Standard Chemotherapy as First-Line Treatment for European Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (EURTAC): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, P.E.N.S.; Gergis, C.; Viray, H.; Varkaris, A.; Fujii, M.; Rangachari, D.; VanderLaan, P.A.; Kobayashi, I.S.; Kobayashi, S.S.; Costa, D.B. EGFR-A763_Y764insFQEA Is a Unique Exon 20 Insertion Mutation That Displays Sensitivity to Approved and In-Development Lung Cancer EGFR Tyrosine Kinase Inhibitors. JTO Clin. Res. Rep. 2020, 1, 100051. [Google Scholar] [CrossRef]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of Resistance to EGFR-Targeted Drugs: Lung Cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR TKI Therapy in 155 Patients with EGFR Mutant Lung Cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M Mutation in EGFR Kinase Causes Drug Resistance by Increasing the Affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef]
- Katakami, N.; Atagi, S.; Goto, K.; Hida, T.; Horai, T.; Inoue, A.; Ichinose, Y.; Koboyashi, K.; Takeda, K.; Kiura, K.; et al. LUX-Lung 4: A Phase II Trial of Afatinib in Patients with Advanced Non-Small-Cell Lung Cancer Who Progressed during Prior Treatment with Erlotinib, Gefitinib, or Both. J. Clin. Oncol. 2013, 31, 3335–3341. [Google Scholar] [CrossRef]
- Miller, V.A.; Hirsh, V.; Cadranel, J.; Chen, Y.-M.; Park, K.; Kim, S.-W.; Zhou, C.; Su, W.-C.; Wang, M.; Sun, Y.; et al. Afatinib versus Placebo for Patients with Advanced, Metastatic Non-Small-Cell Lung Cancer after Failure of Erlotinib, Gefitinib, or Both, and One or Two Lines of Chemotherapy (LUX-Lung 1): A Phase 2b/3 Randomised Trial. Lancet Oncol. 2012, 13, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Haspinger, E.R.; Agustoni, F.; Torri, V.; Gelsomino, F.; Platania, M.; Zilembo, N.; Gallucci, R.; Garassino, M.C.; Cinquini, M. Is There Evidence for Different Effects among EGFR-TKIs? Systematic Review and Meta-Analysis of EGFR Tyrosine Kinase Inhibitors (TKIs) versus Chemotherapy as First-Line Treatment for Patients Harboring EGFR Mutations. Crit. Rev. Oncol. Hematol. 2015, 94, 213–227. [Google Scholar] [CrossRef]
- Yang, Z.; Hackshaw, A.; Feng, Q.; Fu, X.; Zhang, Y.; Mao, C.; Tang, J. Comparison of Gefitinib, Erlotinib and Afatinib in Non-Small Cell Lung Cancer: A Meta-Analysis. Int. J. Cancer 2017, 140, 2805–2819. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.-H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel Mutant-Selective EGFR Kinase Inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Yang, J.C.-H.; Kim, D.-W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.-J.; Kim, S.-W.; Su, W.-C.; Horn, L.; et al. AZD9291 in EGFR Inhibitor–Resistant Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Soria, J.-C.; Goldman, J.W.; Wakelee, H.A.; Gadgeel, S.M.; Varga, A.; Papadimitrakopoulou, V.; Solomon, B.J.; Oxnard, G.R.; Dziadziuszko, R.; et al. Rociletinib in EGFR-Mutated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Van Der Steen, N.; Caparello, C.; Rolfo, C.; Pauwels, P.; Peters, G.J.; Giovannetti, E. New Developments in the Management of Non-Small-Cell Lung Cancer, Focus on Rociletinib: What Went Wrong? Onco Targets Ther. 2016, 9, 6065–6074. [Google Scholar] [CrossRef]
- Jänne, P.A.; Ahn, M.-J.; Kim, D.-W.; Kim, S.-W.; Planchard, D.; Ramalingam, S.S.; Frewer, P.; Cantarini, M.; Ghiorghiu, S.; Yang, J.C.-H. A Phase I Study of AZD9291 in Patients with Egfr-Tki-Resistant Advanced Nsclc—Updated Progression Free Survival and Duration of Response Data. Ann. Oncol. 2015, 26, i57. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.-J.; Han, J.-Y.; Lee, K.H.; Kim, S.-W.; Kim, D.-W.; Lee, Y.-G.; Cho, E.K.; Kim, J.-H.; Lee, G.-W.; Lee, J.-S.; et al. Lazertinib in Patients with EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer: Results from the Dose Escalation and Dose Expansion Parts of a First-in-Human, Open-Label, Multicentre, Phase 1–2 Study. Lancet Oncol. 2019, 20, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.-H.; Camidge, D.R.; Yang, C.-T.; Zhou, J.; Guo, R.; Chiu, C.-H.; Chang, G.-C.; Shiah, H.-S.; Chen, Y.; Wang, C.-C.; et al. Safety, Efficacy, and Pharmacokinetics of Almonertinib (HS-10296) in Pretreated Patients With EGFR-Mutated Advanced NSCLC: A Multicenter, Open-Label, Phase 1 Trial. J. Thorac. Oncol. 2020, 15, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S Mediates Resistance to AZD9291 in Advanced Non-Small Cell Lung Cancer Harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.A.; Wu, Y.-L.; Han, J.-Y.; Ahn, M.-J.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.-H.; Bulusu, K.C.; Laus, G.; et al. Analysis of Resistance Mechanisms to Osimertinib in Patients with EGFR T790M Advanced NSCLC from the AURA3 Study. Ann. Oncol. 2018, 29, viii741. [Google Scholar] [CrossRef]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.-C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of Acquired Resistance to First-Line Osimertinib: Preliminary Data from the Phase III FLAURA Study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non–Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef]
- Zhang, Y.; He, B.; Zhou, D.; Li, M.; Hu, C. Newly Emergent Acquired EGFR Exon 18 G724S Mutation after Resistance of a T790M Specific EGFR Inhibitor Osimertinib in Non-Small-Cell Lung Cancer: A Case Report. Onco Targets Ther. 2018, 12, 51–56. [Google Scholar] [CrossRef]
- Lin, C.-C.; Shih, J.-Y.; Yu, C.-J.; Ho, C.-C.; Liao, W.-Y.; Lee, J.-H.; Tsai, T.-H.; Su, K.-Y.; Hsieh, M.-S.; Chang, Y.-L.; et al. Outcomes in Patients with Non-Small-Cell Lung Cancer and Acquired Thr790Met Mutation Treated with Osimertinib: A Genomic Study. Lancet Respir. Med. 2018, 6, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Jänne, P.A.; Mok, T.; Peters, S. Overcoming Therapy Resistance in EGFR-Mutant Lung Cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, Z.; Isozaki, H.; Lennerz, J.K.; Gainor, J.F.; Lennes, I.T.; Zhu, V.W.; Marcoux, N.; Banwait, M.K.; Digumarthy, S.R.; Su, W.; et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov. 2018, 8, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.-H.; Chen, L.-Y.; Lin, Y.-C.; Shih, J.-Y.; Lin, Y.-C.; Tseng, R.-Y.; Chiu, A.-C.; Yeh, Y.-H.; Liu, C.; Lin, Y.-T.; et al. Epithelial-Mesenchymal Transition (EMT) beyond EGFR Mutations per Se Is a Common Mechanism for Acquired Resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) Resistance with Mutant-Selective Allosteric Inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- To, C.; Beyett, T.S.; Jang, J.; Feng, W.W.; Bahcall, M.; Haikala, H.M.; Shin, B.H.; Heppner, D.E.; Rana, J.K.; Leeper, B.A.; et al. An Allosteric Inhibitor against the Therapy-Resistant Mutant Forms of EGFR in Non-Small Cell Lung Cancer. Nat. Cancer 2022, 3, 402–417. [Google Scholar] [CrossRef]
- Ferlenghi, F.; Scalvini, L.; Vacondio, F.; Castelli, R.; Bozza, N.; Marseglia, G.; Rivara, S.; Lodola, A.; La Monica, S.; Minari, R.; et al. A Sulfonyl Fluoride Derivative Inhibits EGFRL858R/T790M/C797S by Covalent Modification of the Catalytic Lysine. Eur. J. Med. Chem. 2021, 225, 113786. [Google Scholar] [CrossRef]
- Spigel, D.; Goto, K.; Camidge, D.R.; Elamin, Y.; de Langen, A.J.; Leighl, N.B.; Minchom, A.; Piotrowska, Z.; Planchard, D.; Reckamp, K.; et al. Abstract P230: A Phase 1/2 Study of BLU-945, a Highly Potent and Selective Inhibitor of Epidermal Growth Factor Receptor (EGFR) Resistance Mutations, in Patients with EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC). Mol. Cancer Ther. 2021, 20, P230. [Google Scholar] [CrossRef]
- Lim, S.M.; Kim, D.-W.; Jung, J.E.; Lee, G.; Ryou, J.-H.; Kang, S.-U.; Lee, Y.-H.; Shin, H.-J.; Yum, S.Y.; Yim, E.; et al. 1365TiP A Phase I/II, Open-Label Study of BBT-176, a Triple Mutation Targeting EGFR TKI, in Patients with NSCLC Who Progressed after Prior EGFR TKI Therapy. Ann. Oncol. 2021, 32, S1035. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, X.; Yang, L.; Tian, X.; Dong, T.; Ding, C.Z.; Hu, L.; Wu, L.; Zhao, L.; Mao, J.; et al. Abstract 1320: Preclinical Evaluation of TQB3804, a Potent EGFR C797S Inhibitor. Cancer Res. 2019, 79, 1320. [Google Scholar] [CrossRef]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef]
- Dale, B.; Cheng, M.; Park, K.-S.; Kaniskan, H.Ü.; Xiong, Y.; Jin, J. Advancing Targeted Protein Degradation for Cancer Therapy. Nat. Rev. Cancer 2021, 21, 638–654. [Google Scholar] [CrossRef]
- Qu, X.; Liu, H.; Song, X.; Sun, N.; Zhong, H.; Qiu, X.; Yang, X.; Jiang, B. Effective Degradation of EGFRL858R+T790M Mutant Proteins by CRBN-Based PROTACs through Both Proteosome and Autophagy/Lysosome Degradation Systems. Eur. J. Med. Chem. 2021, 218, 113328. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, H.-Y.; Xi, X.-X.; Liu, Y.-J.; Xin, M.; Mao, S.; Zhang, J.-J.; Lu, A.-X.; Zhang, S.-Q. Discovery of Potent Epidermal Growth Factor Receptor (EGFR) Degraders by Proteolysis Targeting Chimera (PROTAC). Eur. J. Med. Chem. 2020, 189, 112061. [Google Scholar] [CrossRef]
- Zhao, H.-Y.; Yang, X.-Y.; Lei, H.; Xi, X.-X.; Lu, S.-M.; Zhang, J.-J.; Xin, M.; Zhang, S.-Q. Discovery of Potent Small Molecule PROTACs Targeting Mutant EGFR. Eur. J. Med. Chem. 2020, 208, 112781. [Google Scholar] [CrossRef]
- Du, Y.; Chen, Y.; Wang, Y.; Chen, J.; Lu, X.; Zhang, L.; Li, Y.; Wang, Z.; Ye, G.; Zhang, G. HJM-561, a Potent, Selective, and Orally Bioavailable EGFR PROTAC That Overcomes Osimertinib-Resistant EGFR Triple Mutations. Mol. Cancer Ther. 2022, 21, 1060–1066. [Google Scholar] [CrossRef]
- Bhang, H.C.; Ruddy, D.A.; Krishnamurthy Radhakrishna, V.; Caushi, J.X.; Zhao, R.; Hims, M.M.; Singh, A.P.; Kao, I.; Rakiec, D.; Shaw, P.; et al. Studying Clonal Dynamics in Response to Cancer Therapy Using High-Complexity Barcoding. Nat. Med. 2015, 21, 440–448. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.C.; Krishnamurthy Radhakrishna, V.; et al. Tumor Cells Can Follow Distinct Evolutionary Paths to Become Resistant to Epidermal Growth Factor Receptor Inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Pre-Existence and Clonal Selection of MET Amplification in EGFR Mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef]
- Marine, J.-C.; Dawson, S.-J.; Dawson, M.A. Non-Genetic Mechanisms of Therapeutic Resistance in Cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef]
- Shen, S.; Vagner, S.; Robert, C. Persistent Cancer Cells: The Deadly Survivors. Cell 2020, 183, 860–874. [Google Scholar] [CrossRef]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent Bacterial Infections and Persister Cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef]
- Russo, M.; Sogari, A.; Bardelli, A. Adaptive Evolution: How Bacteria and Cancer Cells Survive Stressful Conditions and Drug Treatment. Cancer Discov. 2021, 11, 1886–1895. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.-H.; Thurley, K.; et al. Diverse Drug-Resistance Mechanisms Can Emerge from Drug-Tolerant Cancer Persister Cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- Touil, Y.; Igoudjil, W.; Corvaisier, M.; Dessein, A.-F.; Vandomme, J.; Monté, D.; Stechly, L.; Skrypek, N.; Langlois, C.; Grard, G.; et al. Colon Cancer Cells Escape 5FU Chemotherapy-Induced Cell Death by Entering Stemness and Quiescence Associated with the c-Yes/YAP Axis. Clin. Cancer Res. 2014, 20, 837–846. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and Adaptive Resistance to BRAF(V600E) Inhibition in Melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e7. [Google Scholar] [CrossRef]
- Swayden, M.; Chhouri, H.; Anouar, Y.; Grumolato, L. Tolerant/Persister Cancer Cells and the Path to Resistance to Targeted Therapy. Cells 2020, 9, 2601. [Google Scholar] [CrossRef]
- Guler, G.D.; Tindell, C.A.; Pitti, R.; Wilson, C.; Nichols, K.; KaiWai Cheung, T.; Kim, H.-J.; Wongchenko, M.; Yan, Y.; Haley, B.; et al. Repression of Stress-Induced LINE-1 Expression Protects Cancer Cell Subpopulations from Lethal Drug Exposure. Cancer Cell 2017, 32, 221–237.e13. [Google Scholar] [CrossRef]
- Ercan, D.; Xu, C.; Yanagita, M.; Monast, C.S.; Pratilas, C.A.; Montero, J.; Butaney, M.; Shimamura, T.; Sholl, L.; Ivanova, E.V.; et al. Reactivation of ERK Signaling Causes Resistance to EGFR Kinase Inhibitors. Cancer Discov. 2012, 2, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Tricker, E.M.; Xu, C.; Uddin, S.; Capelletti, M.; Ercan, D.; Ogino, A.; Pratilas, C.A.; Rosen, N.; Gray, N.S.; Wong, K.-K.; et al. Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer. Cancer Discov. 2015, 5, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL Confers Intrinsic Resistance to Osimertinib and Advances the Emergence of Tolerant Cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL Kinase Causes Resistance to EGFR-Targeted Therapy in Lung Cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Lu, Z.; Zhang, Y.; Xue, D.; Xia, H.; She, J.; Li, F. Integrin Β3 Promotes Resistance to EGFR-TKI in Non-Small-Cell Lung Cancer by Upregulating AXL through the YAP Pathway. Cells 2022, 11, 2078. [Google Scholar] [CrossRef] [PubMed]
- Noronha, A.; Belugali Nataraj, N.; Lee, J.S.; Zhitomirsky, B.; Oren, Y.; Oster, S.; Lindzen, M.; Mukherjee, S.; Will, R.; Ghosh, S.; et al. AXL and Error-Prone DNA Replication Confer Drug Resistance and Offer Strategies to Treat EGFR-Mutant Lung Cancer. Cancer Discov. 2022, 12, 2666–2683. [Google Scholar] [CrossRef]
- Shah, K.N.; Bhatt, R.; Rotow, J.; Rohrberg, J.; Olivas, V.; Wang, V.E.; Hemmati, G.; Martins, M.M.; Maynard, A.; Kuhn, J.; et al. Aurora Kinase A Drives the Evolution of Resistance to Third-Generation EGFR Inhibitors in Lung Cancer. Nat. Med. 2019, 25, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Yu, H.A.; Yang, S.; Han, S.; Selcuklu, S.D.; Kim, K.; Ramani, S.; Ganesan, Y.T.; Moyer, A.; Sinha, S.; et al. Targeting Aurora B Kinase Prevents and Overcomes Resistance to EGFR Inhibitors in Lung Cancer by Enhancing BIM- and PUMA-Mediated Apoptosis. Cancer Cell 2021, 39, 1245–1261.e6. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e12. [Google Scholar] [CrossRef]
- Arasada, R.R.; Shilo, K.; Yamada, T.; Zhang, J.; Yano, S.; Ghanem, R.; Wang, W.; Takeuchi, S.; Fukuda, K.; Katakami, N.; et al. Notch3-Dependent β-Catenin Signaling Mediates EGFR TKI Drug Persistence in EGFR Mutant NSCLC. Nat. Commun. 2018, 9, 3198. [Google Scholar] [CrossRef]
- Maynard, A.; McCoach, C.E.; Rotow, J.K.; Harris, L.; Haderk, F.; Kerr, D.L.; Yu, E.A.; Schenk, E.L.; Tan, W.; Zee, A.; et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 2020, 182, 1232–1251.e22. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Mikubo, M.; Inoue, Y.; Liu, G.; Tsao, M.-S. Mechanism of Drug Tolerant Persister Cancer Cells: The Landscape and Clinical Implication for Therapy. J. Thorac. Oncol. 2021, 16, 1798–1809. [Google Scholar] [CrossRef]
- Raha, D.; Wilson, T.R.; Peng, J.; Peterson, D.; Yue, P.; Evangelista, M.; Wilson, C.; Merchant, M.; Settleman, J. The Cancer Stem Cell Marker Aldehyde Dehydrogenase Is Required to Maintain a Drug-Tolerant Tumor Cell Subpopulation. Cancer Res. 2014, 74, 3579–3590. [Google Scholar] [CrossRef] [PubMed]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-Tolerant Persister Cancer Cells Are Vulnerable to GPX4 Inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare Cell Variability and Drug-Induced Reprogramming as a Mode of Cancer Drug Resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Torre, E.A.; Arai, E.; Bayatpour, S.; Jiang, C.L.; Beck, L.E.; Emert, B.L.; Shaffer, S.M.; Mellis, I.A.; Fane, M.E.; Alicea, G.M.; et al. Genetic Screening for Single-Cell Variability Modulators Driving Therapy Resistance. Nat. Genet. 2021, 53, 76–85. [Google Scholar] [CrossRef]
- Goyal, Y.; Dardani, I.P.; Busch, G.T.; Emert, B.; Fingerman, D.; Kaur, A.; Jain, N.; Mellis, I.A.; Li, J.; Kiani, K.; et al. Pre-Determined Diversity in Resistant Fates Emerges from Homogenous Cells after Anti-Cancer Drug Treatment. bioRxiv 2021, 471833. [Google Scholar] [CrossRef]
- Pisco, A.O.; Brock, A.; Zhou, J.; Moor, A.; Mojtahedi, M.; Jackson, D.; Huang, S. Non-Darwinian Dynamics in Therapy-Induced Cancer Drug Resistance. Nat. Commun. 2013, 4, 2467. [Google Scholar] [CrossRef]
- Su, Y.; Wei, W.; Robert, L.; Xue, M.; Tsoi, J.; Garcia-Diaz, A.; Moreno, B.H.; Kim, J.; Ng, R.H.; Lee, J.W.; et al. Single-Cell Analysis Resolves the Cell State Transition and Signaling Dynamics Associated with Melanoma Drug-Induced Resistance. Proc. Natl. Acad. Sci. USA 2017, 114, 13679–13684. [Google Scholar] [CrossRef] [PubMed]
- Moris, N.; Pina, C.; Arias, A.M. Transition States and Cell Fate Decisions in Epigenetic Landscapes. Nat. Rev. Genet. 2016, 17, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; van Oudenaarden, A. Nature, Nurture, or Chance: Stochastic Gene Expression and Its Consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef]
- Pauklin, S.; Vallier, L. The Cell-Cycle State of Stem Cells Determines Cell Fate Propensity. Cell 2013, 155, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hütter, J.-C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling Cancer Persister Cells Arise from Lineages with Distinct Programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef]
- Dhimolea, E.; de Matos Simoes, R.; Kansara, D.; Al’Khafaji, A.; Bouyssou, J.; Weng, X.; Sharma, S.; Raja, J.; Awate, P.; Shirasaki, R.; et al. An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence. Cancer Cell 2021, 39, 240–256.e11. [Google Scholar] [CrossRef]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.L.; Bruce, J.P.; Wintersinger, J.A.; Mer, A.S.; Lo, E.B.L.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242.e21. [Google Scholar] [CrossRef]
- Bramlett, C.; Jiang, D.; Nogalska, A.; Eerdeng, J.; Contreras, J.; Lu, R. Clonal Tracking Using Embedded Viral Barcoding and High-Throughput Sequencing. Nat. Protoc. 2020, 15, 1436–1458. [Google Scholar] [CrossRef]
- Kebschull, J.M.; Zador, A.M. Cellular Barcoding: Lineage Tracing, Screening and Beyond. Nat. Methods 2018, 15, 871–879. [Google Scholar] [CrossRef]
- Biddy, B.A.; Kong, W.; Kamimoto, K.; Guo, C.; Waye, S.E.; Sun, T.; Morris, S.A. Single-Cell Mapping of Lineage and Identity in Direct Reprogramming. Nature 2018, 564, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, C.; Rodriguez-Fraticelli, A.; Camargo, F.; Klein, A.M. Lineage Tracing on Transcriptional Landscapes Links State to Fate during Differentiation. Science 2020, 367, eaaw3381. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.T.; Shanahan, F.; Nguyen, T.T.T.; Staben, S.T.; Gazzard, L.; Yamazoe, S.; Wertz, I.E.; Piskol, R.; Yang, Y.A.; Modrusan, Z.; et al. Identifying Transcriptional Programs Underlying Cancer Drug Response with TraCe-Seq. Nat. Biotechnol. 2022, 40, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Guernet, A.; Mungamuri, S.K.; Cartier, D.; Sachidanandam, R.; Jayaprakash, A.; Adriouch, S.; Vezain, M.; Charbonnier, F.; Rohkin, G.; Coutant, S.; et al. CRISPR-Barcoding for Intratumor Genetic Heterogeneity Modeling and Functional Analysis of Oncogenic Driver Mutations. Mol. Cell 2016, 63, 526–538. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Drugs | Clinical Trial | Approval | CSF Concentration | EGFR Sensitizing Mutations | EGFR Binding |
|---|---|---|---|---|---|---|
| First generation | Gefitinib | NCT01203917 | FDA/EMA approved | Low | Del19/L858R | Reversible Competitive |
| Erlotinib | NCT00446225 | FDA/EMA approved | ||||
| Icotinib | NCT01040780 | Approved in China | ||||
| Second generation | Afatinib | NCT01466660 | FDA/EMA approved | Low | Del19/L858R/ T790M | Irreversible Covalent |
| Dacomitinib | NCT01774721 | FDA/EMA approved | ||||
| Third generation | WZ4002 | NA | Preclinical | NA | Del19/L858R/ T790M | Irreversible Covalent |
| Rociletinib | NCT01526928 | Rejected | Low | |||
| Osimertinib | NCT02296125 | FDA/EMA approved | High | |||
| Lazertinib | NCT03046992 | Approved in South Korea | High | |||
| Olmutinib | NCT01588145 | Approved in South Korea * | NA | |||
| Avitinib | NCT02330367 | Phase I/II (Active) | Low | |||
| Nazartinib | NCT02108964 | Phase I/II (Active) | NA | |||
| Mavelertinib | NCT02349633 | Phase I/II (Terminated) | NA | |||
| Naquotinib | NCT02588261 | Phase III (Terminated) | NA | |||
| Almonertinib | NCT02981108 | Approved in China | Effective in patient with BM | Del19/L858R/ G719X/L861Q/ T790M | ||
| Alflutinib | NCT03127449 | Approved in China | ||||
| Fourth generation | EAI001 | NA | Preclinical | NA | L858R/T790M/ C797S | Reversible Allosteric |
| EAI045 | NA | Preclinical | NA | |||
| JBJ-09-063 | NA | Preclinical | NA | |||
| BLU945 | NCT04862780 | Phase I/II (Recruiting) | NA | Del19/L858R/ T790M/C797S | Unknown | |
| BBT176 | NCT04820023 | Phase I/II (Recruiting) | NA | |||
| TQB3804 | NCT04128085 | Phase I (Unknown) | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chhouri, H.; Alexandre, D.; Grumolato, L. Mechanisms of Acquired Resistance and Tolerance to EGFR Targeted Therapy in Non-Small Cell Lung Cancer. Cancers 2023, 15, 504. https://doi.org/10.3390/cancers15020504
Chhouri H, Alexandre D, Grumolato L. Mechanisms of Acquired Resistance and Tolerance to EGFR Targeted Therapy in Non-Small Cell Lung Cancer. Cancers. 2023; 15(2):504. https://doi.org/10.3390/cancers15020504
Chicago/Turabian StyleChhouri, Houssein, David Alexandre, and Luca Grumolato. 2023. "Mechanisms of Acquired Resistance and Tolerance to EGFR Targeted Therapy in Non-Small Cell Lung Cancer" Cancers 15, no. 2: 504. https://doi.org/10.3390/cancers15020504
APA StyleChhouri, H., Alexandre, D., & Grumolato, L. (2023). Mechanisms of Acquired Resistance and Tolerance to EGFR Targeted Therapy in Non-Small Cell Lung Cancer. Cancers, 15(2), 504. https://doi.org/10.3390/cancers15020504