
TP53 Mutation in Acute Myeloid Leukemia: An Old Foe Revisited


Abstract
Simple Summary
Abstract
1. TP53—The First Tumor Suppressor Gene to Be Discovered
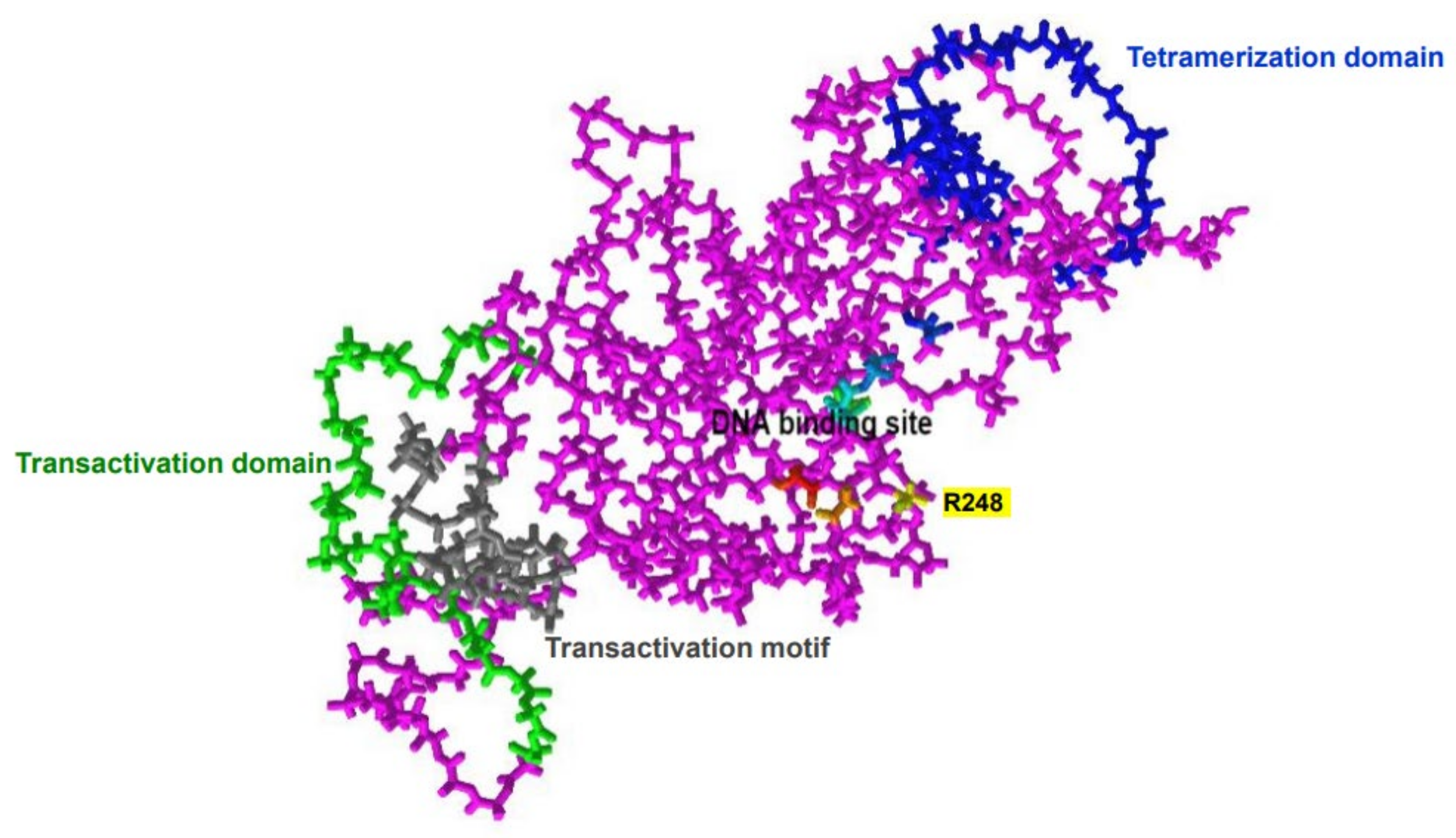
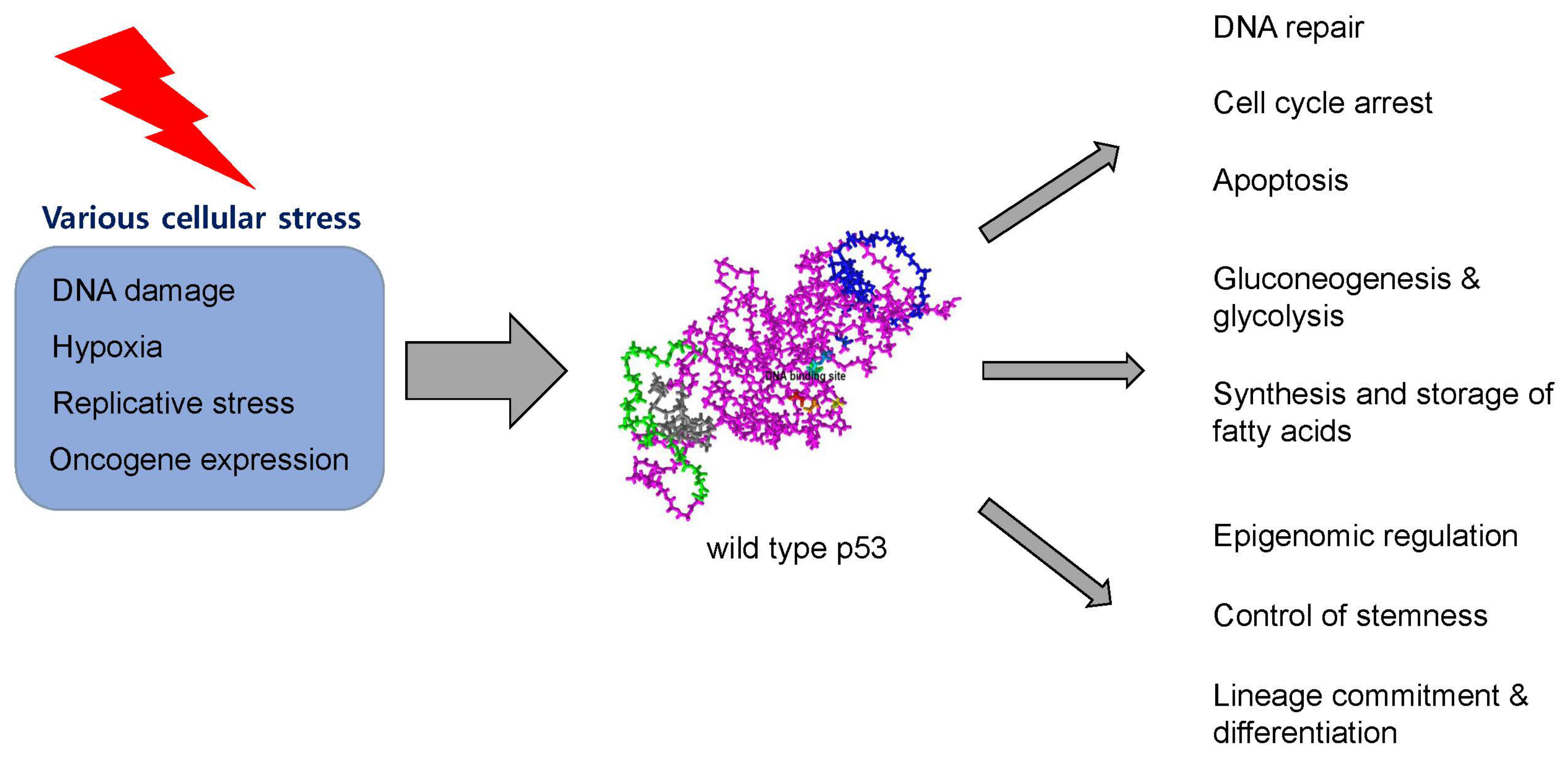
2. Structure and Physiologic Function of p53 Protein
3. Types of TP53 Mutations and Their Effects on p53 Proteins
4. Scoring Systems of TP53 Mutations for Clinical Application
5. Evolution of TP53-Mutated Preleukemic HSPCs to AML
6. Genetic Characteristics of TP53-Mutated AML
7. Despair and Hope in TP53-Mutated AML: Is the Future Bright?
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Finlay, C.A.; Hinds, P.W.; Levine, A.J. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989, 57, 1083–1093. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Strasser, A.; Kelly, G.L. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026062. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.-L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A Global Map of p53 Transcription-Factor Binding Sites in the Human Genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef]
- Nicholls, C.D.; McLure, K.G.; Shields, M.A.; Lee, P.W.K. Biogenesis of p53 Involves Cotranslational Dimerization of Monomers and Posttranslational Dimerization of Dimers. Implications on the dominant negative effect. J. Biol. Chem. 2002, 277, 12937–12945. [Google Scholar] [CrossRef] [PubMed]
- Anbarasan, T.; Bourdon, J.-C. The Emerging Landscape of p53 Isoforms in Physiology, Cancer and Degenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6257. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.-C.; Fernandez, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xiromandis, D.P.; Saville, M.K.; Lane, D.P. Faculty Opinions recommendation of p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef]
- Marcel, V.; Perrier, S.; Aoubala, M.; Ageorges, S.; Groves, M.J.; Diot, A.; Fernandez, K.; Tauro, S.; Bourdon, J.-C. Delta160p53 is a novel N-terminal p53 isoform encoded by Delta133p53 transcript. FEBS Lett. 2010, 584, 4463–4468. [Google Scholar] [CrossRef] [PubMed]
- Steffens Reinhardt, L.; Zhang, X.; Wawruszak, A.; Groen, K.; De Iuliis, G.N.; Avery-Kiejda, K.A. Good Cop, Bad Cop: Defining the Roles of Delta40p53 in Cancer and Aging. Cancers 2020, 12, 1659. [Google Scholar] [CrossRef]
- Wei, J.; Zaika, E.; Zaika, A. p53 Family: Role of Protein Isoforms in Human Cancer. J. Nucleic Acids 2012, 2012, 687359. [Google Scholar] [CrossRef]
- Joruiz, S.M.; Bourdon, J.-C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2019, 33, 2–22. [Google Scholar] [CrossRef]
- Laptenko, O.; Prives, C. p53: Master of life, death, and the epigenome. Genes Dev. 2017, 31, 955–956. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Puzio-Kuter, A.M.; Chan, C.S.; Hainaut, P. The Role of the p53 Protein in Stem-Cell Biology and Epigenetic Regulation. Cold Spring Harb. Perspect. Med. 2016, 6, a026153. [Google Scholar] [CrossRef]
- Monti, P.; Ciribilli, Y.; Jordan, J.; Menichini, P.; Umbach, D.M.; Resnick, M.A.; Luzzatto, L.; Inga, A.; Fronza, G. Transcriptional functionality of germ line p53 mutants influences cancer phenotype. Clin. Cancer Res. 2007, 13, 3789–3795. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Perfumo, C.; Bisio, A.; Ciribilli, Y.; Menichini, P.; Russo, D.; Umbach, D.M.; Resnick, M.A.; Inga, A.; Fronza, G. Dominant-negative features of mutant TP53 in germline carriers have limited impact on cancer outcomes. Mol. Cancer Res. 2011, 9, 271–279. [Google Scholar] [CrossRef]
- Natan, E.; Hirschberg, D.; Morgner, N.; Robinson, C.V.; Fersht, A.R. Ultraslow oligomerization equilibria of p53 and its implications. Proc. Natl. Acad. Sci. USA 2009, 106, 14327–14332. [Google Scholar] [CrossRef]
- Donehower, L.A. Insights into Wild-Type and Mutant p53 Functions Provided by Genetically Engineered Mice. Hum. Mutat. 2014, 35, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.-A.; Liu, G.; Rao, V.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of Function of a p53 Hot Spot Mutation in a Mouse Model of Li-Fraumeni Syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 Gain of Function in Two Mouse Models of Li-Fraumeni Syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Inga, A.; Resnick, M.A. The Biological Impact of the Human Master Regulator p53 Can Be Altered by Mutations That Change the Spectrum and Expression of Its Target Genes. Mol. Cell. Biol. 2006, 26, 2297–2308. [Google Scholar] [CrossRef]
- Saller, E.; Tom, E.; Brunori, M.; Otter, M.; Estreicher, A.; Mack, D.H.; Iggo, R. Increased apoptosis induction by 121F mutant p53. EMBO J. 1999, 18, 4424–4437. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Han, S.-Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef]
- Monti, P.; Bosco, B.; Gomes, S.; Saraiva, L.; Fronza, G.; Inga, A. Yeast As a Chassis for Developing Functional Assays to Study Human P53. J. Vis. Exp. 2019, 150, e59071. [Google Scholar]
- Resnick, M.A.; Inga, A. Functional mutants of the sequence-specific transcription factor p53 and implications for master genes of diversity. Proc. Natl. Acad. Sci. USA 2003, 100, 9934–9939. [Google Scholar] [CrossRef]
- Šmardová, J.; Šmarda, J.; Koptíková, J. Functional analysis of p53 tumor suppressor in yeast. Differentiation 2005, 73, 261–277. [Google Scholar] [CrossRef]
- Monti, P.; Menichini, P.; Speciale, A.; Cutrona, G.; Fais, F.; Taiana, E.; Neri, A.; Bomben, R.; Gentile, M.; Ferrarini, A.; et al. Heterogeneity of TP53 Mutations and P53 Protein Residual Function in Cancer: Does It Matter? Front. Oncol. 2020, 10, 593383. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Inga, A.; Snipe, J.; Krysiak, O.; Schönfelder, G.; Resnick, M.A. A Single-Nucleotide Polymorphism in a Half-Binding Site Creates p53 and Estrogen Receptor Control of Vascular Endothelial Growth Factor Receptor 1. Mol. Cell Biol. 2007, 27, 2590–2600. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Poeta, M.L.; Manola, J.; Goldwasser, M.A.; Forastiere, A.; Benoit, N.; Califano, J.A.; Ridge, J.A.; Goodwin, J.; Kenady, D.; Saunders, J.; et al. TP53Mutations and Survival in Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2007, 357, 2552–2561. [Google Scholar] [CrossRef]
- Neskey, D.M.; Osman, A.A.; Ow, T.J.; Katsonis, P.; McDonald, T.; Hicks, S.C.; Hsu, T.-K.; Pickering, C.R.; Ward, A.; Patel, A.; et al. Evolutionary Action Score of TP53 Identifies High-Risk Mutations Associated with Decreased Survival and Increased Distant Metastases in Head and Neck Cancer. Cancer Res. 2015, 75, 1527–1536. [Google Scholar] [CrossRef]
- Kotler, E.; Shani, O.; Goldfeld, G.; Lotan-Pompan, M.; Tarcic, O.; Gershoni, A.; Hopf, A.; Marks, D.S.; Oren, M.; Segal, E. A Systematic p53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation. Mol. Cell. 2018, 71, 873. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Pregartner, G.; Rücker, F.G.; Heitzer, E.; Zebisch, A.; Bullinger, L.; Berghold, A.; Döhner, K.; Sill, H. Functional Classification of TP53 Mutations in Acute Myeloid Leukemia. Cancers 2020, 12, 637. [Google Scholar] [CrossRef]
- Prochazka, K.T.; Pregartner, G.; Rücker, F.G.; Heitzer, E.; Pabst, G.; Wölfler, A.; Zebisch, A.; Berghold, A.; Döhner, K.; Sill, H. Clinical implications of subclonal TP53 mutations in acute myeloid leukemia. Haematologica 2019, 104, 516–523. [Google Scholar] [CrossRef]
- Smith, S.M.; Le Beau, M.M.; Huo, D.; Karrison, T.; Sobecks, R.M.; Anastasi, J.; Vardiman, J.W.; Rowley, J.D.; Larson, R.A. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: The University of Chicago series. Blood 2003, 102, 43–52. [Google Scholar] [CrossRef]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S. Faculty Opinions recommendation of Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 551–555. [Google Scholar] [CrossRef]
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V. Faculty Opinions recommendation of Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef]
- Yan, B.; Claxton, D.; Huang, S.; Qiu, Y. AML chemoresistance: The role of mutant TP53 subclonal expansion and therapy strategy. Exp. Hematol. 2020, 87, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsey, R.C.; Mermel, C.R.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Linton, P.J.; Dorshkind, K. Age-related changes in lymphocyte development and function. Nat. Immunol. 2004, 5, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Guralnik, J.M.; Eisenstaedt, R.S.; Ferrucci, L.; Klein, H.G.; Woodman, R.C. Prevalence of anemia in persons 65 years and older in the United States: Evidence for a high rate of unexplained anemia. Blood 2004, 104, 2263–2268. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Klesse, S.; Köhler, L.; Gabdoulline, R.; Kloos, A.; Liebich, A.; Wichmann, M.; Chaturvedi, A.; Fabisch, J.; Gaidzik, V.I.; et al. Acute myeloid leukemia derived from lympho-myeloid clonal hematopoiesis. Leukemia 2017, 31, 1286–1295. [Google Scholar] [CrossRef]
- Han, H.; Byun, J.M.; Shin, D.-Y.; Yoon, S.-S.; Koh, Y.; Hong, J.; Kim, I.; Lee, C.; Yoo, H.; Yun, H.; et al. Leukemic stem cell phenotype is associated with mutational profile in acute myeloid leukemia. Korean J. Intern. Med. 2021, 36, 401–412. [Google Scholar] [CrossRef]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal Evolution of Preleukemic Hematopoietic Stem Cells Precedes Human Acute Myeloid Leukemia. Sci. Transl. Med. 2012, 4, 149ra118. [Google Scholar] [CrossRef]
- Majeti, R. Clonal evolution of pre-leukemic hematopoietic stem cells precedes human acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2014, 27, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Saeed, B.R.; Manta, L.; Raffel, S.; Pyl, P.T.; Buss, E.C.; Wang, W.; Eckstein, V.; Jauch, A.; Trumpp, A.; Huber, W.; et al. Analysis of nonleukemic cellular subcompartments reconstructs clonal evolution of acute myeloid leukemia and identifies therapy-resistant preleukemic clones. Int. J. Cancer 2021, 148, 2825–2838. [Google Scholar] [CrossRef]
- Shin, D.-Y. Human acute myeloid leukemia stem cells: Evolution of concept. Blood Res. 2022, 57, S67–S74. [Google Scholar] [CrossRef]
- Stengel, A.; Baer, C.; Walter, W.; Meggendorfer, M.; Kern, W.; Haferlach, T.; Haferlach, C. Mutational patterns and their correlation to CHIP-related mutations and age in hematological malignancies. Blood Adv. 2021, 5, 4426–4434. [Google Scholar] [CrossRef]
- Volkert, S.; Kohlmann, A.; Schnittger, S.; Kern, W.; Haferlach, T.; Haferlach, C. Association of the type of 5q loss with complex karyotype, clonal evolution, TP53 mutation status, and prognosis in acute myeloid leukemia and myelodysplastic syndrome. Genes Chromosom. Cancer 2014, 53, 402–410. [Google Scholar] [CrossRef]
- Weinberg, O.K.; Siddon, A.J.; Madanat, Y.F.; Gagan, J.; Arber, D.A.; Cin, P.D.; Narayanan, D.; Ouseph, M.M.; Kurzer, J.H.; Hasserjian, R.P. TP53 mutation defines a unique subgroup within complex karyotype de novo and therapy-related MDS/AML. Blood Adv. 2022, 6, 2847–2853. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Tashakori, M.; Kadia, T.M.; Loghavi, S.; Daver, N.G.; Kanagal-Shamanna, R.; Pierce, S.R.; Sui, D.; Wei, P.; Khodakarami, F.; Tang, Z.; et al. TP53 copy number and protein expression inform mutation status across risk categories in acute myeloid leukemia. Blood 2022, 140, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.; Takahashi, K.; Borthkaur, G.; Jabbour, E.; Konopoleva, M.; Daver, N.G.; et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer 2016, 122, 3484–3491. [Google Scholar] [CrossRef]
- Tolomeo, D.; L’Abbate, A.; Lonoce, A.; D’Addabbo, P.; Miccoli, M.; Cunsolo, C.L.; Iuzzolino, P.; Palumbo, O.; Carella, M.; Racanelli, V.; et al. Concurrent chromothripsis events in a case of TP53 depleted acute myeloid leukemia with myelodysplasia-related changes. Cancer Genet. 2019, 237, 63–68. [Google Scholar] [CrossRef]
- Abel, H.J.; Oetjen, K.A.; Miller, C.A.; Ramakrishnan, S.M.; Day, R.B.; Helton, N.M.; Fronick, C.C.; Fulton, R.S.; Heath, S.E.; Tarnawsky, S.P.; et al. Genomic landscape of TP53-mutated myeloid malignancies. Blood Adv. 2023, 7, 4586–4598. [Google Scholar] [CrossRef]
- Vadakekolathu, J.; Lai, C.; Reeder, S.; Church, S.E.; Hood, T.; Lourdusamy, A.; Rettig, M.P.; Aldoss, I.; Advani, A.S.; Godwin, J.; et al. TP53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy in AML. Blood Adv. 2020, 4, 5011–5024. [Google Scholar] [CrossRef] [PubMed]
- Abolhalaj, M.; Sincic, V.; Lilljebjorn, H.; Sanden, C.; Aab, A.; Hagerbrand, K.; Ellmark, P.; Borrebaeck, C.A.K.; Fioretos, T.; Lundberg, K. Transcriptional profiling demonstrates altered characteristics of CD8(+) cytotoxic T-cells and regulatory T-cells in TP53-mutated acute myeloid leukemia. Cancer Med. 2022, 11, 3023–3032. [Google Scholar] [CrossRef]
- Li, L.; Li, M.; Wang, X. Cancer type-dependent correlations between TP53 mutations and antitumor immunity. DNA Repair 2020, 88, 102785. [Google Scholar] [CrossRef]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopoleva, M.; Ravandi- Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Boss, I.W.; Beach, C.L.; Copeland, W.B.; Thompson, E.G.; Fox, B.A.; Hasle, V.E.; Hellmann, A.; Taussig, D.C.; Tormo, M.; et al. A randomized phase 2 trial of azacitidine with or without durvalumab as first-line therapy for older patients with AML. Blood Adv. 2022, 6, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Konopleva, M.; Maiti, A.; Kadia, T.M.; DiNardo, C.D.; Loghavi, S.; Pemmaraju, N.; Jabbour, E.J.; Montalban-Bravo, G.; Tang, G.; et al. Phase I/II Study of Azacitidine (AZA) with Venetoclax (VEN) and Magrolimab (Magro) in Patients (pts) with Newly Diagnosed Older/Unfit or High-Risk Acute Myeloid Leukemia (AML) and Relapsed/Refractory (R/R) AML. Blood 2021, 138, 371. [Google Scholar] [CrossRef]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B. Faculty Opinions recommendation of TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Maiti, A.; Loghavi, S.; Pourebrahim, R.; Kadia, T.M.; Rausch, C.R.; Furudate, K.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; et al. Outcomes of TP53 -mutant acute myeloid leukemia with decitabine and venetoclax. Cancer 2021, 127, 3772–3781. [Google Scholar] [CrossRef]
- Roboz, G.J.; Mandrekar, S.J.; Desai, P.; Laumann, K.; Walker, A.R.; Wang, E.S.; Kolitz, E.S.; Powell, B.E.; Attar, E.C.; Stock, W.; et al. Randomized trial of 10 days of decitabine +/− bortezomib in untreated older patients with AML: CALGB 11002 (Alliance). Blood Adv. 2018, 2, 3608–3617. [Google Scholar] [CrossRef] [PubMed]
- Duong, V.H.; Ruppert, A.S.; Mims, A.S.; Borate, U.; Stein, E.M.; Baer, M.R.; Stock, W.; Kovacsovics, T.; Blum, W.; Arellano, M.L.; et al. Entospletinib with decitabine in acute myeloid leukemia with mutant TP53 or complex karyotype: A phase 2 substudy of the Beat AML Master Trial. Cancer 2023, 129, 2308–2320. [Google Scholar] [CrossRef]
- Huls, G.; Chitu, D.A.; Pabst, T.; Klein, S.K.; Stussi, G.; Griskevicius, L.; Valk, P.J.M.; Cloos, J.; van de Loosdrecht, A.A.; Breems, D.; et al. Ibrutinib added to 10-day decitabine for older patients with AML and higher risk MDS. Blood Adv. 2020, 4, 4267–4277. [Google Scholar] [CrossRef]
- Kadia, T.M.; Cortes, J.; Ravandi, F.; Jabbour, E.; Konopleva, M.; Benton, C.B.; Burger, J.; Sasaki, K.; Borthakur, G.; DiNardo, C.D.; et al. Cladribine and low-dose cytarabine alternating with decitabine as front-line therapy for elderly patients with acute myeloid leukaemia: A phase 2 single-arm trial. Lancet Haematol. 2018, 5, e411–e421. [Google Scholar] [CrossRef] [PubMed]
- Bally, C.; Adès, L.; Renneville, A.; Sebert, M.; Eclache, V.; Preudhomme, C.; Mozziconacci, M.-J.; de The, H.; Lehmann-Che, J.; Fenaux, P. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk. Res. 2014, 38, 751–755. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Saliba, A.N.; Kaufmann, S.H.; Stein, E.M.; Patel, P.A.; Baer, M.R.; Stock, W.; Deininger, M.; Blum, W.; Schiller, G.J.; Olin, R.L.; et al. Pevonedistat with azacitidine in older patients with TP53-mutated AML: A phase 2 study with laboratory correlates. Blood Adv. 2023, 7, 2360–2363. [Google Scholar] [CrossRef]
- Yee, K.; Papayannidis, C.; Vey, N.; Dickinson, M.J.; Kelly, K.R.; Assouline, S.; Kasner, M.; Seiter, K.; Drummond, M.K.; Yoon, S.Y.; et al. Murine double minute 2 inhibition alone or with cytarabine in acute myeloid leukemia: Results from an idasanutlin phase 1/1b study small star, filled. Leuk. Res. 2021, 100, 106489. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen, Y.-B.; Deeg, H.J.; Sallman, D.; Gallacher, P.; Wennborg, A.; et al. Eprenetapopt Plus Azacitidine After Allogeneic Hematopoietic Stem-Cell Transplantation for TP53-Mutant Acute Myeloid Leukemia and Myelodysplastic Syndromes. J. Clin. Oncol. 2022, 40, 3985–3993. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Goldberg, A.D.; Winer, E.S.; Altman, J.K.; Fathi, A.T.; Odenike, O.; Roboz, G.J.; Sweet, K.; Miller, C.; Wennborg, A.; et al. Eprenetapopt combined with venetoclax and azacitidine in TP53-mutated acute myeloid leukaemia: A phase 1, dose-finding and expansion study. Lancet Haematol. 2023, 10, e272–e283. [Google Scholar] [CrossRef]
- Penter, L.; Liu, Y.; Wolff, J.O.; Yang, L.; Taing, L.; Jhaveri, A.; Southard, J.; Patel, M.; Cullen, N.M.; Pfaff, K.L.; et al. Mechanisms of response and resistance to combined decitabine and ipilimumab for advanced myeloid disease. Blood 2023, 141, 1817–1830. [Google Scholar] [CrossRef]
- Garcia, J.S.; Flamand, Y.; Penter, L.; Keng, M.; Tomlinson, B.K.; Mendez, L.M.; Koller, P.; Cullen, N.; Arihara, Y.; Pfaff, K.; et al. Ipilimumab plus decitabine for patients with MDS or AML in posttransplant or transplant-naïve settings. Blood 2023, 141, 1884–1888. [Google Scholar] [CrossRef]
- Qin, G.; Han, X. The Prognostic Value of TP53 Mutations in Adult Acute Myeloid Leukemia: A Meta-Analysis. Transfus. Med. Hemother. 2022, 50, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Zarif, M.; Zhou, Q.; Capo-Chichi, J.-M.; Schuh, A.; Minden, M.D.; Atenafu, E.G.; Kumar, R.; Chang, H. TP53 Mutations in AML Patients Are Associated with Dismal Clinical Outcome Irrespective of Frontline Induction Regimen and Allogeneic Hematopoietic Cell Transplantation. Cancers 2023, 15, 3210. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Montalban-Bravo, G.; Hwang, H.; Ning, J.; Franquiz, M.J.; Kanagal-Shamanna, R.; Patel, K.P.; DiNardo, C.D.; Ravandi, F.; Garcia-Manero, G.; et al. Prognostic and therapeutic impacts of mutant TP53 variant allelic frequency in newly diagnosed acute myeloid leukemia. Blood Adv. 2020, 4, 5681–5689. [Google Scholar] [CrossRef]
- Venugopal, S.; Shoukier, M.; Konopleva, M.; Dinardo, C.D.; Ravandi, F.; Short, N.J.; Andreeff, M.; Borthakur, G.; Daver, N.; Pemmaraju, N.; et al. Outcomes in patients with newly diagnosed TP53-mutated acute myeloid leukemia with or without venetoclax-based therapy. Cancer 2021, 127, 3541–3551. [Google Scholar] [CrossRef]
- Lambert, J.M.R.; Grozov, F.; Veprintsev, D.B.; Soderqvist, M.; Sagerback, D.; Bergman, J.; Fresht, A.E.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. Faculty Opinions recommendation of PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef]
- Salim, K.Y.; Vareki, S.M.; Danter, W.R.; San-Marina, S.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.-L.; Liang, Y.; Tang, Y.-G.; Song, H.-X.; Wu, L.-L.; Xing, Y.-F.; Yan, N.; Li, Y.-T.; Wang, Z.-Y.; et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2021, 39, 225–239.e8. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Braicu, I.; Berger, R.; Mahner, S.; Sehouli, J.; Pujade-Lauraine, E.; Cassier, P.A.; Moll, U.M.; Ulmer, H.; Leunen, K.; et al. Part I of GANNET53: A European Multicenter Phase I/II Trial of the Hsp90 Inhibitor Ganetespib Combined with Weekly Paclitaxel in Women with High-Grade, Platinum-Resistant Epithelial Ovarian Cancer—A Study of the GANNET53 Consortium. Front. Oncol. 2019, 9, 832. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.J.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients with TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Badar, T.; Atallah, E.; Shallis, R.; Saliba, A.N.; Patel, A.; Bewersdorf, J.P.; Grenet, J.; Stahl, M.; Duvall, A.; Burkart, M.; et al. Survival of TP53-mutated acute myeloid leukemia patients receiving allogeneic stem cell transplantation after first induction or salvage therapy: Results from the Consortium on Myeloid Malignancies and Neoplastic Diseases (COMMAND). Leukemia 2023, 37, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Iqbal, S.; Huang, J.; Renard, C.; Lin, J.; Pan, Y.; Williamson, M.; Ramsingh, G. Clinical characteristics and overall survival among acute myeloid leukemia patients with TP53 gene mutation or chromosome 17p deletion. Am. J. Hematol. 2023, 98, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Hsiue, E.H.-C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Phase | Study Population | Treatment and Mechanism of Action | TP53-Mutated AML Sub-Population | Response | Survival | Trial ID | Ref. |
|---|---|---|---|---|---|---|---|---|
| Hypomethylating Agents and Their Combinations | ||||||||
| Decitabine | II | AML/MDS with adverse-risk karyotype (n = 116) | 10-day decitabine (Hypomethylating agent) | 10% (12 TP53-mutated AML, 9 TP53-mutated MDS) | CR/CRi/MLFS 46% in all MDS/AML, CR/CRi/MLFS 100% (21/21) in TP53-mutated MDS/AML | mOS 12.7 months in TP53-mutated MDS/AML, mOS 15.4 months in TP53-WT MDS/AML | NCT01687400 | [67] |
| Decitabine + venetoclax | II | Newly diagnosed AML (n = 118) | 10-day decitabine + venetoclax (BCL-2 inhibitor) | 30% (35 TP53-mutated AML) | ORR 66% and CR/CRi 57% in mTP53 AML, ORR 89% and CR/CRi 77% in TP53-WT AML | mOS 5.2 months in TP53-mutated AML, 19.4 months in TP53-WT AML | NCT03404193 | [68] |
| Decitabine ± bortezomib | II, randomized | Newly diagnosed elderly AML (n = 163) | 10-day decitabine vs. decitabine + bortezomib (proteasome inhibitor) | 22% | CR 21% in TP53-mutated AML treated with decitabine, CR 17% in TP53-mutated AML treated with decitabine plus bortezomib | 1 yr OS 18% in TP53-mutated AML, 51% in TP53-WT AML | NCT014230926 | [69] |
| Decitabine + entospletinib | II | Newly diagnosed elderly AML with TP53 mutation (cohort A, n = 45) or complex karyotype (cohort B, n = 13) | 10-day decitabine + entospletinib (Syk inhibitor) | 78% (45/58) | CR/CRi and CR/CRi/MLFS 33.3% and 48.9% in TP53-mutated AML, CR/CRi and CR/CRi/MLFS 61.5% and 76.9% in TP53-WT and complex karyotype | mOS 6.5 months in TP53-mutated AML, mOS 11.5 months in TP53-WT and complex karyotype | NCT03013998 | [70] |
| Decitabine ± ibrutinib | II, randomized | Untreated AML (n = 144) | 10-day decitabine (n = 72) vs. decitabine + ibrutinib (n = 72) (BTK inhibitor) | 19% (27/144) | CR/CRi 56% in TP53-mutated AML treated with decitabine +- ibrutinib | mOS about 6 months in TP53-mutated AML, mOS about 12 months in TP53-WT AML | EudraCT 2015-002855-85 | [71] |
| Decitabine + cladribine/LDAC | II | Untreated elderly AML | 5-decitabine alternating with cladribine and LDAC | 17% (20/118) | CR/CRi 40% | mOS 5.4 months | NCT01515527 | [72] |
| Azacytidine | Prospective observational | MDS/AML/CMMoL (n = 62) | Azacytidine (hypomethylating agent) | 15% (9 TP53-mutated AML, 14 TP53-mutated MDS, 39 TP53-WT) | CR 22% and ORR 44% in TP53-mutated MDS/AML CR 38% and ORR 51% in TP53-WT MDS/AML | mOS 12.4 months in TP53-mutated AML/MDS, 23.7 months in TP53-WT AML/MDS | N/A | [73] |
| Azacytidine ± venetoclax | III | Untreated AML (n = 431) | Azacytidine + venetoclax vs. azacytidine | 12% (52 TP53-mutated AML, 379 TP53-WT AML) | CR/CRi 40.8% and ORR 55.3% in TP53-mutated AML with poor cytogenetics treated with azacytidine and venetoclax | mOS 5.17 and 4.9 months in TP53-mutated AML with poor-risk cytogenetics treated with azacytidine ± venetoclax | NCT02993523 | [74] |
| Azacytidine + pevonedistat | II | TP53-mutated AML (n = 10) | Azacytidine + pevonedistat (NEDD8-activating enzyme inhibitor) | 100% (10/10) | No CR, 2 PR, 7 SD | mOS 6.3 months | NCT03013998 | [75] |
| TP53-targeting agents and their combinations | ||||||||
| Idasanutlin ± cytarabine | I | R/R AML (n = 122) | Idasanutlin (MDM2 inhibitor) (n = 46), idasanutlin + cytarabine (n = 76) | 20% (25/122) | CRc = 18.9% and 35.6% in idasanutlin and + cytarabine. CRc = 4.0% (1/25) in TP53-mutated AML | DoR = 7.7 and 8.5 months in idasanutlin and + cytarabine arm in TP53-WT AML (data of TP53-mutated AML are N/A) | NCT017773408 | [76] |
| APR-246 + azacytidine | Ib/II | HMA-naive MDS/AML (n = 55) | APR-246 (TP53 reactivator) + azacytidine | 20% (11 AML, 40 MDS 4 MDS/MPN) | ORR 64% CR 36% in AML, ORR 73% and CR 50% in MDS | mOS 10.8 months for AML, 10.4 months for MDS | NCT 03072043 | [77] |
| APR- 246 + azacytidine | II | Treatment-naive TP53-mutated MDS/AML (n = 52) | APR-246 + azacytidine | 35% (18 AML, 34 MDS) | CR/CRi (47% in MDS, 45% in 20–30% blast AML, 14% in >30% blast AML) | mOS 12.1 months for MDS, 10.4 months for AML | NCT 03072043 | [78] |
| APR-246 + azacytidine | II | TP53-mutated MDS/AML receiving allogeneic HCT (n = 33) | APR + azacytidine maintenance for 12 cycles | 42% (14 AML, 19 MDS) | mRFS 12.5 months mOS 20.6 months (for MDS/AML) | NCT 03931291 | [79] | |
| APR-246 + azacytidine + venetoclax | I | TP53-mutated AML (n = 49; APR-246 + venetoclax 6, APR-246 + venetoclax + azacytidine n = 43) | APR-246 + venetoclax + azacytidine | 100% (de novo 45%, secondary 55%) | CR/CRi/MLFS 59% | Duration of CR 4.9 months mOS 7.3 months mOS (bridged to allogeneic HCT) 20.0 months | NCT 04214860 | [80] |
| Azacytidine ± APR-246 | III | TP53-mutated MDS/AML (n = 154) | Azacytidine vs. azacytidine + APR-246 | N/A | CR 33.3% in azacytidine + APR-246 vs. CR 22.4% in azacytidine (p = 0.13) | N/A | NCT 03745716 | Unpublished (press release only) |
| Immunologic agents and their combinations | ||||||||
| Ipilimumab + decitabine | I | R/R MDS/AML, (n = 54; 23 AML, 2 MDS, post-transplant (arm A) + 15 AML, 8 MDS, transplant-naive (arm B) + 6 N/E pts) | Ipilimumab (anti-CTLA-4 moAb) + Decitabine | 22.2% TP53-mutated MDS/AML (12/54 including 8 pts without NGS data) | ORR 20% in post-transplant MDS/AML, 52.1% in transplant-naive pts (Data in TP53-mutated subgroup are N/A) | DOR = 4.46 and 6.14 months in with and without prior HSCT (data in TP53-mutated subgroup are N/A) | NCT02890329 | [81,82] |
| Nivolumab + azacytidine | II | R/R AML (n = 70) | Nivolumab (anti-PD-1 MoAb) + azacytidine | 23% (16/70) | ORR 18.8% in TP53-mutated AML, ORR 33% in R/R AML (all patients) | mOS 5.98 months in TP53-mutated AML, mOS 6.3 months in R/R AML (all patients) | NCT02397720 | [64] |
| Durvalumab + azacytidine | II | Untreated MDS/AML (n = 213) | Durvalumab (anti-PD-L1 MoAb) + azacytidine | 27.7% (59/213), 45.7% among AML (59/129) | CR/CRi 20% in aza/durvalumab arm, CR/CRi 23% in azacytidine arm ORR 35% in TP53-mutated AML, ORR 34% in TP53-WT AML | mOS and RFS 13.0 and 9.5 months in aza/durvalumab arm, mOS and RFS = 14.4 and 12.2 in azacytidine alone arm | NCT02775903 | [65] |
| Flotetuzumab | I/II | R/R AML/MDS | Flotetuzumab (Anti-CD3/CD123 bispecific DART) | 33% (15/45 R/R AML) | CR 47% (7/15) in TP53-mutated AML | mOS 10.3 months in TP53-mutated AML | NCT02152956 | [60] |
| Magrolimab + azacytidine | Ib | Untreated, venetoclax-naive R/R or -exposed R/R | Magrolimab (anti-CD47 MoAb) + azacytidine | ORR 63% (27/43) in de novo AML ORR 69% (20/29) in TP53-mutated AML | mOS 12.9 months for TP53-mutated AML mOS 18.9 months for TP53 WT AML | NCT04435691 | ASH 2021 abstract | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, D.-Y. TP53 Mutation in Acute Myeloid Leukemia: An Old Foe Revisited. Cancers 2023, 15, 4816. https://doi.org/10.3390/cancers15194816
Shin D-Y. TP53 Mutation in Acute Myeloid Leukemia: An Old Foe Revisited. Cancers. 2023; 15(19):4816. https://doi.org/10.3390/cancers15194816
Chicago/Turabian StyleShin, Dong-Yeop. 2023. "TP53 Mutation in Acute Myeloid Leukemia: An Old Foe Revisited" Cancers 15, no. 19: 4816. https://doi.org/10.3390/cancers15194816
APA StyleShin, D.-Y. (2023). TP53 Mutation in Acute Myeloid Leukemia: An Old Foe Revisited. Cancers, 15(19), 4816. https://doi.org/10.3390/cancers15194816