
World’s First Long-Term Colorectal Cancer Model by 3D Bioprinting as a Mechanism for Screening Oncolytic Viruses
 , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
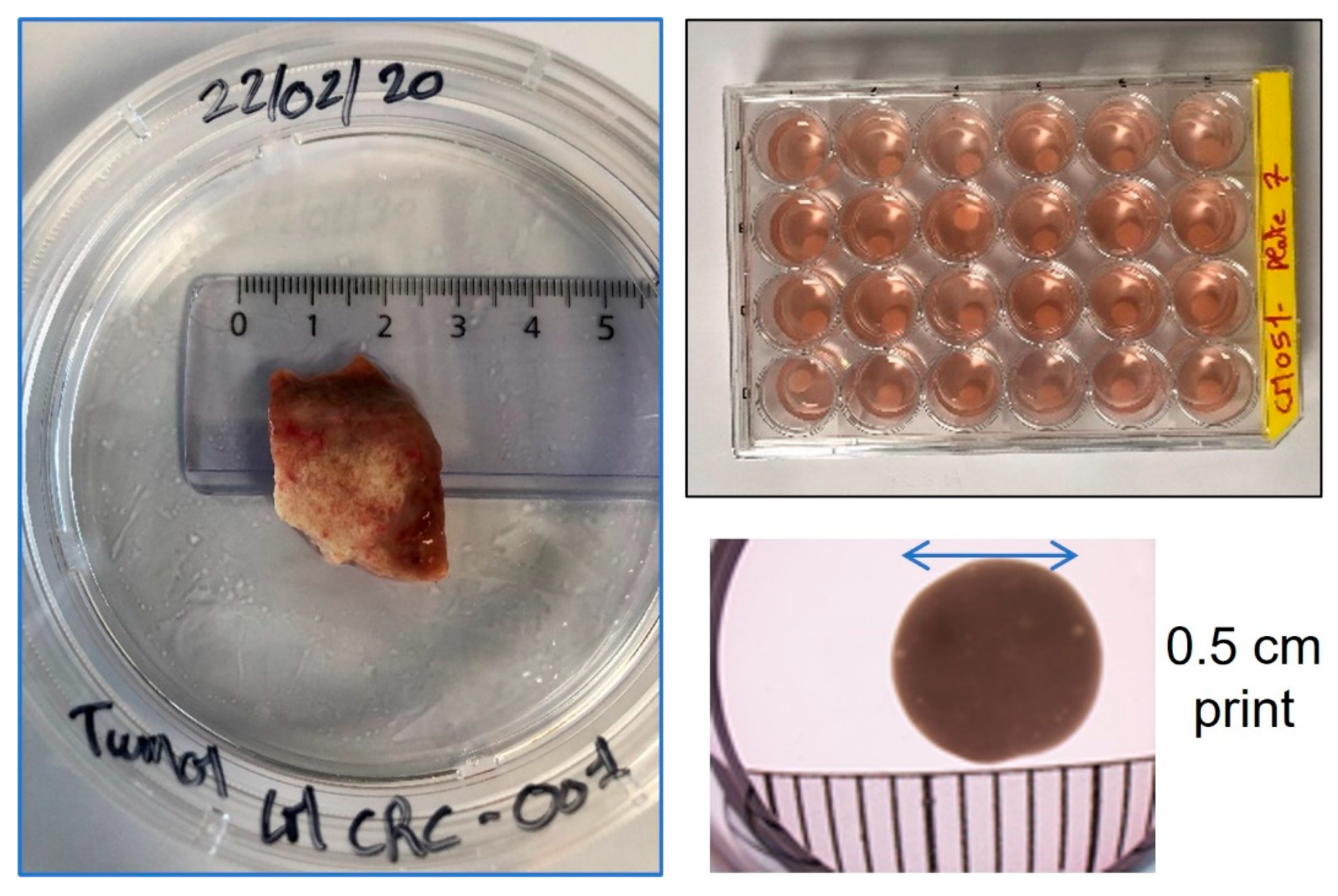
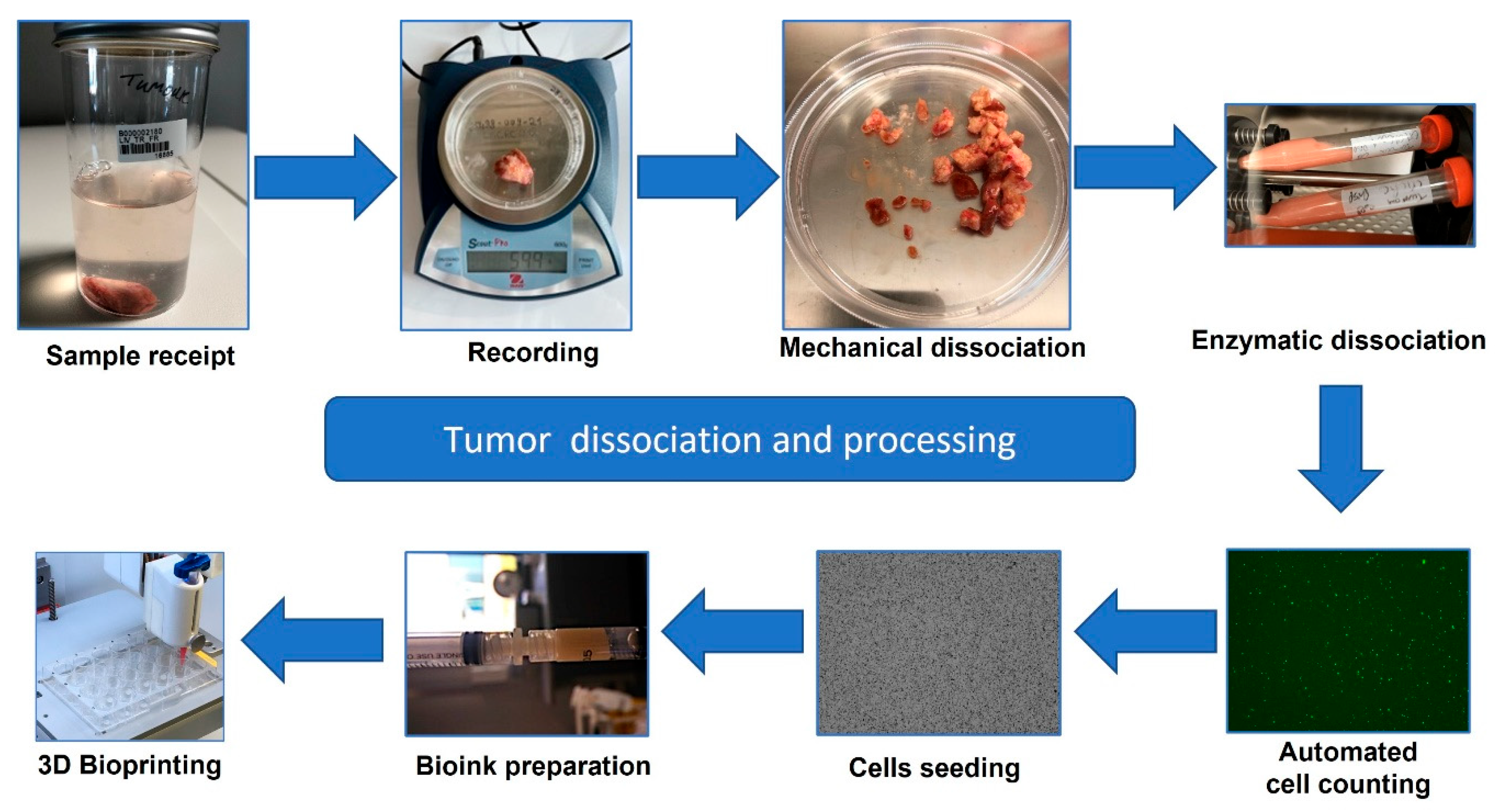
2.1. Sample Collection and Processing
2.2. Cell Culture and 3D Bioprinting
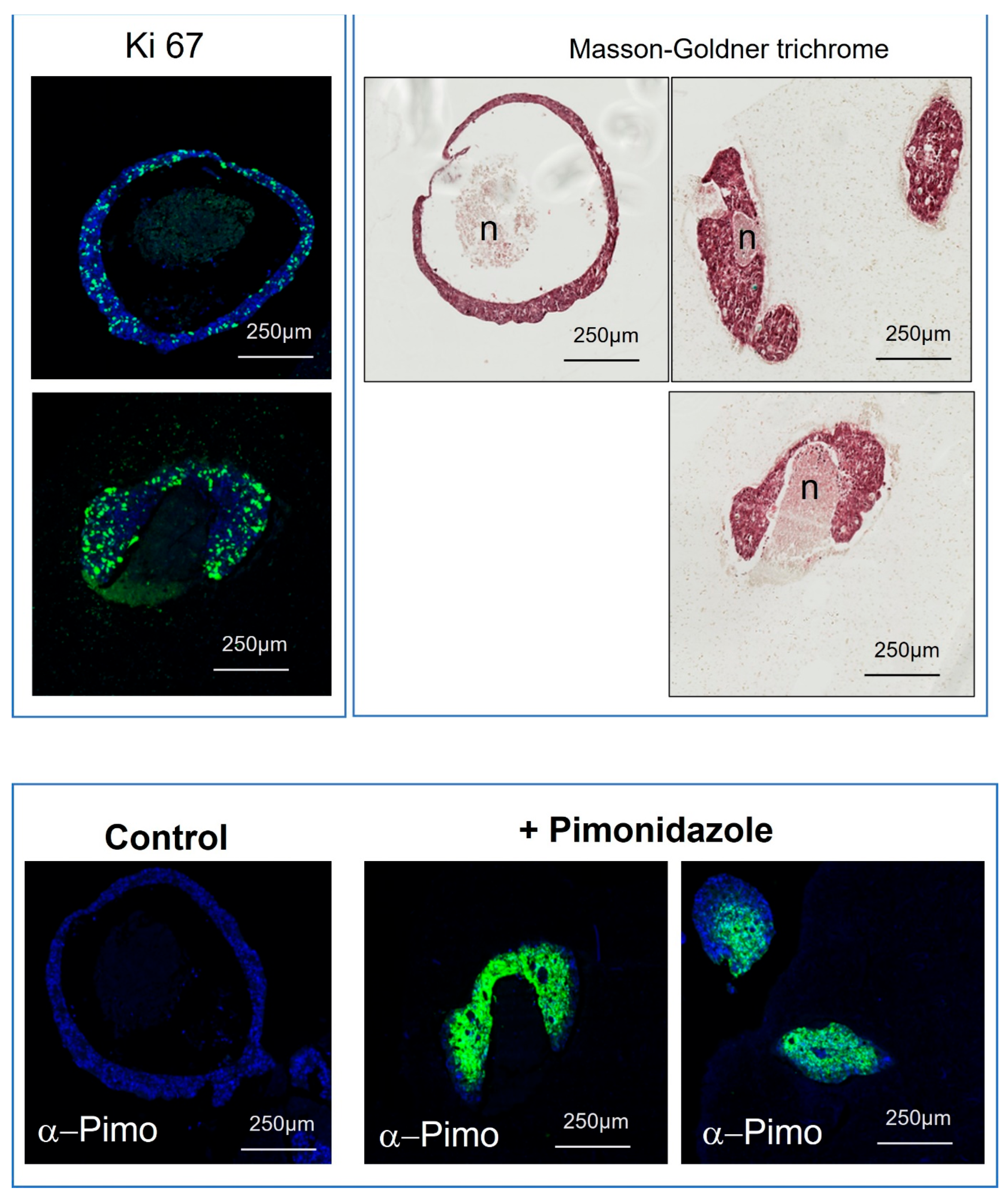
2.3. Cell Viability and Microtumor Analysis
2.4. Hypoxia Assessment of 3D Bioprinted Microtumors
2.5. Oncolytic Viral Infection and Analysis of Microtumors
3. Results
3.1. Patient-Derived Cellular Expansion from Small Donations Can Be Selective or Multi-Phenotype
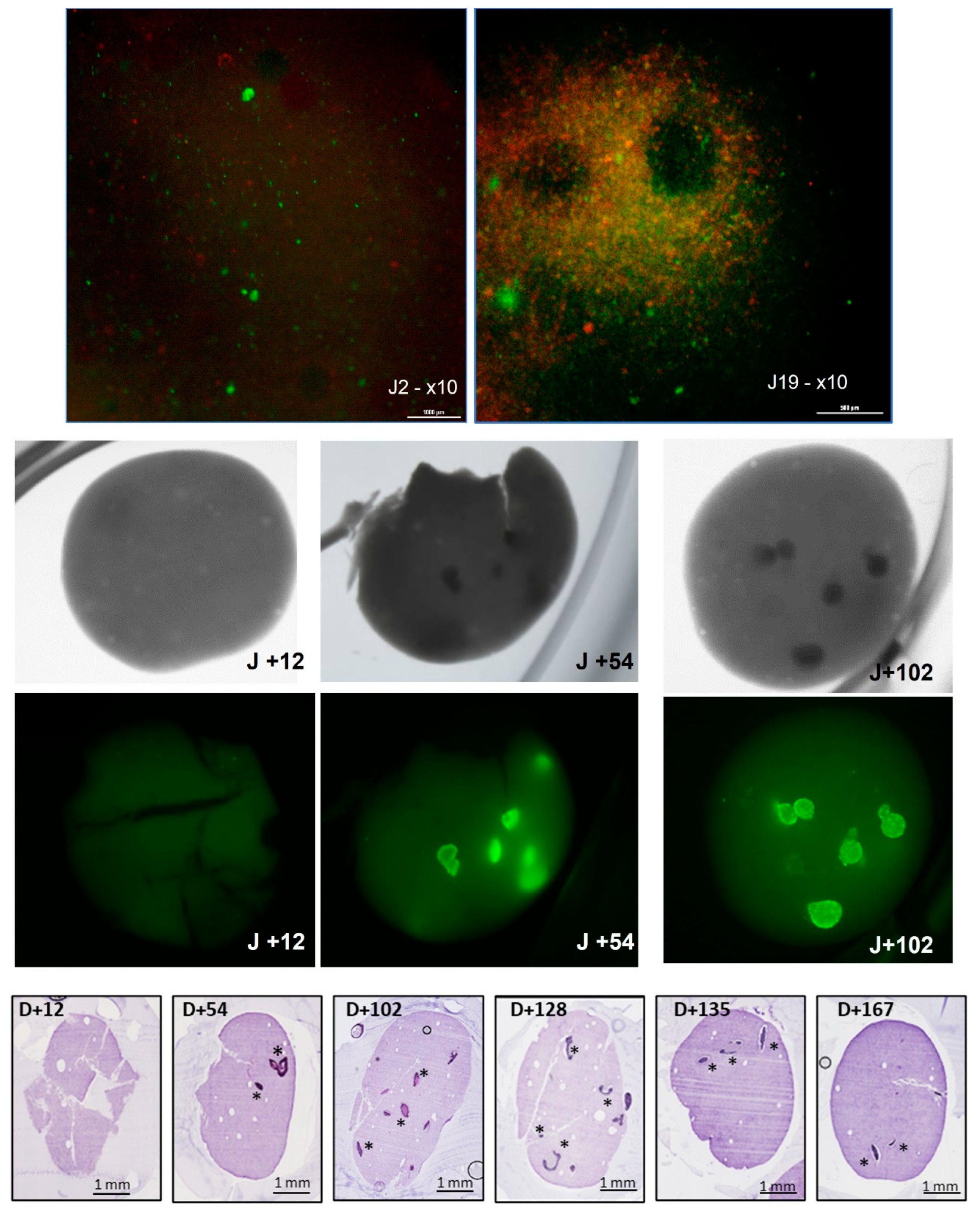
3.2. Development of Microtumors for Short-Term and Longitudinal Studies Is Achieved with 3D Bioprinting
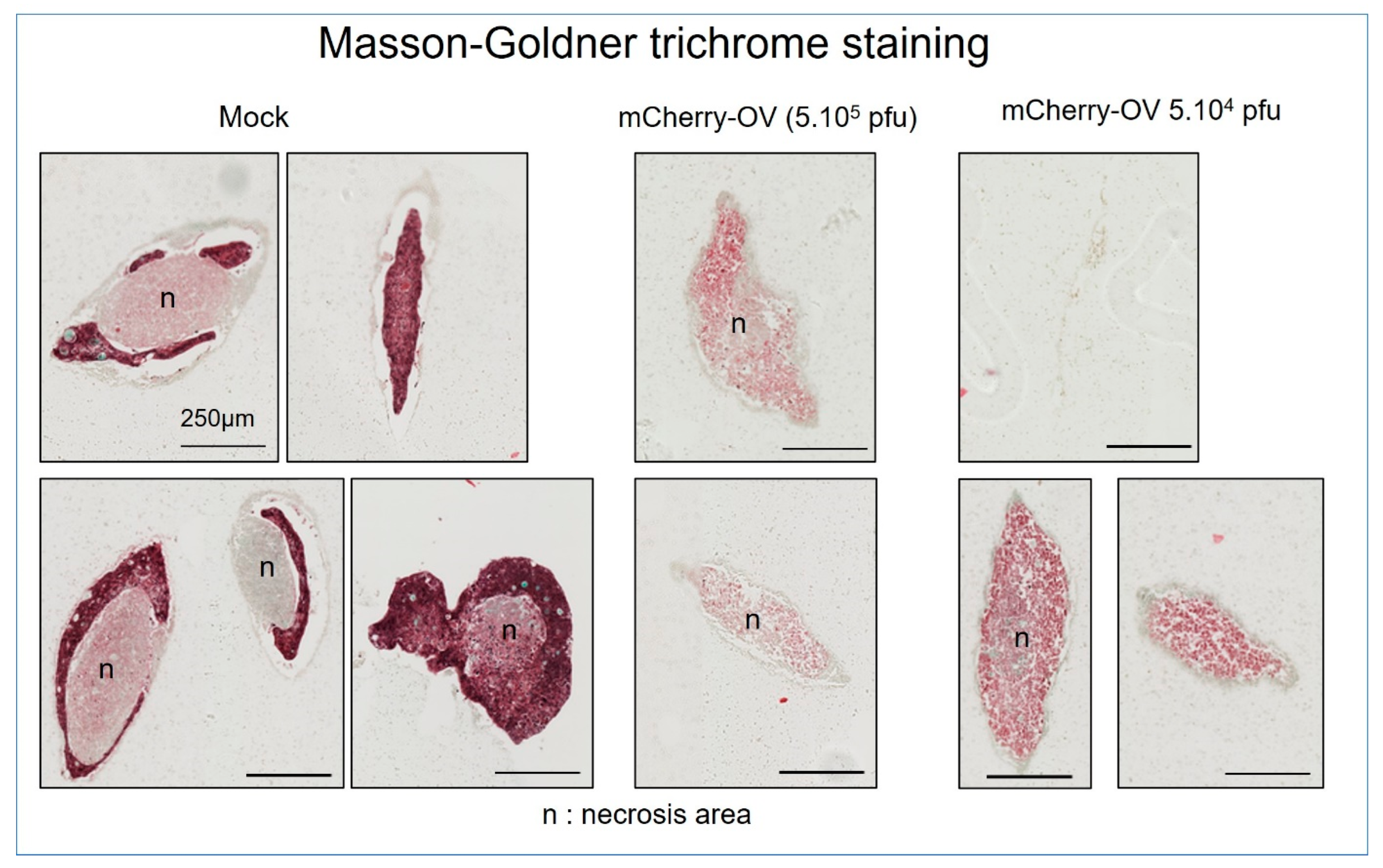
3.3. Replication of In Vivo Heterogeneous Tumor Areas with 3D Bioprinted Microtumors
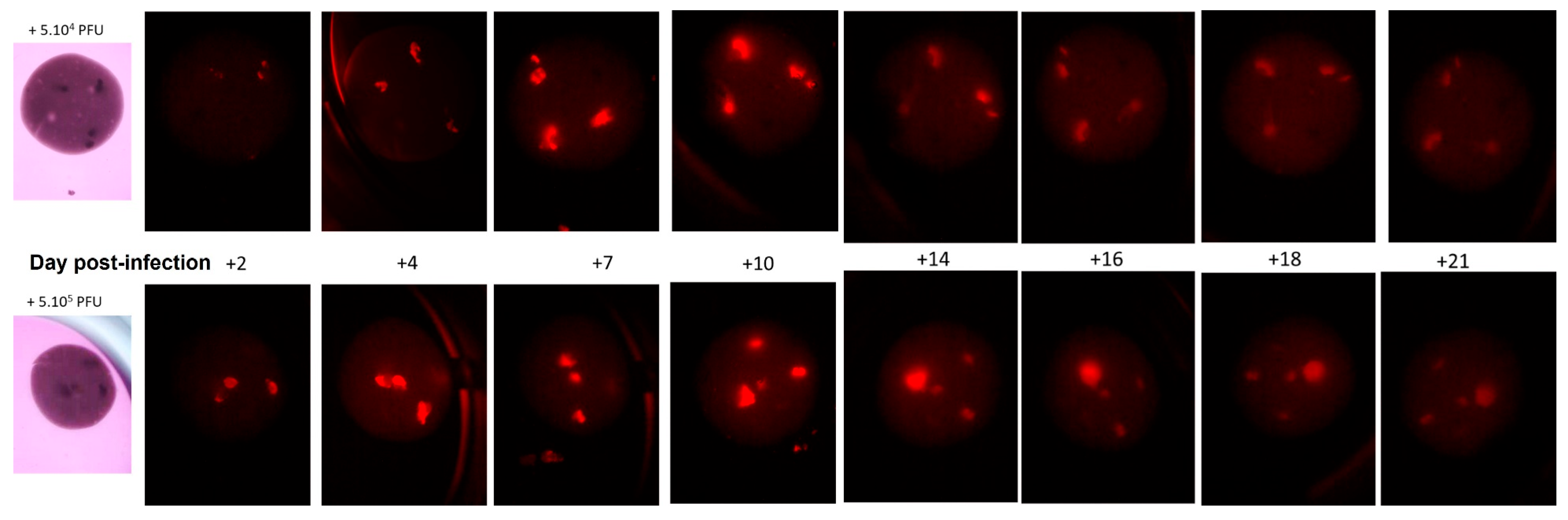
3.4. Accurate Sophisticated Screening of Advanced Biologics Can Be Achieved in Microtumors
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Russomano, T.; Cardoso, R.; Falcao, F.P.; Dalmarco, G.; dos Santos, C.R.V.; dos Santos, L.G.F.; de Azevedo, D.F.G.; dos Santos, M.A.; Martinelli, L.; Motta, J.D.; et al. Development and Validation of a 3D Clinostat for the Study of Cells during Microgravity Simulation. In Proceedings of the 2005 IEEE Engineering in Medicine and Biology 27th Annual Conference, Shanghai, China, 17–18 January 2006; pp. 564–566. [Google Scholar] [CrossRef]
- Gupta, S.; Bit, A. 3D bioprinting in tissue engineering and regenerative medicine. Cell Tissue Bank. 2022, 23, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Sung HFerlay, F.; Siegel, R.L.; Laversanne, M.; Soergomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Loomans-Kropp, H.A.; Umar, A. Increasing Incidence of Colorectal Cancer in Young Adults. J. Cancer Epidemiol. 2019, 11, 9841295. [Google Scholar] [CrossRef] [PubMed]
- Creasy, J.M.; Sadot, E.; Koerkamp, B.G.; Chou, J.F.; Gonen, M.; Kemeny, N.E.; Balachandran, V.P.; Kingham, T.P.; DeMatteo, R.P.; Allen, P.J.; et al. Actual 10-year survival after hepatic resection of colorectal liver metastases: What factors preclude cure? Surgery 2018, 163, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Engstrand, J.; Nilsson, H.; Strömberg, C.; Jonas, E.; Freedman, J. Colorectal cancer liver metastases—A population-based study on incidence, management and survival. BMC Cancer 2018, 18, 78. [Google Scholar] [CrossRef]
- Crooke, H.; Kobayashi, M.; Mitchell, B.; Nwokeji, E.; Laurie, M.; Kamble, S.; McKenna, M.; Masood, A.; Korytowsky, B. Estimating 1-and 5-year relative survival trends in colorectal cancer (CRC) in the United States: 2004 to 2014. J. Clin. Oncol. 2018, 36, 587. [Google Scholar] [CrossRef]
- Chao, C.; Carmical, J.R.; Ives, K.L.; Ives, K.L.; Wood, T.G.; Aronson, J.F.; Gomez, G.A.; Djukom, C.D.; Hellmich, M.R. CD133+ colon cancer cells are more interactive with the tumor microenvironment than CD133- cells. Lab. Investig. 2012, 92, 420–436. [Google Scholar] [CrossRef]
- Reidy, E.; Leonard, N.A.; Treacy, O.; Ryan, A.E. A 3D View of Colorectal Cancer Models in Predicting Therapeutic Responses and Resistance. Cancers 2021, 13, 227. [Google Scholar] [CrossRef]
- Shakibaei, M.; Kraehe, P.; Popper, B.; Shayan, P.; Goel, A.; Buhrmann, C. Curcumin potentiates antitumor activity of 5-fluorouracil in a 3D alginate tumor microenvironment of colorectal cancer. BMC Cancer 2015, 15, 250. [Google Scholar] [CrossRef] [PubMed]
- Loessner, D.; Stok, K.S.; Lutolf, M.; Hutmacher, D.W.; Clements, J.A.; Rizzi, S.C. Bioengineered 3D platform to explore cell-ECM interactions and drug resistance of epithelial ovarian cancer cells. Biomaterials 2010, 31, 8494–8506. [Google Scholar] [CrossRef] [PubMed]
- Maloney, E.; Clark, C.; Sivakumar, H.; Yoo, K.; Aleman, J.; Rajan, S.A.P.; Forsythe, S.; Mazzocchi, A.; Laxton, A.W.; Tatter, S.B. Immersion Bioprinting of Tumor Organoids in Multi-Well Plates for Increasing Chemotherapy Screening Throughput. Micromachines 2020, 11, 208. [Google Scholar] [CrossRef] [PubMed]
- Jurga, M.; Dainiak, M.B.; Sarnowska, A.; Jablonska, A.; Tripathi, A.; Plieva, F.M.; Savina, I.N.; Strojek, L.; Lungvid, H.; Kumar, A.; et al. The performance of laminin-containing cryogel scaffolds in neural tissue regeneration. Biomaterials 2011, 32, 3423–3434. [Google Scholar] [CrossRef]
- Mueller, A.A.; Forraz, N.; Gueven, S.; Atzeni, G.; Degoul, O.; Pagnon-Minot, A.; Hartmann, D.; Martin, I.; Scherberich, A.; McGuckin, C. Osteoblastic differentiation of Wharton jelly biopsy specimens and their mesenchymal stromal cells after serum-free culture. Plast. Reconstr. Surg. 2014, 134, 59e–69e. [Google Scholar] [CrossRef]
- McGuckin, C.P.; Jurga, M.; Miller, A.M.; Sarnowska, A.; Wiedner, M.; Boyle, N.T.; Lynch, M.A.; Jablonska, A.; Drela, K.; Lukomska, B.; et al. Ischemic brain injury: A consortium analysis of key factors involved in mesenchymal stem cell-mediated inflammatory reduction. Arch. Biochem. Biophys. 2013, 534, 88–97. [Google Scholar] [CrossRef]
- Sarnowska, A.; Jablonska, A.; Jurga, M.; Dainiak, M.; Strojek, L.; Drela, K.; Wright, K.; Tripathi, A.; Kumar, A.; Jungvid, H.; et al. Encapsulation of mesenchymal stem cells by bioscaffolds protects cell survival and attenuates neuroinflammatory reaction in injured brain tissue after transplantation. Cell Transplant. 2013, 1, S67–S82. [Google Scholar] [CrossRef]
- Sbirkov, Y.; Molander, D.; Milet, C.; Bodurov, I.; Atanasov, B.; Penkov, R.; Belev, N.; Forraz, N.; McGuckin, C.; Sarafian, V. A Colorectal Cancer 3D Bioprinting Workflow as a Platform for Disease Modeling and Chemotherapeutic Screening. Front. Bioeng. Biotechnol. 2021, 18, 755563. [Google Scholar] [CrossRef]
- Zhou XZhu, W.; Nowicki, M.; Miao, S.; Cui, H.; Holmes, B.; Glazer, R.I.; Zhang, L.G. 3D Bioprinting a Cell-Laden Bone Matrix for Breast Cancer Metastasis Study. ACS Appl. Mater. Interfaces 2016, 8, 30017–30026. [Google Scholar] [CrossRef]
- Zhao, Y.; Yao, R.; Ouyang, L.; Ding, H.; Zhang, T.; Zhang, K.; Cheng, S.; Sun, W. Three-dimensional printing of Hela cells for cervical tumor model in vitro. Biofabrication 2014, 6, 035001. [Google Scholar] [CrossRef]
- Dai, X.; Ma, C.; Lan, Q.; Xu, T. 3D bioprinted glioma stem cells for brain tumor model and applications of drug susceptibility. Biofabrication 2016, 8, 045005. [Google Scholar] [CrossRef] [PubMed]
- Mencattini, A.; Lansche, C.; Veith, I.; Erbs, P.; Balloul, J.-M.; Quemeneur, E.; Descroix, S.; Mechta-Grigoriou, F.; Zalcman, G.; Zaupa, C.; et al. Direct imaging and automatic analysis in tumor-on-chip reveal cooperative antitumoral activity of immune cells and oncolytic vaccinia virus. Biosens. Bioelectron. 2022, 215, 114571. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, K.Y.; Brekken, R.A. Hypoxia Studies with Pimonidazole in vivo. Bio-Protocol 2014, 4, e1254. [Google Scholar] [CrossRef]
- Seyhan, A.A. Lost in translation: The valley of death across preclinical and clinical divide—Identification of problems and overcoming obstacles. Transl. Med. Commun. 2019, 4, 18. [Google Scholar] [CrossRef]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.-B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef]
- Boutin, A.T.; Liao, W.-T.; Wang, W.; Hwang, S.S.; Karpinets, T.V.; Cheung, H.; Chu, G.C.; Jiang, S.; Hu, J.; Chang, K.; et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev. 2017, 31, 370–382. [Google Scholar] [CrossRef]
- Rios de la Rosa, J.M.; Wubetu, J.; Tirelli, N.; Tirella, A. Colorectal tumor 3D in vitro models: Advantages of biofabrication for the recapitulation of early stages of tumour development. Biomed. Phys. Eng. Express 2018, 4, 045010. [Google Scholar] [CrossRef]
- Augustine, R.; Kalva, S.N.; Ahmad, R.; Zahid, A.A.; Hasan, S.; Nayeem, A.; McClements, L.; Hasan, A. 3D Bioprinted cancer models: Revolutionizing personalized cancer therapy. Transl. Oncol. 2021, 14, 101015. [Google Scholar] [CrossRef]
- Chen, H.; Cheng, Y.; Wang, X.; Wang, J.; Shi, X.; Tan, W.; Tan, Z. 3D printed in vitro tumor tissue model of colorectal cancer. Theranostics 2020, 10, 12127–12143. [Google Scholar] [CrossRef] [PubMed]
- Cascinu, S.; Berardi, R.; Salvagni, S.; Beretta, G.D.; Catalano, V.; Pucci, F.; Sobrero, A.; Tagliaferri, P.; Labianca, R.; Scartozzi, M.; et al. A combination of gefitinib and FOLFOX-4 as first-line treatment in advanced colorectal cancer patients. A GISCAD multicentre phase II study including a biological analysis of EGFR overexpression, amplification and NF-κB activation. Br. J. Cancer 2008, 98, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Joyce, J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Blondy, S.; David, V.; Verdier, M.; Mathonnet, M.; Perraud, A.; Christou, N. 5-Fluorouracil resistance mechanisms in colorectal cancer: From classical pathways to promising processes. Cancer Sci. 2020, 111, 3142–3154. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, Z.; Zhu, Y.; Pan, Q.; Lui, Y.; Qi, X.; Jin, L.; Jin, J.; Ma, X.; Hua, D. Inhibition of transient receptor potential channel 5 reverses 5-Fluorouracil resistance in human colorectal cancer cells. J. Biol. Chem. 2015, 290, 448–456. [Google Scholar] [CrossRef]
- Hoare, O.; Fraunhoffer, N.; Elkaoutari, A.; Gayet, O.; Bigonnet, M.; Roques, J.; Nicolle, R.; McGuckin, C.; Forraz, N.; Sohier, E.; et al. Exploring the Complementarity of Pancreatic Ductal Adenocarcinoma Preclinical Models. Cancers 2021, 13, 2473. [Google Scholar] [CrossRef]
- Inamura, K. Colorectal Cancers: An Update on Their Molecular Pathology. Cancers 2018, 10, 26. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Result |
|---|---|
| Sex | Male |
| Age | 74 |
| Weight | 80.78 kg |
| BMI | 28.49 |
| Diagnosis | Primary colorectal adenocarcinoma |
| Extent | Metastatic |
| Tumor sample size | 3.66 g |
| Models printed per hour | 173 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGuckin, C.; Forraz, N.; Milet, C.; Lacroix, M.; Sbirkov, Y.; Sarafian, V.; Ebel, C.; Spindler, A.; Koerper, V.; Balloul, J.-M.; et al. World’s First Long-Term Colorectal Cancer Model by 3D Bioprinting as a Mechanism for Screening Oncolytic Viruses. Cancers 2023, 15, 4724. https://doi.org/10.3390/cancers15194724
McGuckin C, Forraz N, Milet C, Lacroix M, Sbirkov Y, Sarafian V, Ebel C, Spindler A, Koerper V, Balloul J-M, et al. World’s First Long-Term Colorectal Cancer Model by 3D Bioprinting as a Mechanism for Screening Oncolytic Viruses. Cancers. 2023; 15(19):4724. https://doi.org/10.3390/cancers15194724
Chicago/Turabian StyleMcGuckin, Colin, Nico Forraz, Clément Milet, Mathieu Lacroix, Yordan Sbirkov, Victoria Sarafian, Caroline Ebel, Anita Spindler, Véronique Koerper, Jean-Marc Balloul, and et al. 2023. "World’s First Long-Term Colorectal Cancer Model by 3D Bioprinting as a Mechanism for Screening Oncolytic Viruses" Cancers 15, no. 19: 4724. https://doi.org/10.3390/cancers15194724
APA StyleMcGuckin, C., Forraz, N., Milet, C., Lacroix, M., Sbirkov, Y., Sarafian, V., Ebel, C., Spindler, A., Koerper, V., Balloul, J.-M., Quéméneur, E., & Zaupa, C. (2023). World’s First Long-Term Colorectal Cancer Model by 3D Bioprinting as a Mechanism for Screening Oncolytic Viruses. Cancers, 15(19), 4724. https://doi.org/10.3390/cancers15194724