1. Introduction
Cutaneous melanoma (M) represents 1.7% of all global cancer diagnoses [
1]. Over the last 10 years, M incidence has consistently risen. However, the M-specific mortality decreased by more than 30% due to the introduction of novel therapeutic agents and their use in combinations [
2]. Approximately half of the patients with M harbor an activating mutation in the serine-threonine kinase BRAF, 90% of which occur at codon 600 in exon 15 V600E (substituting valine to glutamine) [
3]. Other BRAF-activating mutations are rare [
4]. BRAF acts via the RAS/MAPK pathway, which regulates cell proliferation, differentiation, migration, and apoptosis. BRAF mutation (BRAFv600) activates BRAF and upregulates the downstream signal transduction in the MAP kinase pathway involving different mechanisms of M carcinogenesis, growth, progression, and immune escape [
5,
6,
7]. The BRAF mutation is associated with reduced survival compared to wild-type melanoma [
8]. Targeting BRAF, with the small selective molecule inhibitors vemurafenib (V), dabrafenib (D), and encorafenib (E), showed clinical efficacy and improved survival outcomes in untreated BRAFV600 metastatic melanomas (MM) compared to standard chemotherapy [
9,
10,
11,
12]. Despite encouraging initial response rates, almost 50% of patients treated with BRAF inhibitors (BRAFi) monotherapies relapse within six months, with a median progression-free survival (PFS) from 5 to 10 months in landmark phase III trials (BRIM-3, BREAK-3) [
11,
12]. Failure of treatment with BRAFi is related to a paradoxical hyperactivation of the MEK-mediated signaling cascade, leading to the rapid development of drug resistance [
13,
14,
15,
16,
17]. In addition, using BRAFi may result in the development of secondary early squamous cell carcinomas induced by the paradoxical activation of the MAP kinase pathway occurring in non-cancer cells in which the oncogenic BRAFV600 is lacking [
18,
19]. Inhibitors of MEK (MEKi), a signaling molecule downstream of BRAF, when co-administrated with BRAFi, were demonstrated to dramatically enhance BRAFi activity and delay the development of biological resistance [
20,
21,
22,
23]. Considering this synergistic effect, the oral small molecules trametinib (T), cobimetinib (C), and binimetinib (B) have been investigated in association with BRAFi in different first-line phase III trials enrolling untreated MM patients with V600 mutation [
20,
21,
22,
23]. The D+T combination showed superior efficacy to D and V monotherapies in COMBI-D and COMBI-V trials, respectively [
24,
25]. Likewise, in the coBRIM trial, V+C improved PFS, overall survival (OS), and response rate versus V alone [
22,
26]. Subsequently, a phase III study (COLUMBUS) compared E+B vs. single agents in patients with BRAFV600 MM who had not received prior therapies or had progressed on or after previous first-line immunotherapy [
23,
27]. BRAFi/MEKi combination therapy has been shown to have superior PFS and OS compared to V alone. At the same time, the improvements in survival outcomes did not reach statistical significance when BRAF/MEK association was compared to E monotherapy [
28]. Treatment with BRAFi/MEKi was associated with adverse events (AEs) of any grade in almost all patients (>90%) across phase III trials. Grade 3–4 AEs occurred in 46–56% of patients treated with D-T (COMBI-D, COMBI-V) and 69% of patients undergoing V plus C (coBRIM), respectively. Furthermore, 58% of pts in the E-B arm in the COLUMBUS study (part I) experienced a severe AE (G3–4) [
29]. BRAFi/MEKi combinations shared similar “class effects” including gastrointestinal toxicity, impaired liver function, and skin toxicity [
29]. On the other hand, in addition to squamous cell carcinoma, arthralgia was a specific BRAFi class reaction. At the same time, ocular edema and cardiovascular toxicity were mainly associated with MEKi therapy as well as with induction in almost all treated patients of a papulopustular exanthema.
Neuro-retinal detachment, muscular problems, hypertension, and ventricular ejection fraction decrease were also related to MEK inhibition. Moreover, specific side effects were related to specific combinations. Skin toxicities, including Stevens–Johnson syndrome, photosensitivity, and acute kidney injury, were higher using the V+C combination. Pyrexia and elevated C-reactive protein were significantly associated with D+T, whereas gastrointestinal AEs, arthralgia, peripheral neuropathies, renal disorder, and an increase in Guillain–Barrè syndrome were significantly associated with E+B [
30]. Based on this heavy toxicity profile, proactive management of toxicities is needed to ensure better clinical outcomes avoiding unnecessary treatment discontinuation. Moreover, serious AEs and cumulative toxicities negatively impact the patient’s quality of life [
31]. The toxicity spectrum of the combo BRAFi/MEKi treatment can potentially be even worse for all pharmacokinetic interactions related to the intake of foods, dietary supplements, complementary alternative therapies, excipients, and drugs that can interfere with the pharmacodynamics and pharmacokinetics of the BRAFi/MEKi treatment combinations [
32]. Environmental factors also affect the absorption, distribution, metabolism, and excretion of the drug, which may also be affected by interpatient variability, given the potential impact of age, gender, genetics, and comorbidity conditions on drug handling [
33,
34]. In this complex scenario, drug–drug interactions (DDIs), inducing metabolic interference, can result in drug toxicities, reduced pharmacological effects, and adverse drug reactions. DDIs can substantially influence the drug activity of BRAFi/MEKi anticancer therapy in either a beneficial or detrimental manner, reducing treatment efficacy or promoting the development of adverse drug reactions [
35]. Drugs undergo multiple metabolic pathways and modifications, which could affect plasma drug concentrations [
36]. DDIs usually involve all cytochromes of the P450 (CYPs) enzyme superfamily, P-glycoprotein, ATP-binding cassette transporters, as well as detoxifying and DNA-repair enzymes, fundamental in drug metabolism and central to the occurrence of drugs interaction [
37,
38,
39,
40,
41,
42,
43,
44].
Although the risk of DDI is potentially significant and quite common, DDIs in cancer treatment were investigated only in a few retrospective studies in which no patients with MM treated with target therapies were included [
45,
46,
47]. This multicentric retrospective study aims to describe the role of DDIs on the toxicity profile and clinical outcomes in a real-world population treated with BRAFi/MEKi therapy using Drug-PIN
® (Personalized Interactions Network), a medical software able to detect and improve drug interactions in combination with patient profiles including demographic, clinical and biochemical data [
35,
48].
The main objective of this study was to retrospectively assess and define the risk of drug interactions in clinical practice and their impact on the toxicity spectrum of patients with BRAFV600 MM treated with BRAFi/MEKi inhibitors, with the intent to reduce potentially avoidable toxicities by improving the tolerability of treatment and the quality of life of patients.
4. Discussion
Our study highlights that, in patients with advanced melanoma treated with BRAFi/MEKi, the presence of DDIs favors the development of adverse drug reactions; in particular, DDIs appear to be significantly associated with an increased risk of cardiological toxicity. Moreover, DDIs also have a prognostic value as they are associated with worse oncological outcomes in terms of OS and PFS, probably because they affect both the efficacy of anti-BRAFi/MEKi drugs and treatment adherence.
As expected, most patients (66.7%) had comorbidities, 99 of which (55.9%) required a specific concomitant pharmacological treatment. Thus, more than half of the patients were exposed to complex drug regimens, which increased the risk of potential drug–drug interactions and, consequently, reduced the efficacy and safety of oncological therapies. Patients were at risk of being undertreated for their comorbidities since concomitant treatment may be reduced by DDIs [
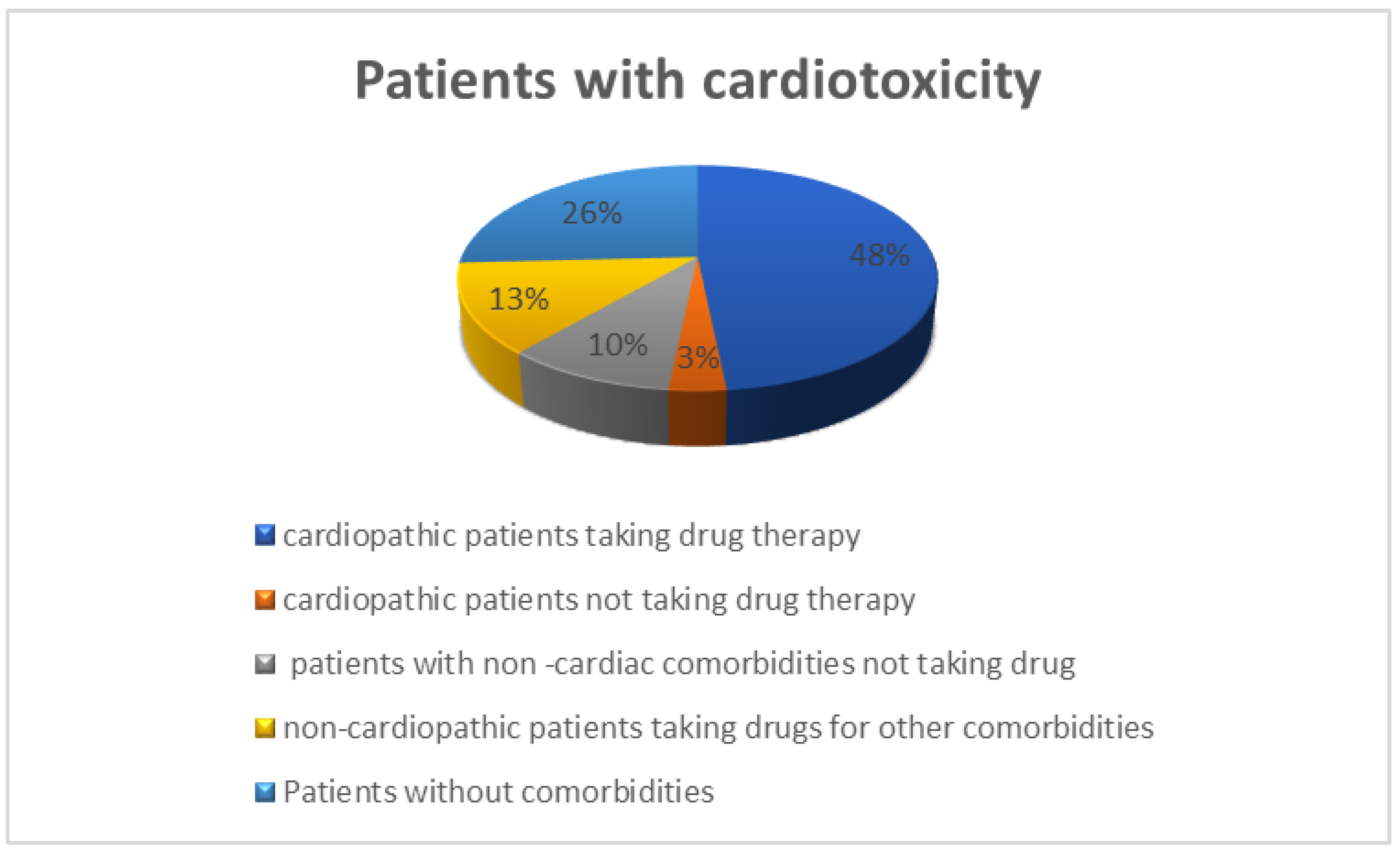
51]. It is worthy of note that ten patients had potentially dangerous DDIs (dark yellow (8), orange (1), and red (1) Drug-PIN light) before the start of BRAFi/MEKi treatment. Furthermore, most of these patients (8/10) developed toxicity when BRAFi/MEKi treatment was added.
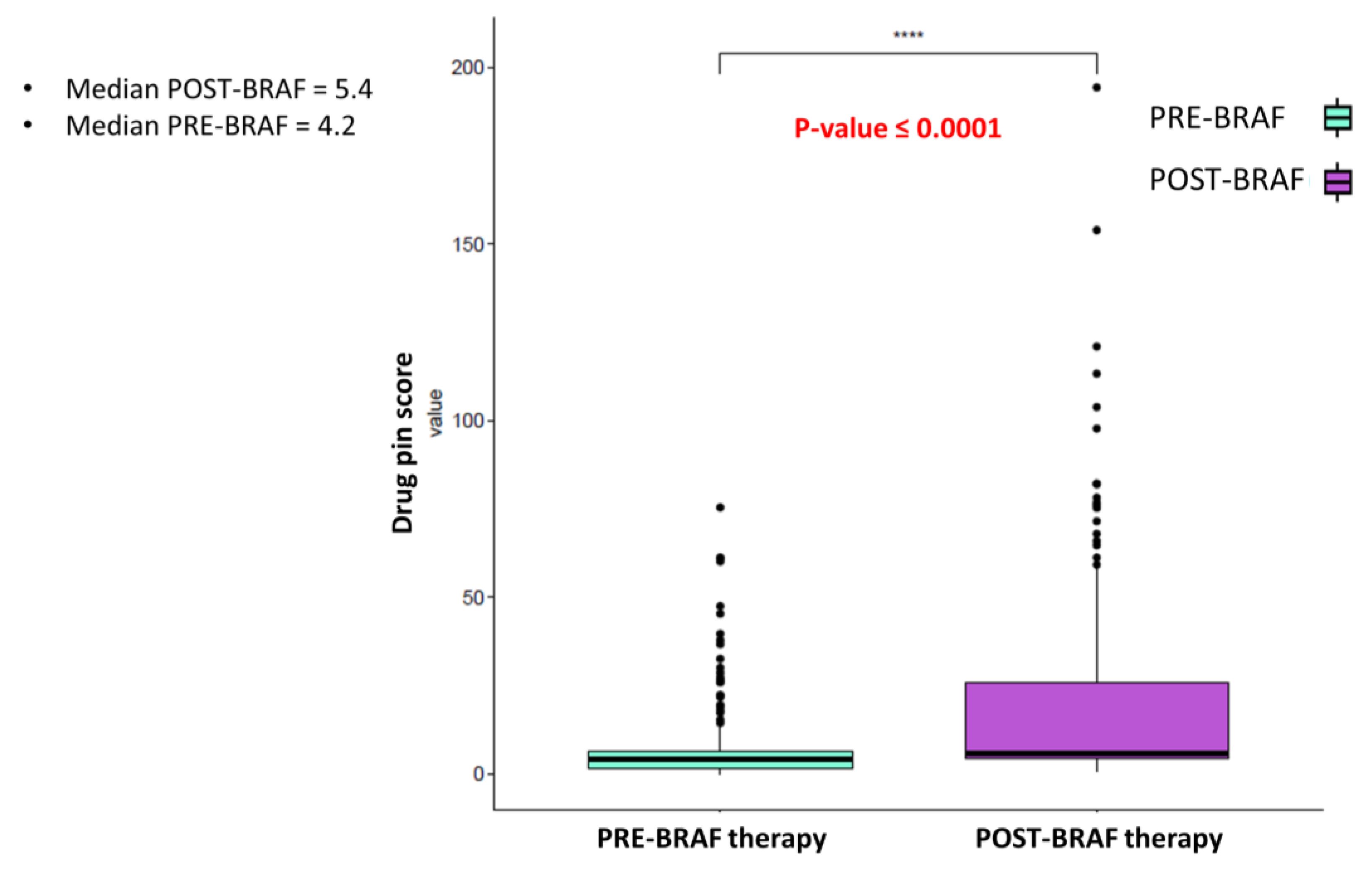
Interestingly, cardiological toxicity occurred in half of these cases, confirming the central role of DDIs in the development of AEs and the onset of cardiotoxicity. In this scenario, adding BRAFi/MEKi treatment contributed to the increase in the DDIs score. When the treatment with BRAFi/MEKi was added, the median Drug-PIN score increased from the basal score of 4.2 to 5.4 (
p-value < 0.0001). A relevant change in the Drug-PIN traffic light was also reported in 54 patients, with an increase in potentially dangerous interactions. The increased risk of toxicities related to DDIs could translate to a critical clinical impact, especially when the patients are exposed to a treatment burdened by a high toxicity profile. In phase III clinical trials, related adverse effects were experienced in nearly all patients (97%) treated with BRAFi/MEKi [
29]. During BRAFi/MEKi treatment, 78% of the patients experienced toxicity of any grade. This lower-than-expected incidence of AEs may be partially related to lower reports of low-grade toxicities, requiring no action or dose adjustments in clinical practice compared to clinical trials. However, pathologies currently directly linked to BRAFi/MEKi treatment, such as arterial hypertension, were treated pharmacologically and monitored before therapy in all patients. Antihypertensive treatment was started or pursued according to the existing guidelines. This proactive attitude could partially explain the very low rate of newly-onset arterial hypertension reported compared to that evidenced in clinical trials (ranging from 11% to 29%). High-grade toxicities, occurring in 19.2% (34 patients), also had a lower rate [
29]. Nevertheless, their management required dose reduction and treatment discontinuation in 16.6% and 8.5% of cases, respectively. It should also be stressed that the toxicity profile was similar to that emerging from clinical trials, with no unexpected toxicity, even in patients with a high DDI score.
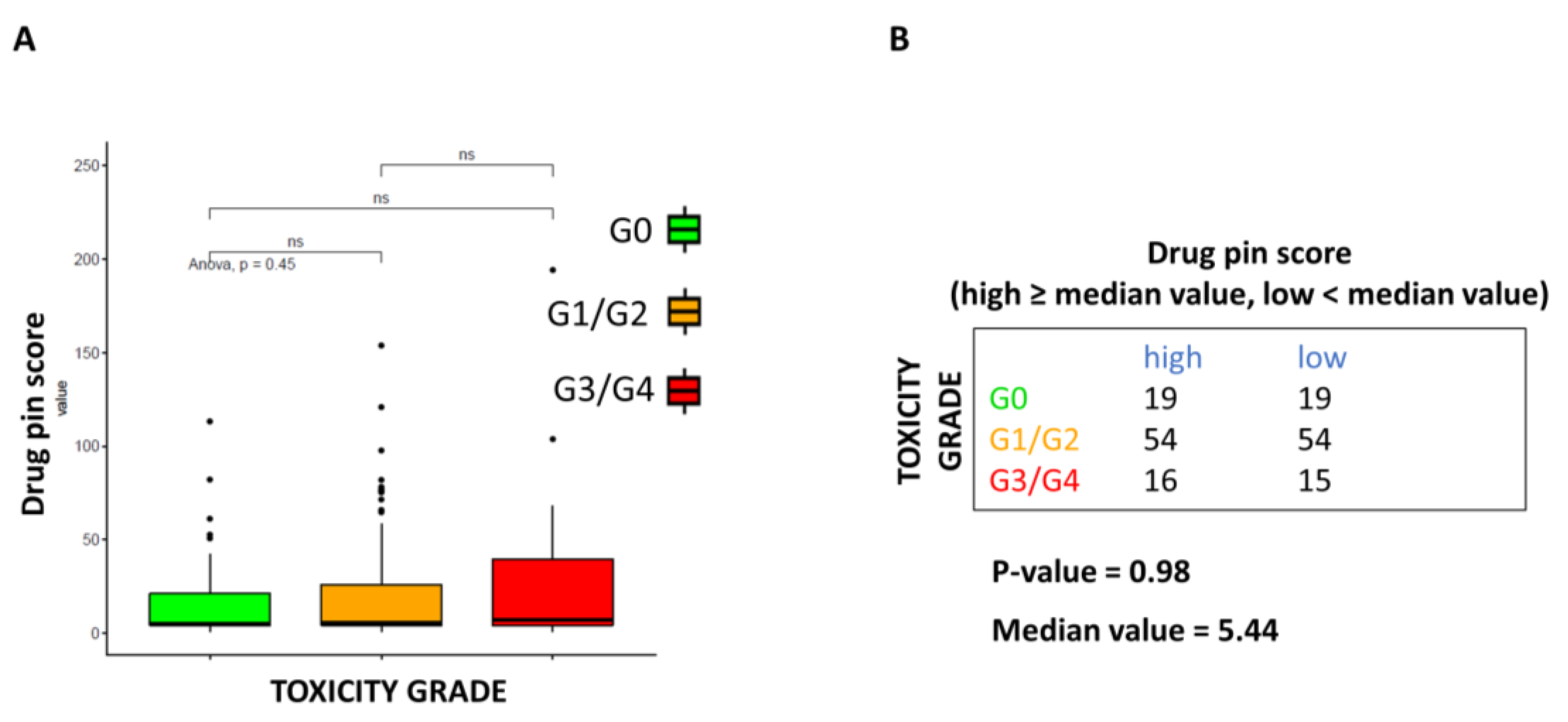
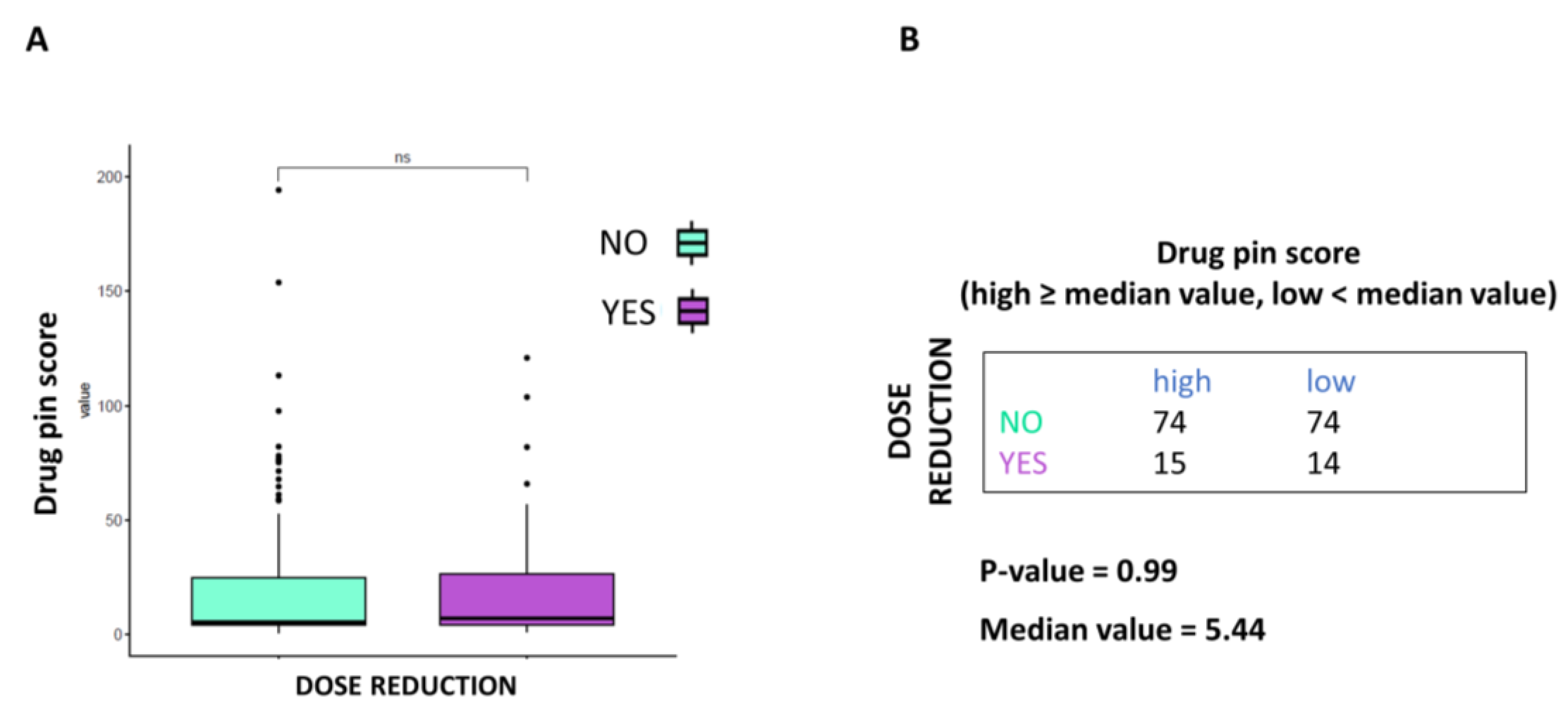
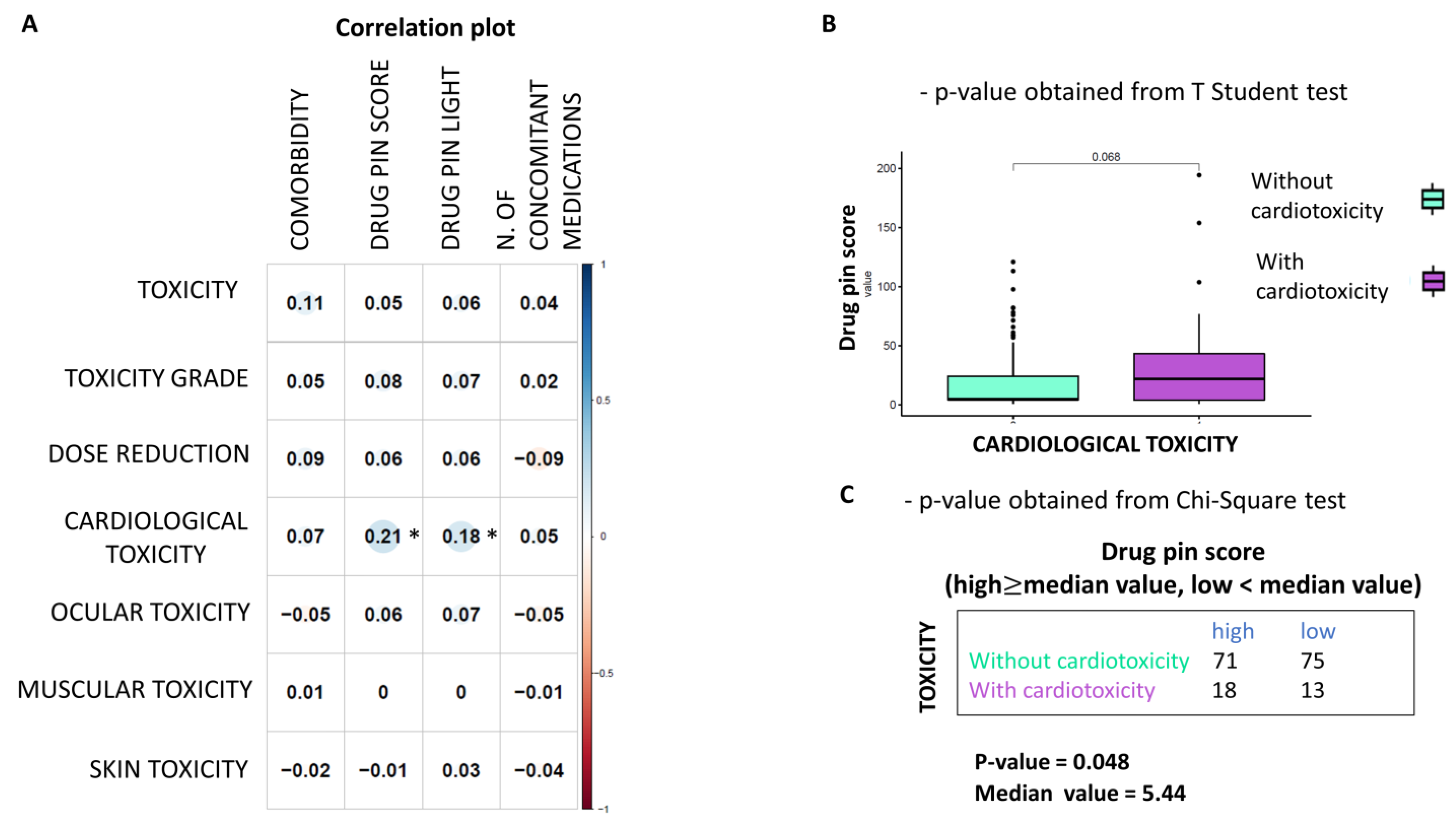
This study is not exempt from limitations. It failed to demonstrate that higher Drug-PIN scores and traffic lights are related to a higher number of toxicities of all types, high-grade toxicities, unexpected toxicities, and dose reduction. Nevertheless, even when the data did not reach statistical significance, patients with higher Drug-PIN scores tended to experience more toxicities in this series. Any significant associations between toxicity and the number of concomitant medications were found, suggesting that the type of interactions between drugs is more relevant than the number of interactions in the onset of the toxicity profile. Conversely, this study evidenced that a high Drug-PIN score was significantly associated with cardiovascular toxicity, predominantly constituted by acute or subacute clinical manifestations; more frequently, they were represented by disturbances of the repolarization, QT-interval prolongation, ventricular and supraventricular arrhythmias, conduction disorders and acute heart failure. Multiple regression analysis also confirmed the predictive role of Drug-PIN traffic light and score concerning the onset of cardiological toxicity, compared to several other clinical parameters (age, gender, performance status, comorbidity, and several concomitant drugs).
Cytochromes of the P450 superfamily of enzymes (CYPs) play a central role in the metabolism of BRAF-i/MEK-i drugs and drug–drug interaction onset. Numerous classes of drugs (cardiological and non), cytochromes inhibitors or inducers, can influence plasma concentrations of drugs and consequently treatment efficacy, related toxicity profile, and treatment adherence.
Both BRAFi and MEKi showed a cardiovascular toxicity profile and cardiovascular toxicity, which is more frequent when they are used in a combination therapy rather than in a monotherapy setting [
52]. BRAFi treatment is associated with hypertension and QT-interval prolongation, while hypertension, peripheral edema, and cardiomyopathy with decreased cardiac ejection fraction have been usually associated with MEKi treatment [
53,
54,
55,
56]. Cardiovascular AEs may be directly related to the effect that BRAFi and MEKi exert on the selective inhibition of the pathway in the heart. Cardiovascular side effects can be considered an on-target effect of BRAFi/MEKi treatment. In 15/31 patients, those who did not have overt cardiological comorbidities, did not take drugs with a cardiotoxic profile, and did not present any high DDI, cardiotoxicity onset could be considered predominantly related to the cardiotoxic effect of the BRAFi/MEKi treatments.
A second mechanism for the onset of cardiotoxicity could be ascribed to a synergistic effect between drugs, which have the same cardiac adverse effect, QT prolongation occurring in the 16 patients (9%) in our study. QT prolongation was observed in up to 3–7% of patients treated with V and 2% treated with V+C. Grade ≥ 3 QT-interval prolongation occurred in 1% of patients treated with V monotherapy or V+C. Due to an additional fluorinated phenyl ring, negative effects on QT prolongation were not seen with D or E. In this series, high-grade QT prolongation was observed in four patients (2.3%). This drug interaction could lead to early depolarizations, triggering the initiation of torsade de pointes ventricular tachycardia. A potential synergist effect related to DDI was underlined in QT prolongation during BRAFi/MEKi treatment in 15 patients who did not have cardiovascular disease at baseline and developed cardiotoxicity. The involved medication could be any other QT-prolonging drug (e.g., pantoprazole, antibacterial and antifungal agent ciprofloxacin) and ethanol, antiemetic and prokinetic agents, ondansetron, and psychotropic agents such as tricyclic antidepressants, citalopram, escitalopram, haloperidol, methadone, pimozide, and thioridazine, which were taken concomitantly enhancing DDI scores [
51].
In addition, the MAPK pathway in cardiomyocytes is a protective signaling pathway, and its pharmacological inhibition interferes with intramyocytic repair mechanisms by inhibiting the extracellular signal-regulated kinases ERK 1⁄2 [
51,
56,
57,
58,
59]. An impaired protective effect of the pathway may expose the myocardium more easily to damage. In this scenario, concomitant medications with high/intermediate DDIs may mediate subclinical cardiotoxicity or damage until significant left ventricular systolic dysfunction or even heart failure induced by the co-exposure of drugs with a potentially cardiological toxicity profile ensue. Non-steroidal anti-inflammatory drugs (NSAIDs) consumption for pain management may increase the risk of myocardial infarction [
60]. The increased risk is attributed to the imbalance caused by the inhibition of COX-2 cyclooxygenases and prostacyclin without inhibition of COX-1 and, therefore, thromboxane. They are also connected with platelet inhibition, oxidative stress, and impaired regulation of renal perfusion.
Similarly, some neurological and psychiatric drugs, cortisones, bisphosphonates, and some antibiotics (erythromycin) may have a potential cardiological toxicity profile that could become critical when intramyocyte repair mechanisms are impaired. Furthermore, a DDIs-dependent reduced availability of cardiac drugs may lead, in patients requiring concomitant cardiac treatment, to an under treatment of the cardiac comorbidity, which could become critical when cardiomyocytes exhibit pharmacological inhibition of the cardioprotective pathway of MEK. DDIs in these patients increased the risk for significant left ventricular systolic dysfunction or even heart failure induced by the concomitant or subsequent use of BRAFi and MEKi.
In cancer patients with concomitant cardiovascular pathologies, left ventricular dysfunction, ranging from asymptomatic changes (best diagnosed by echocardiographic strain analysis) to severe cardiac failure, is related to several factors. Both metabolic (diabetes, arterial hypertension, arterial disease, metabolic syndrome/obesity, BMI > 30 kg/m
2, etc.) and cardiological (genetic factors, age > 65 years, gender, underlying impaired myocardial function, and vascular status) comorbidities increase the risk of impaired myocardial function during treatment. Even here, the difference between therapeutic doses and doses evoking DDIs-related deleterious effects might not be clear, and the interindividual susceptibility difference is high. Furthermore, previous treatments, including immunotherapy-mediated subclinical cardiotoxicity or damage induced by radiotherapy, may enhance the risk of developing cardiotoxicity during therapy with BRAF/MEK inhibitors. Notably, other factors occurring during treatment, like electrolyte imbalance due to diarrhea, stomatitis, or long QT syndrome, could potentiate the AEs and should be promptly corrected [
29]. Therefore, the risks and benefits of BRAFi/MEKi therapy should be carefully evaluated in patients with significant heart disease and managed in a multidisciplinary consensus.
This study evidenced that DDI must be considered an additional risk factor for cardiological adverse reaction onset in this complex and multifactorial scenario. The risk of cardiological adverse reactions during treatment with BRAFi/MEKi increases when the Drug-PIN score rises, and the risk of cardiovascular AEs is significantly associated with drug interactions. Therefore, any DDIs must be assessed before cancer therapy, and then drug–drug interactions promptly removed when possible. It is crucial to remove an additional cardiac toxicity risk factor and promptly detect left ventricular dysfunction during treatment with BRAF/MEK inhibitors since it often results in discontinuation of the oncological therapy and, while most of the cardiac side effects could be adequately managed and are reversible with the interruption of treatment, fatal events considered to be due to arrhythmias or sudden cardiac death may occur.
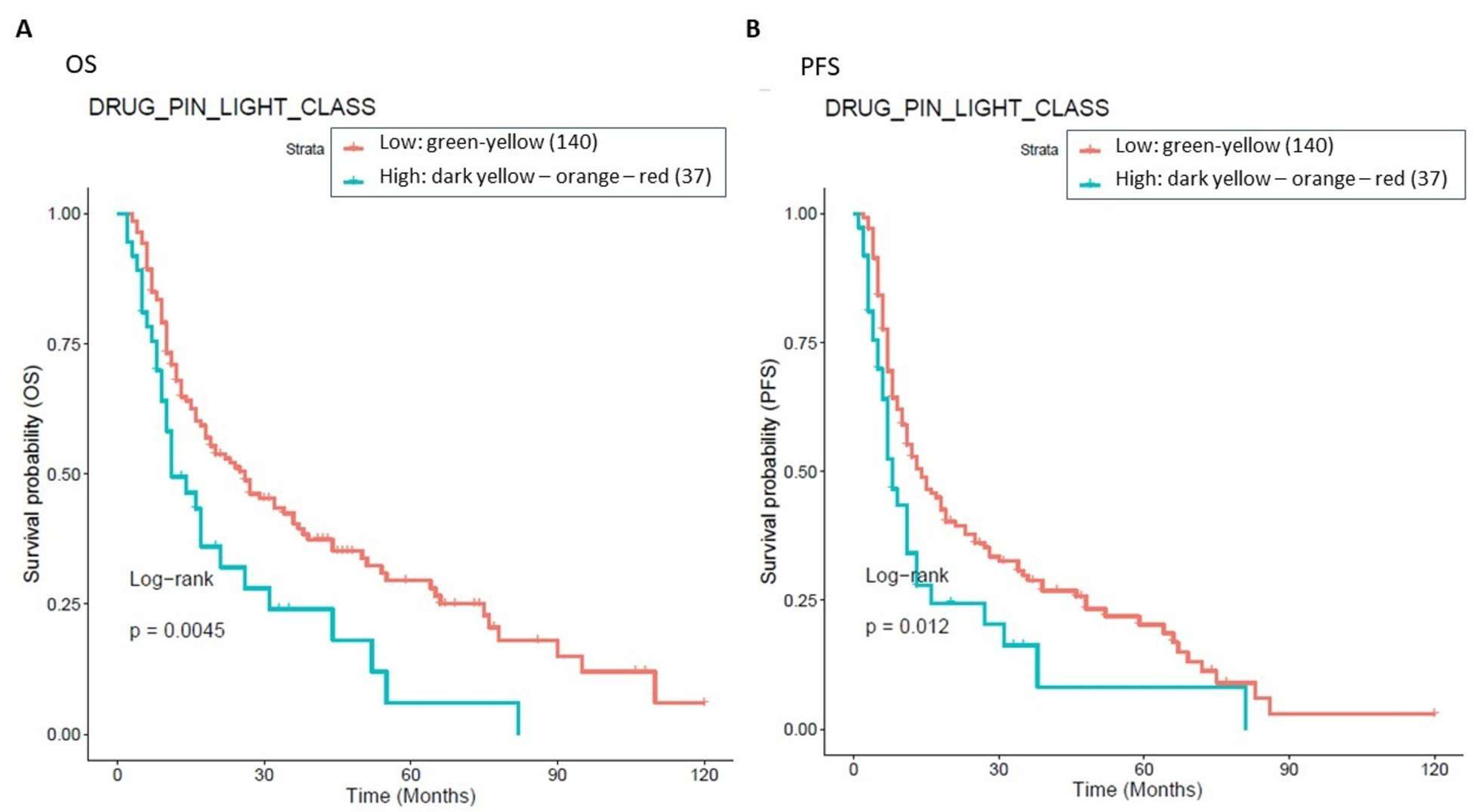
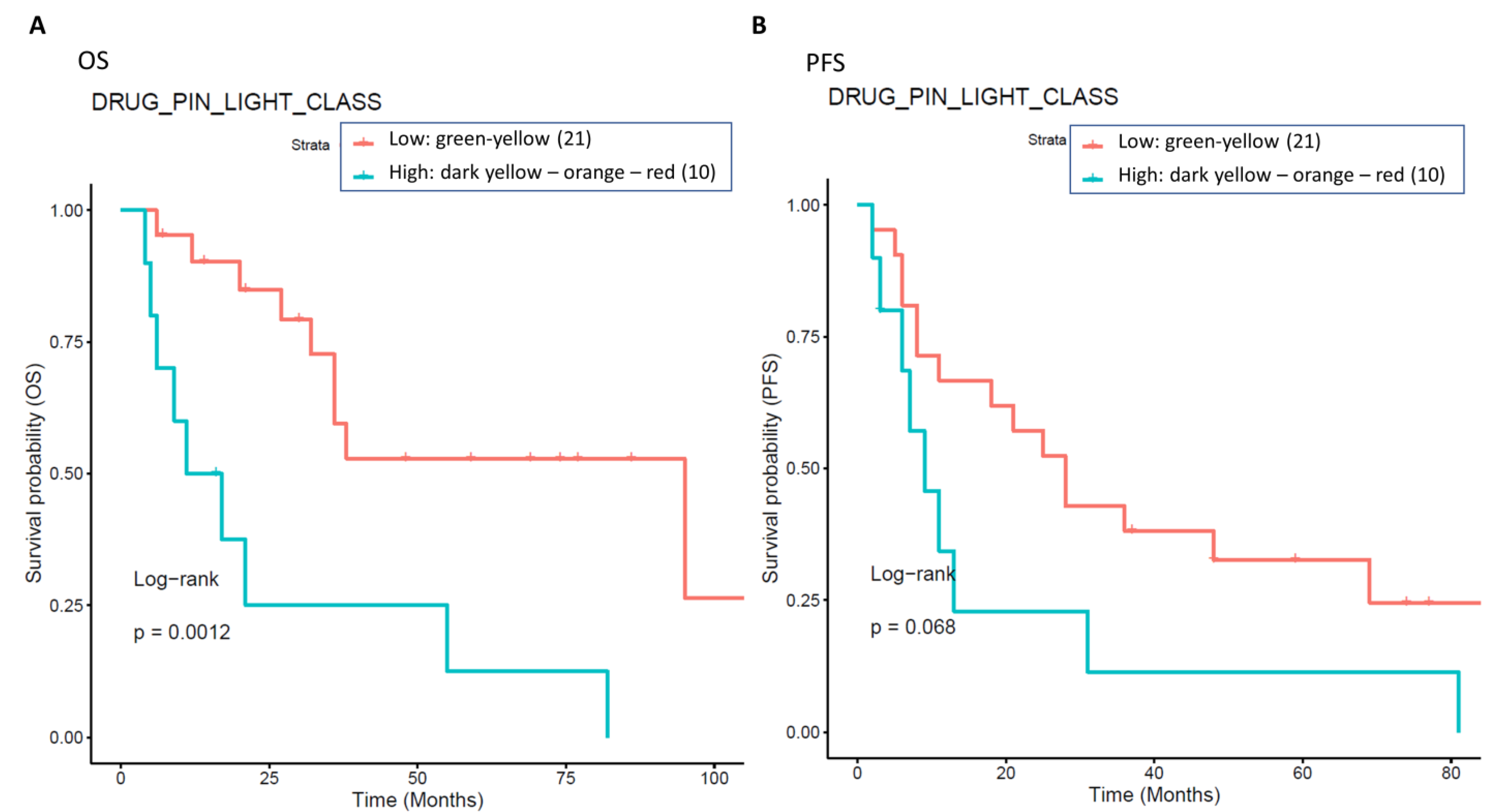
The presence of high-grade DDIs in approximately 30.5% (54 patients with Drug-PIN traffic light from dark yellow to red) of patients receiving BRAFi/MEKi does correlate with a significant reduction in OS and PFS. Survival analysis showed that in the overall population, a green-yellow Drug-PIN traffic light correlates with a better OS and PFS than a dark yellow, orange, or red Drug-PIN one. In addition, considering only patients who develop cardiological toxicity, the green-yellow Drug-PIN traffic light correlates with a better OS and PFS, confirming that a low number DDIs is likely to allow better adherence to treatment and ensure better efficacy of anti-BRAFi/MEKi therapy. On the other hand, the presence of any grade toxicity does not correlate with both OS and PFS, in contrast with the available literature. Recent evidence suggests that the development of immune-related toxicities such as vitiligo, keratitis, uveitis, and erythema nodosum under BRAFi/MEKi could be associated with long-term benefits in terms of oncological outcomes [
61].
The study’s main limitation is its retrospective nature, which only allows for a more in-depth investigation of some aspects of drug interactions. Indeed, these results suggest that the Drug-PIN may have a significant prognostic and predictive value for oncological outcomes and the development of severe toxicities that should be further investigated in prospective, targeted studies.



 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}