



Prognostic Biomarkers of Cell Proliferation in Colorectal Cancer (CRC): From Immunohistochemistry to Molecular Biology Techniques

Abstract
:Simple Summary
Abstract
1. Introduction
2. Molecular Mechanisms of Colorectal Cancerogenesis
3. Cellular Proliferation Models versus Colorectal Carcinogenesis Theories
4. Methods to Assess Cell Proliferation in Colorectal Cancer
4.1. Assessment of Mitosis in Cancer Tissues
4.2. DNA Ploidy and Percentage of Cells in S Phase
4.3. Immunohistochemical Methods for the Detection of Proliferative Markers
4.3.1. Thymidylate Synthase (TS) in CRC
4.3.2. Cyclins in CRC
4.3.3. Proliferating Cell Nuclear Antigen (PCNA) in CRC
4.3.4. Ki-67 Antigen in CRC
Ki-67 and Clinicopathologic Data in CRC Patients
Ki-67 as a Prognostic Marker in CRC
4.4. Modern Molecular Biology Techniques for the Assessment of Proliferative Markers in CRC
4.4.1. PCNA mRNA Expression
4.4.2. Ki-67 mRNA Expression
4.4.3. Non-Coding RNAs (ncRNAs) Expression
MicroRNA (miRNAs, miRs)
Long Non-Coding RNAs (LncRNAs)
4.4.4. Prognostic Genetic and Epigenetic Biomarkers
4.5. Positron Emission Tomography (PET) to Assess Tumor Growth Rate
5. Final Remarks and Future Perspectives
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ABCG2 | ATP-binding cassette super-family G member 2 |
| AC | Adenocarcinoma |
| AD | Adenoma |
| AKT | Serine/threonine kinase Akt, or protein kinase B (PKB) |
| ALDH1 | Aldehyde dehydrogenase 1 |
| APC | Adenomatous polyposis coli |
| BMPR1A | Bone morphogenetic protein receptor, type 1A |
| BRAF | Protooncogene B-Raf; encodes protein called B-Raf |
| Brd/Id/Urd | Bromo-, iodo-deoxyuridine |
| CAMs | Cell adhesion molecules |
| CCND2 | G1/S-specific cyclin-D2 |
| CD44 | CD44 molecule (Indian blood group), a cell-surface glycoprotein |
| Cdks | Cyclin-dependent kinases |
| CEA | Carcinoembryonic antigen |
| CCS | Colon cancer subtype |
| CI | Confidence interval |
| CIMP | CpG island methylator phenotype |
| CIN | Chromosomal instability |
| CMS | Consensus molecular subtype |
| CRC | Colorectal cancer |
| CRLM(s) | Colorectal cancer liver metastasis(es) |
| CSCs | Cancer stem cells |
| CSS | Cancer-specific survival |
| DCC | Deleted in colorectal cancer |
| DFS | Disease-free survival |
| EdU | 5-Ethynyl-2′-deoxyuridine |
| EGFR | Epidermal Growth Factor Receptor (HER1 in humans) |
| EIF3H | Eukaryotic translation initiation factor 3 subunit H |
| EMT | Epithelial-mesenchymal transition |
| ERK | Extracellular signal-regulated kinase or classical MAP kinase (MAPK) |
| FAP | Familial adenomatous polyposis |
| FCM | Flow cytometry |
| FDG | 18-Fluoro-2-deoxy-D-glucose |
| FLT | 18-Fluoro-3-deoxy-3-fluorothymidine |
| GALT | Gut-associated lymphoid tissue |
| GI | Gastrointestinal |
| GLUT1 | Glucose transporter 1 |
| GREM1 | Gremlin 1, DAN family BMP antagonist |
| HIF-1 | Hypoxia-inducible factor 1 |
| HNPCC | Hereditary non-polyposis colorectal cancer |
| HPs | Hyperplastic polyps |
| HPFs | High-power fields |
| HR | Hazard ratio |
| IGF-2 | Insulin growth factor 2 |
| IGFBP2 | IGF binding protein 2 |
| IHC | Immunohistochemistry |
| ISCs | Intestinal stem cells |
| ISH | In situ hybridization |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinase |
| KRAS/K-ras | Kirsten rat sarcoma virus; encodes protein called K-Ras |
| LGR5 | G-protein-coupled receptor 5 |
| LKB1 | Serine/threonine-protein kinase STK11 |
| LI | Labeling index |
| LS | Lynch syndrome |
| mAb(s) | Monoclonal antibody (antibodies) |
| MAPK | Mitogen-activated protein kinase |
| MCC | Colorectal mutant cancer protein |
| MKI67 | Marker of proliferation Ki-67 gene |
| MLH1, 2, 6 | MutL homolog 1, 2, 6 |
| MMR | DNA mismatch repair |
| MSI-H | High microsatellite instability |
| MSH2 | MutS homolog 2 |
| MSS | Microsatellite stable |
| MUTYH | E. coli MutY homolog |
| NCAMs | Neural cell adhesion molecules |
| OS | Overall survival |
| PCNA | Proliferating cell nuclear antigen |
| PFS | Progression-free survival |
| PI | Proliferating index |
| PI3K | Phosphatidylinositol-3-kinase |
| PKM2 | Pyruvate kinase M2 |
| PMS2 | Mismatch repair endonuclease 2 |
| PP1 | Protein phosphatase 1 |
| PTEN | Phosphatase and tensin homolog deleted on chromosome ten |
| RAS | Oncogene “Rat sarcoma virus” from three Ras genes: HRAS, KRAS and NRAS |
| RC | Rectal cancer |
| RFS | Relapse/recurrence-free survival |
| RT | Radiotherapy |
| RT-PCR/qRT-PCR | Reverse transcriptase-polymerase chain reaction; quantitative real-time PCR |
| SMAD4 | SMAD family member 4, Mothers Against DPP Homolog 4 |
| SOX2 | Transcription factor 2, known also as sex determining region Y (SRY)-box 2 |
| SPs | Serrated polyps |
| SPS | Serrated polyposis syndrome |
| SSLs | Sessile serrated lesions |
| STAT3 | Signal transducer and activator of transcription 3 |
| TCGA | The cancer genome atlas |
| TGF-β | Transforming growth factor beta |
| TMA | Tissue microarray |
| TME | Tumor microenvironment |
| TNM | Tumor-node-metastasis |
| TP53/p53 | Tumor gene/protein 53 |
| Tpot | The potential tumor doubling time |
| Ts | Duration of S phase |
| TS | Thymidylate synthase |
| VEGF | Vascular endothelial growth factor |
| Wnt/WNT | Gene wingless + integrated or int-1 |
References
- Ahmed, M. Colon Cancer: A Clinician’s Perspective in 2019. Gastroenterol. Res. 2020, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef]
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Cho, K.R.; Vogelstein, B. Suppressor gene alterations in the colorectal adenoma-carcinoma sequence. J. Cell. Biochem. Suppl. 1992, 16G, 137–141. [Google Scholar] [CrossRef]
- Dariya, B.; Aliya, S.; Merchant, N.; Alam, A.; Nagaraju, G.P. Colorectal Cancer Biology, Diagnosis, and Therapeutic Approaches. Crit. Rev. Oncog. 2020, 25, 71–94. [Google Scholar] [CrossRef]
- Leedham, S.J.; Schier, S.; Thliveris, A.T.; Halberg, R.B.; Newton, M.A.; Wright, N.A. From gene mutations to tumours—Stem cells in gastrointestinal carcinogenesis. Cell Prolif. 2005, 38, 387–405. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Fabrizi, E.; Palio, E.; De Maria, R. Colon cancer stem cells. J. Mol. Med. 2009, 87, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009, 69, 3382–3389. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef]
- Dieter, S.M.; Ball, C.R.; Hoffmann, C.M.; Nowrouzi, A.; Herbst, F.; Zavidij, O.; Abel, U.; Arens, A.; Weichert, W.; Brand, K.; et al. Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell 2011, 9, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Dieter, S.M.; Glimm, H.; Ball, C.R. Colorectal cancer-initiating cells caught in the act. EMBO Mol. Med. 2017, 9, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.; Yan, P. Molecular pathology of colorectal cancer. Pol. J. Pathol. 2014, 65, 257–266. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Cheng, L.; Lai, M.D. Aberrant crypt foci as microscopic precursors of colorectal cancer. World J. Gastroenterol. 2003, 9, 2642–2649. [Google Scholar] [CrossRef]
- Kowalczyk, M.; Orłowski, M.; Klepacki, Ł.; Zinkiewicz, K.; Kurpiewski, W.; Kaczerska, D.; Pesta, W.; Zieliński, E.; Siermontowski, P. Rectal aberrant crypt foci (ACF) as a predictor of benign and malignant neoplastic lesions in the large intestine. BMC Cancer 2020, 20, 133. [Google Scholar] [CrossRef]
- Lynch, H.T.; Kimberling, W.; Albano, W.A.; Lynch, J.F.; Biscone, K.; Schuelke, G.S.; Sandberg, A.A.; Lipkin, M.; Deschner, E.E.; Mikol, Y.B.; et al. Hereditary nonpolyposis colorectal cancer (Lynch syndromes I and II). I. Clinical description of resource. Cancer 1985, 56, 934–938. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kang, G.H. Evolving pathologic concepts of serrated lesions of the colorectum. J. Pathol. Transl. Med. 2020, 54, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Jass, J.R. Molecular heterogeneity of colorectal cancer: Implications for cancer control. Surg. Oncol. 2007, 16 (Suppl. S1), S7–S9. [Google Scholar] [CrossRef]
- Snover, D.C. Update on the serrated pathway to colorectal carcinoma. Hum. Pathol. 2011, 42, 1–10. [Google Scholar] [CrossRef]
- Patai, A.V.; Molnár, B.; Tulassay, Z.; Sipos, F. Serrated pathway: Alternative route to colorectal cancer. World J. Gastroenterol. 2013, 19, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Thorlacius, H.; Takeuchi, Y.; Kanesaka, T.; Ljungberg, O.; Uedo, N.; Toth, E. Serrated polyps—A concealed but prevalent precursor of colorectal cancer. Scand. J. Gastroenterol. 2017, 52, 654–661. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular features and mouse models of colorectal cancer. Trans. Am. Clin. Climatol. Assoc. 2018, 129, 56–62. [Google Scholar]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef]
- Huxley, R.R.; Ansary-Moghaddam, A.; Clifton, P.; Czernichow, S.; Parr, C.L.; Woodward, M. The impact of dietary and lifestyle risk factors on risk of colorectal cancer: A quantitative overview of the epidemiological evidence. Int. J. Cancer. 2009, 125, 171–180. [Google Scholar] [CrossRef]
- Mandic, M.; Li, H.; Safizadeh, F.; Niedermaier, T.; Hoffmeister, M.; Brenner, H. Is the association of overweight and obesity with colorectal cancer underestimated? An umbrella review of systematic reviews and meta-analyses. Eur. J. Epidemiol. 2023, 38, 135–144. [Google Scholar] [CrossRef]
- Polyak, K.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Early alteration of cell-cycle-regulated gene expression in colorectal neoplasia. Am. J. Pathol. 1996, 149, 381–387. [Google Scholar] [PubMed]
- Harada, S.; Morlote, D. Molecular Pathology of Colorectal Cancer. Adv. Anat. Pathol. 2020, 27, 20–26. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, J.; Zellmer, L.; Xu, N.; Liu, M.; Hu, Y.; Ma, H.; Deng, F.; Yang, W.; Liao, D.J. Mutation or not, what directly establishes a neoplastic state, namely cellular immortality and autonomy, still remains unknown and should be prioritized in our research. J. Cancer 2022, 13, 2810–2843. [Google Scholar] [CrossRef] [PubMed]
- Boman, B.M.; Huang, E. Human colon cancer stem cells: A new paradigm in gastrointestinal oncology. J. Clin. Oncol. 2008, 26, 2828–2838. [Google Scholar] [CrossRef] [PubMed]
- Simms, L.; Barraclough, H.; Govindan, R. Biostatistics primer: What a clinician ought to know—Prognostic and predictive factors. J. Thorac. Oncol. 2013, 8, 808–813. [Google Scholar] [CrossRef]
- National Cancer Institute Dictionary of Cancer Terms. Definition of Prognostic Factor. Available online: http://www.cancer.gov/dictionary?CdrID=44245 (accessed on 14 May 2023).
- Powell, S.M.; Zilz, N.; Beazer-Barclay, Y.; Bryan, T.M.; Hamilton, S.R.; Thibodeau, S.N.; Vogelstein, B.; Kinzler, K.W. APC mutations occur early during colorectal tumorigenesis. Nature 1992, 359, 235–237. [Google Scholar] [CrossRef]
- Cottrell, S.; Bicknell, D.; Kaklamanis, L.; Bodmer, W.F. Molecular analysis of APC mutations in familial adenomatous polyposis and sporadic colon carcinomas. Lancet 1992, 340, 626–630. [Google Scholar] [CrossRef]
- Giles, R.H.; van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 2003, 1653, 1–24. [Google Scholar] [CrossRef]
- Hedrick, L.; Cho, K.R.; Fearon, E.R.; Wu, T.C.; Kinzler, K.W.; Vogelstein, B. The DCC gene product in cellular differentiation and colorectal tumorigenesis. Genes Dev. 1994, 8, 1174–1183. [Google Scholar] [CrossRef]
- Fearon, E.R.; Pierceall, W.E. The deleted in colorectal cancer (DCC) gene: A candidate tumour suppressor gene encoding a cell surface protein with similarity to neural cell adhesion molecules. Cancer Surv. 1995, 24, 3–17. [Google Scholar]
- Llambi, F.; Causeret, F.; Bloch-Gallego, E.; Mehlen, P. Netrin-1 acts as a survival factor via its receptors UNC5H and DCC. EMBO J. 2001, 20, 2715–2722. [Google Scholar] [CrossRef]
- Nakayama, H.; Ohnuki, H.; Nakahara, M.; Nishida-Fukuda, H.; Sakaue, T.; Fukuda, S.; Higashiyama, S.; Doi, Y.; Mitsuyoshi, M.; Okimoto, T.; et al. Inactivation of axon guidance molecule netrin-1 in human colorectal cancer by an epigenetic mechanism. Biochem. Biophys. Res. Commun. 2022, 611, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Munro, A.J.; Lain, S.; Lane, D.P. P53 abnormalities and outcomes in colorectal cancer: A systematic review. Br. J. Cancer 2005, 92, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Kothalawala, W.J.; Barták, B.K.; Nagy, Z.B.; Zsigrai, S.; Szigeti, K.A.; Valcz, G.; Takács, I.; Kalmár, A.; Molnár, B. A Detailed Overview About the Single-Cell Analyses of Solid Tumors Focusing on Colorectal Cancer. Pathol. Oncol. Res. 2022, 28, 1610342. [Google Scholar] [CrossRef] [PubMed]
- Cerretelli, G.; Ager, A.; Arends, M.J.; Frayling, I.M. Molecular pathology of Lynch syndrome. J. Pathol. 2020, 250, 518–531. [Google Scholar] [CrossRef]
- Briggs, S.; Tomlinson, I. Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J. Pathol. 2013, 230, 148–153. [Google Scholar] [CrossRef]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A. WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef]
- Teodoridis, J.M.; Hardie, C.; Brown, R. CpG island methylator phenotype (CIMP) in cancer: Causes and implications. Cancer Lett. 2008, 268, 177–186. [Google Scholar] [CrossRef]
- Arnau-Collell, C.; Soares de Lima, Y.; Díaz-Gay, M.; Muñoz, J.; Carballal, S.; Bonjoch, L.; Moreira, L.; Lozano, J.J.; Ocaña, T.; Cuatrecasas, M.; et al. Colorectal cancer genetic variants are also associated with serrated polyposis syndrome susceptibility. J. Med. Genet. 2020, 57, 677–682. [Google Scholar] [CrossRef]
- Hang, D.; Joshi, A.D.; He, X.; Chan, A.T.; Jovani, M.; Gala, M.K.; Ogino, S.; Kraft, P.; Turman, C.; Peters, U.; et al. Colorectal cancer susceptibility variants and risk of conventional adenomas and serrated polyps: Results from three cohort studies. Int. J. Epidemiol. 2020, 49, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Rubio, C.A.; Puppa, G.; de Petris, G.; Kis, L.; Schmidt, P.T. The third pathway of colorectal carcinogenesis. J. Clin. Pathol. 2018, 71, 7–11. [Google Scholar] [CrossRef]
- Sadanandam, A.; Wang, X.; de Sousa E Melo, F.; Gray, J.W.; Vermeulen, L.; Hanahan, D.; Medema, J.P. Reconciliation of classification systems defining molecular subtypes of colorectal cancer: Interrelationships and clinical implications. Cell Cycle 2014, 13, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Kamel, F.; Eltarhoni, K.; Nisar, P.; Soloviev, M. Colorectal Cancer Diagnosis: The Obstacles We Face in Determining a Non-Invasive Test and Current Advances in Biomarker Detection. Cancers 2022, 14, 1889. [Google Scholar] [CrossRef] [PubMed]
- Suryadinata, R.; Sadowski, M.; Sarcevic, B. Control of cell cycle progression by phosphorylation of cyclin-dependent kinase (CDK) substrates. Biosci. Rep. 2010, 30, 243–255. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Alonso, D.; Malumbres, M. Mammalian cell cycle cyclins. Semin. Cell Dev. Biol. 2020, 107, 28–35. [Google Scholar] [CrossRef]
- Dang, F.; Nie, L.; Wei, W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2021, 28, 427–438. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef]
- Doonan, J.H.; Kitsios, G. Functional evolution of cyclin-dependent kinases. Mol. Biotechnol. 2009, 42, 14–29. [Google Scholar] [CrossRef]
- Loyer, P.; Trembley, J.H. Roles of CDK/Cyclin complexes in transcription and pre-mRNA splicing: Cyclins L and CDK11 at the cross-roads of cell cycle and regulation of gene expression. Semin. Cell Dev. Biol. 2020, 107, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef] [PubMed]
- Sonnenschein, C.; Soto, A.M.; Rangarajan, A.; Kulkarni, P. Competing views on cancer. J. Biosci. 2014, 39, 281–302. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Pardee, A.B. A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. USA 1974, 71, 1286–1290. [Google Scholar] [CrossRef]
- Wilson, G.D. Proliferation models in tumours. Int. J. Radiat. Biol. 2003, 79, 525–530. [Google Scholar] [CrossRef]
- Baisch, H.; Otto, U.; Hatje, U.; Fack, H. Heterogeneous cell kinetics in tumors analyzed with a simulation model for bromodeoxyuridine single and multiple labeling. Cytometry 1995, 21, 52–61. [Google Scholar] [CrossRef]
- Shackney, S.E.; Shankey, T.V. Cell cycle models for molecular biology and molecular oncology: Exploring new dimensions. Cytometry 1999, 35, 97–116. [Google Scholar] [CrossRef]
- Blanpain, C.; Horsley, V.; Fuchs, E. Epithelial stem cells: Turning over new leaves. Cell 2007, 128, 445–458. [Google Scholar] [CrossRef]
- Sender, R.; Milo, R. The distribution of cellular turnover in the human body. Nat. Med. 2021, 7, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Hammarlund, E.U.; Amend, S.R.; Pienta, K.J. The issues with tissues: The wide range of cell fate separation enables the evolution of multicellularity and cancer. Med. Oncol. 2020, 37, 62. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Huels, D.J.; Sansom, O.J. Stem vs non-stem cell origin of colorectal cancer. Br. J. Cancer 2015, 113, 1–5. [Google Scholar] [CrossRef]
- Shih, I.M.; Wang, T.L.; Traverso, G.; Romans, K.; Hamilton, S.R.; Ben-Sasson, S.; Kinzler, K.W.; Vogelstein, B. Top-down morphogenesis of colorectal tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 2640–2645. [Google Scholar] [CrossRef]
- Preston, S.L.; Wong, W.M.; Chan, A.O.; Poulsom, R.; Jeffery, R.; Goodlad, R.A.; Mandir, N.; Elia, G.; Novelli, M.; Bodmer, W.F.; et al. Bottom-up histogenesis of colorectal adenomas: Origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res. 2003, 63, 3819–3825. [Google Scholar]
- Cortina, C.; Turon, G.; Stork, D.; Hernando-Momblona, X.; Sevillano, M.; Aguilera, M.; Tosi, S.; Merlos-Suárez, A.; Stephan-Otto Attolini, C.; Sancho, E.; et al. A genome editing approach to study cancer stem cells in human tumors. EMBO Mol. Med. 2017, 9, 869–879. [Google Scholar] [CrossRef]
- De Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef]
- Szaryńska, M.; Olejniczak, A.; Kobiela, J.; Spychalski, P.; Kmieć, Z. Therapeutic strategies against cancer stem cells in human colorectal cancer. Oncol. Lett. 2017, 14, 7653–7668. [Google Scholar] [CrossRef]
- Alowaidi, F.; Hashimi, S.M.; Alqurashi, N.; Alhulais, R.; Ivanovski, S.; Bellette, B.; Meedenyia, A.; Lam, A.; Wood, S. Assessing stemness and proliferation properties of the newly established colon cancer ‘stem’ cell line, CSC480 and novel approaches to identify dormant cancer cells. Oncol. Rep. 2018, 39, 2881–2891. [Google Scholar] [CrossRef] [PubMed]
- Thirlwell, C.; Will, O.C.; Domingo, E.; Graham, T.A.; McDonald, S.A.; Oukrif, D.; Jeffrey, R.; Gorman, M.; Rodriguez-Justo, M.; Chin-Aleong, J.; et al. Clonality assessment and clonal ordering of individual neoplastic crypts shows polyclonality of colorectal adenomas. Gastroenterology 2010, 138, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Thliveris, A.T.; Clipson, L.; White, A.; Waggoner, J.; Plesh, L.; Skinner, B.L.; Zahm, C.D.; Sullivan, R.; Dove, W.F.; Newton, M.A.; et al. Clonal structure of carcinogen-induced intestinal tumors in mice. Cancer Prev. Res. 2011, 4, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Paganelli, G.M.; Faivre, J.; Biasco, G.; Scheppach, W.; Saldanha, M.H.; Beckly, D.E. Pattern of epithelial cell proliferation in colorectal mucosa of patients with large bowel adenoma or cancer: An ECP case-control study. European cancer prevention. Eur. J. Cancer Prev. 1999, 8, 401–407. [Google Scholar] [CrossRef]
- Tubiana, M. The kinetics of tumour cell proliferation and radiotherapy. Br. J. Radiol. 1971, 44, 325–347. [Google Scholar] [CrossRef]
- Colak, S.; Medema, J.P. Cancer stem cells—Important players in tumor therapy resistance. FEBS J. 2014, 281, 4779–4791. [Google Scholar] [CrossRef]
- Gasinska, A.; Adamczyk, A.; Niemiec, J.; Biesaga, B.; Darasz, Z.; Skolyszewski, J. Gender-related differences in pathological and clinical tumor response based on immunohistochemical proteins expression in rectal cancer patients treated with short course of preoperative radiotherapy. J. Gastrointest. Surg. 2014, 18, 1306–1318. [Google Scholar] [CrossRef]
- Trott, K.R.; Kummermehr, J. What is known about tumour proliferation rates to choose between accelerated fractionation or hyperfractionation? Radiother. Oncol. 1985, 3, 1–9. [Google Scholar] [CrossRef]
- Wilson, G.D. Cell kinetics. Clin. Oncol. R. Coll. Radiol. 2007, 19, 370–384. [Google Scholar] [CrossRef]
- Jin, F.; Luo, H.; Zhou, J.; Wu, Y.; Sun, H.; Liu, H.; Zheng, X.; Wang, Y. Dose-time fractionation schedules of preoperative radiotherapy and timing to surgery for rectal cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920907537. [Google Scholar] [CrossRef]
- Das, P.K.; Islam, F.; Lam, A.K. The Roles of Cancer Stem Cells and Therapy Resistance in Colorectal Carcinoma. Cells 2020, 9, 1392. [Google Scholar] [CrossRef] [PubMed]
- Hen, O.; Barkan, D. Dormant disseminated tumor cells and cancer stem/progenitor-like cells: Similarities and opportunities. Semin. Cancer Biol. 2020, 60, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Zimberlin, C.D.; Fessler, E.; Hogdal, L.; Prasetyanti, P.R.; Grandela, C.M.; Letai, A.; Medema, J.P. Decreased mitochondrial priming determines chemoresistance of colon cancer stem cells. Cell Death Differ. 2014, 21, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ishii, H.; Nishida, N.; Takemasa, I.; Mizushima, T.; Ikeda, M.; Yokobori, T.; Mimori, K.; Yamamoto, H.; Sekimoto, M.; et al. Significance of Lgr5(+ve) cancer stem cells in the colon and rectum. Ann. Surg. Oncol. 2011, 18, 1166–1174. [Google Scholar] [CrossRef]
- Oshima, C.T.; Iriya, K.; Forones, N.M. Ki-67 as a prognostic marker in colorectal cancer but not in gastric cancer. Neoplasma 2005, 52, 420–424. [Google Scholar]
- Ahadi, M.; Sokolova, A.; Brown, I.; Chou, A.; Gill, A.J. The 2019 World Health Organization Classification of appendiceal, colorectal and anal canal tumours: An update and critical assessment. Pathology 2021, 53, 454–461. [Google Scholar] [CrossRef]
- Cree, I.A.; Tan, P.H.; Travis, W.D.; Wesseling, P.; Yagi, Y.; White, V.A.; Lokuhetty, D.; Scolyer, R.A. Counting mitoses: SI(ze) matters! Mod. Pathol. 2021, 34, 1651–1657. [Google Scholar] [CrossRef]
- Potten, C.S.; Kellett, M.; Roberts, S.A.; Rew, D.A.; Wilson, G.D. Measurement of in vivo proliferation in human colorectal mucosa using bromodeoxyuridine. Gut 1992, 33, 71–78. [Google Scholar] [CrossRef]
- Palmqvist, R.; Oberg, A.; Bergström, C.; Rutegård, J.N.; Zackrisson, B.; Stenling, R. Systematic heterogeneity and prognostic significance of cell proliferation in colorectal cancer. Br. J. Cancer 1998, 77, 917–925. [Google Scholar] [CrossRef]
- Salud, A.; Porcel, J.M.; Raikundalia, B.; Camplejohn, R.S.; Taub, N.A. Prognostic significance of DNA ploidy, S-phase fraction, and P-glycoprotein expression in colorectal cancer. J. Surg. Oncol. 1999, 72, 167–174. [Google Scholar] [CrossRef]
- Rew, D.A.; Wilson, G.D. Cell production rates in human tissues and tumours and their significance. Part 1: An introduction to the techniques of measurement and their limitations. Eur. J. Surg. Oncol. 2000, 26, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Biasco, G.; Paganelli, G.M.; Santucci, R.; Brandi, G.; Barbara, L. Methodological problems in the use of rectal cell proliferation as a biomarker of colorectal cancer risk. J. Cell. Biochem. Suppl. 1994, 19, 55–60. [Google Scholar] [PubMed]
- Paganelli, G.M.; Lalli, E.; Facchini, A.; Biasco, G.; Santucci, R.; Brandi, G.; Barbara, L. Flow cytometry and in vitro tritiated thymidine labeling in normal rectal mucosa of patients at high risk of colorectal cancer. Am. J. Gastroenterol. 1994, 89, 220–224. [Google Scholar] [PubMed]
- Gerdes, J.; Lemke, H.; Baisch, H.; Wacker, H.H.; Schwab, U.; Stein, H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J. Immunol. 1984, 133, 1710–1715. [Google Scholar] [CrossRef] [PubMed]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Hall, P.A.; Levison, D.A. Review: Assessment of cell proliferation in histological material. J. Clin. Pathol. 1990, 43, 184–192. [Google Scholar] [CrossRef]
- Sawtell, R.M.; Rew, D.A.; Stradling, R.N.; Wilson, G.D. Pan cycle expression of proliferating cell nuclear antigen in human colorectal cancer and its proliferative correlations. Cytometry 1995, 22, 190–199. [Google Scholar] [CrossRef]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef]
- Wang, Y.L.; Zhang, H.W.; Mo, Y.Q.; Zhong, H.; Liu, W.M.; Lei, Y.; Lin, F. Application of dual-layer spectral detector computed tomography to evaluate the expression of Ki-67 in colorectal cancer. J. Chin. Med. Assoc. 2022, 85, 610–616. [Google Scholar] [CrossRef]
- Francis, D.L.; Freeman, A.; Visvikis, D.; Costa, D.C.; Luthra, S.K.; Novelli, M.; Taylor, I.; Ell, P.J. In vivo imaging of cellular proliferation in colorectal cancer using positron emission tomography. Gut 2003, 52, 1602–1606. [Google Scholar] [CrossRef]
- Francis, D.L.; Visvikis, D.; Costa, D.C.; Arulampalam, T.H.; Townsend, C.; Luthra, S.K.; Taylor, I.; Ell, P.J. Potential impact of [18F]3′-deoxy-3′-fluorothymidine versus [18F]fluoro-2-deoxy-D-glucose in positron emission tomography for colorectal cancer. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Harimoto, N.; Yokobori, T.; Araki, K.; Kubo, N.; Igarashi, T.; Tsukagoshi, M.; Ishii, N.; Yamanaka, T.; Handa, T.; et al. FDG-PET reflects tumor viability on SUV in colorectal cancer liver metastasis. Int. J. Clin. Oncol. 2020, 25, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Biasco, G.; Paganelli, G.M.; Miglioli, M.; Barbara, L. Cell proliferation biomarkers in the gastrointestinal tract. J. Cell. Biochem. Suppl. 1992, 16G, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Anjomshoaa, A.; Nasri, S.; Humar, B.; McCall, J.L.; Chatterjee, A.; Yoon, H.S.; McNoe, L.; Black, M.A.; Reeve, A.E. Slow proliferation as a biological feature of colorectal cancer metastasis. Br. J. Cancer 2009, 101, 822–828. [Google Scholar] [CrossRef]
- Tomita, T. DNA ploidy and proliferating cell nuclear antigen in colonic adenomas and adenocarcinomas. Dig. Dis. Sci. 1995, 40, 996–1004. [Google Scholar] [CrossRef]
- Barletta, A.; Marzullo, F.; Pellecchia, A.; Montemurro, S.; Labriola, A.; Lomonaco, R.; Grammatica, L.; Paradiso, A. DNA flow cytometry, p53 levels and proliferative cell nuclear antigen in human colon dysplastic, precancerous and cancerous tissues. Anticancer Res. 1998, 18, 1677–1682. [Google Scholar]
- Yigit, N.; Gunal, A.; Kucukodaci, Z.; Karslioglu, Y.; Onguru, O.; Ozcan, A. Are we counting mitoses correctly? Ann. Diagn. Pathol. 2013, 17, 536–539. [Google Scholar] [CrossRef]
- Kim, D.; Pantanowitz, L.; Schüffler, P.; Yarlagadda, D.V.K.; Ardon, O.; Reuter, V.E.; Hameed, M.; Klimstra, D.S.; Hanna, M.G. (Re) Defining the High-Power Field for Digital Pathology. J. Pathol. Inform. 2020, 11, 33. [Google Scholar] [CrossRef]
- Orr, B.; Godek, K.M.; Compton, D. Aneuploidy. Curr. Biol. 2015, 25, R538–R542. [Google Scholar] [CrossRef]
- Brás, R.; Sunkel, C.E.; Resende, L.P. Tissue stem cells: The new actors in the aneuploidy field. Cell Cycle 2019, 18, 1813–1823. [Google Scholar] [CrossRef]
- Ben-David, U.; Amon, A. Context is everything: Aneuploidy in cancer. Nat. Rev. Genet. 2020, 21, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Lazaris, A.C.; Davaris, P.; Nakopoulou, L.; Theodoropoulos, G.E.; Koullias, G.; Golematis, B.C. Correlation between immunohistochemical expression of proliferating cell nuclear antigen and flow cytometry parameters in colorectal neoplasia. Dis. Colon Rectum 1994, 37, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Anderson, E.; Bell, J.C.; Pearson, J.M.; Haboubi, N.Y.; James, R.D.; Schofield, P.F. An evaluation of five different methods for estimating proliferation in human colorectal adenocarcinomas. Surg. Oncol. 1994, 3, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Henk, M.J.; Carney, K.J.; Wong, W.D.; Rothenberger, D.A.; Zheng, T.; Feygin, M.; Madoff, R.D. Prognostic significance of tumor markers in colorectal cancer patients: DNA index, S-phase fraction, p53 expression, and Ki-67 index. J. Gastrointest. Surg. 1997, 1, 266–272, discussion 273. [Google Scholar] [CrossRef]
- Le Pessot, F.; Michel, P.; Paresy, M.; Lemoine, F.; Hellot, M.F.; Paillot, B.; Scotte, M.; Peillon, C.; Hemet, J. Cell proliferation in colorectal adenocarcinomas: Comparison between Ki-67 immunostaining and bromodeoxyuridine uptake detected by immunohistochemistry and flow cytometry. Pathol. Res. Pract. 2001, 197, 411–418. [Google Scholar] [CrossRef]
- Laubert, T.; Freitag-Wolf, S.; Linnebacher, M.; König, A.; Vollmar, B.; Habermann, J.K. North German Tumorbank of Colorectal Cancer (ColoNet) consortium. Stage-specific frequency and prognostic significance of aneuploidy in patients with sporadic colorectal cancer—A meta-analysis and current overview. Int. J. Color. Dis. 2015, 30, 1015–1028. [Google Scholar] [CrossRef]
- Lin, J.K.; Chang, S.C.; Yang, S.H.; Jiang, J.K.; Chen, W.C.; Lin, T.C. Prognostic value of DNA ploidy patterns of colorectal adenocarcinoma. Hepatogastroenterology 2003, 50, 1927–1932. [Google Scholar]
- Araujo, S.E.; Bernardo, W.M.; Habr-Gama, A.; Kiss, D.R.; Cecconello, I. DNA ploidy status and prognosis in colorectal cancer: A meta-analysis of published data. Dis. Colon Rectum 2007, 50, 1800–1810. [Google Scholar] [CrossRef]
- Crissman, J.D.; Zarbo, R.J.; Ma, C.K.; Visscher, D.W. Histopathologic parameters and DNA analysis in colorectal adenocarcinomas. Pathol. Annu. 1989, 24, 103–147. [Google Scholar]
- Michel, P.; Paresy, M.; Lepessot, F.; Hellot, M.F.; Paillot, B.; Scotte, M.; Peillon, C.; Ducrotté, P.; Hemet, J. Pre-operative kinetic parameter determination of colorectal adenocarcinomas. Prognostic significance. Eur. J. Gastroenterol. Hepatol. 2000, 12, 275–280. [Google Scholar] [CrossRef]
- Zarbo, R.J.; Nakhleh, R.E.; Brown, R.D.; Kubus, J.J.; Ma, C.K.; Mackowiak, P. Prognostic significance of DNA ploidy and proliferation in 309 colorectal carcinomas as determined by two-color multiparametric DNA flow cytometry. Cancer 1997, 79, 2073–2086. [Google Scholar] [CrossRef]
- Rew, D.A.; Wilson, G.D.; Taylor, I.; Weaver, P.C. Proliferation characteristics of human colorectal carcinomas measured in vivo. Br. J. Surg. 1991, 78, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Gasinska, A.; Skolyszewski, J.; Popiela, T.; Richter, P.; Darasz, Z.; Nowak, K.; Niemiec, J.; Biesaga, B.; Adamczyk, A.; Bucki, K.; et al. Bromodeoxyuridine labeling index as an indicator of early tumor response to preoperative radiotherapy in patients with rectal cancer. J. Gastrointest. Surg. 2007, 11, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Magaki, S.; Hojat, S.A.; Wei, B.; So, A.; Yong, W.H. An Introduction to the Performance of Immunohistochemistry. Methods Mol. Biol. 2019, 1897, 289–298. [Google Scholar] [CrossRef]
- Mauriello, S.; Treglia, M.; Pallocci, M.; Bonfiglio, R.; Giacobbi, E.; Passalacqua, P.; Cammarano, A.; D’Ovidio, C.; Marsella, L.T.; Scimeca, M. Antigenicity Preservation Is Related to Tissue Characteristics and the Post-Mortem Interval: Immunohistochemical Study and Literature Review. Healthcare 2022, 10, 1495. [Google Scholar] [CrossRef]
- Koo, M.; Squires, J.M.; Ying, D.; Huang, J. Making a Tissue Microarray. Methods Mol. Biol. 2019, 1897, 313–323. [Google Scholar] [CrossRef]
- Johnston, P.G.; Liang, C.M.; Henry, S.; Chabner, B.A.; Allegra, C.J. Production and characterization of monoclonal antibodies that localize human thymidylate synthase in the cytoplasm of human cells and tissue. Cancer Res. 1991, 51, 6668–6676. [Google Scholar]
- Showalter, S.L.; Showalter, T.N.; Witkiewicz, A.; Havens, R.; Kennedy, E.P.; Hucl, T.; Kern, S.E.; Yeo, C.J.; Brody, J.R. Evaluating the drug-target relationship between thymidylate synthase expression and tumor response to 5-fluorouracil. Is it time to move forward? Cancer Biol. Ther. 2008, 7, 986–994. [Google Scholar] [CrossRef]
- Rahman, L.; Voeller, D.; Rahman, M.; Lipkowitz, S.; Allegra, C.; Barrett, J.C.; Kaye, F.J.; Zajac-Kaye, M. Thymidylate synthase as an oncogene: A novel role for an essential DNA synthesis enzyme. Cancer Cell 2004, 5, 341–351. [Google Scholar] [CrossRef]
- Edler, D.; Glimelius, B.; Hallström, M.; Jakobsen, A.; Johnston, P.G.; Magnusson, I.; Ragnhammar, P.; Blomgren, H. Thymidylate synthase expression in colorectal cancer: A prognostic and predictive marker of benefit from adjuvant fluorouracil-based chemotherapy. J. Clin. Oncol. 2002, 20, 1721–1728. [Google Scholar] [CrossRef]
- Allegra, C.J.; Parr, A.L.; Wold, L.E.; Mahoney, M.R.; Sargent, D.J.; Johnston, P.; Klein, P.; Behan, K.; O’Connell, M.J.; Levitt, R.; et al. Investigation of the prognostic and predictive value of thymidylate synthase, p53, and Ki-67 in patients with locally advanced colon cancer. J. Clin. Oncol. 2002, 20, 1735–1743. [Google Scholar] [CrossRef]
- Badary, D.M.; Elkabsh, M.M.; Mady, H.H.; Gabr, A.; Kroosh, S.S. Prognostic and Predictive Role of Excision Repair Cross-complementation Group 1 and Thymidylate Synthase in Colorectal Carcinoma Patients Received FOLFOX Chemotherapy: An Immunohistochemical Study. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.K.; Singh, H.; Thareja, S.; Kumar, P. Regulation of thymidylate synthase: An approach to overcome 5-FU resistance in colorectal cancer. Med. Oncol. 2022, 40, 3. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.G.; Fisher, E.R.; Rockette, H.E.; Fisher, B.; Wolmark, N.; Drake, J.C.; Chabner, B.A.; Allegra, C.J. The role of thymidylate synthase expression in prognosis and outcome of adjuvant chemotherapy in patients with rectal cancer. J. Clin. Oncol. 1994, 12, 2640–2647. [Google Scholar] [CrossRef] [PubMed]
- Van Triest, B.; Pinedo, H.M.; Blaauwgeers, J.L.; van Diest, P.J.; Schoenmakers, P.S.; Voorn, D.A.; Smid, K.; Hoekman, K.; Hoitsma, H.F.; Peters, G.J. Prognostic role of thymidylate synthase, thymidine phosphorylase/platelet-derived endothelial cell growth factor, and proliferation markers in colorectal cancer. Clin. Cancer Res. 2000, 6, 1063–1072. [Google Scholar] [PubMed]
- Allegra, C.J.; Paik, S.; Colangelo, L.H.; Parr, A.L.; Kirsch, I.; Kim, G.; Klein, P.; Johnston, P.G.; Wolmark, N.; Wieand, H.S. Prognostic value of thymidylate synthase, Ki-67, and p53 in patients with Dukes’ B and C colon cancer: A National Cancer Institute-National Surgical Adjuvant Breast and Bowel Project collaborative study. J. Clin. Oncol. 2003, 21, 241–250. [Google Scholar] [CrossRef]
- Ciaparrone, M.; Quirino, M.; Schinzari, G.; Zannoni, G.; Corsi, D.C.; Vecchio, F.M.; Cassano, A.; La Torre, G.; Barone, C. Predictive role of thymidylate synthase, dihydropyrimidine dehydrogenase and thymidine phosphorylase expression in colorectal cancer patients receiving adjuvant 5-fluorouracil. Oncology 2006, 70, 366–377. [Google Scholar] [CrossRef]
- Kim, S.H.; Kwon, H.C.; Oh, S.Y.; Lee, D.M.; Lee, S.; Lee, J.H.; Roh, M.S.; Kim, D.C.; Park, K.; Choi, H.J.; et al. Prognostic value of ERCC1, thymidylate synthase, and glutathione S-transferase pi for 5-FU/oxaliplatin chemotherapy in advanced colorectal cancer. Am. J. Clin. Oncol. 2009, 32, 38–43. [Google Scholar] [CrossRef]
- Tsourouflis, G.; Theocharis, S.E.; Sampani, A.; Giagini, A.; Kostakis, A.; Kouraklis, G. Prognostic and predictive value of thymidylate synthase expression in colon cancer. Dig. Dis. Sci. 2008, 53, 1289–1296. [Google Scholar] [CrossRef]
- Popat, S.; Matakidou, A.; Houlston, R.S. Thymidylate synthase expression and prognosis in colorectal cancer: A systematic review and meta-analysis. J. Clin. Oncol. 2004, 22, 529–536. [Google Scholar] [CrossRef]
- Aschele, C.; Debernardis, D.; Tunesi, G.; Maley, F.; Sobrero, A. Thymidylate synthase protein expression in primary colorectal cancer compared with the corresponding distant metastases and relationship with the clinical response to 5-fluorouracil. Clin. Cancer Res. 2000, 6, 4797–4802. [Google Scholar] [PubMed]
- Niedzwiecki, D.; Hasson, R.M.; Lenz, H.J.; Ye, C.; Redston, M.; Ogino, S.; Fuchs, C.S.; Compton, C.C.; Mayer, R.J.; Goldberg, R.M.; et al. A Study of Thymidylate Synthase Expression as a Biomarker for Resectable Colon Cancer: Alliance (Cancer and Leukemia Group B) 9581 and 89803. Oncologist 2017, 22, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wu, Y.; Jin, J.; Yan, J.; Kuang, S.; Zhou, M.; Zhang, Y.; Guo, A.Y. Phylogenetic analysis reveals the evolution and diversification of cyclins in eukaryotes. Mol. Phylogenet. Evol. 2013, 66, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Bondi, J.; Husdal, A.; Bukholm, G.; Nesland, J.M.; Bakka, A.; Bukholm, I.R. Expression and gene amplification of primary (A, B1, D1, D3, and E) and secondary (C and H) cyclins in colon adenocarcinomas and correlation with patient outcome. J. Clin. Pathol. 2005, 58, 509–514. [Google Scholar] [CrossRef]
- Yam, C.H.; Fung, T.K.; Poon, R.Y. Cyclin A in cell cycle control and cancer. Cell. Mol. Life Sci. 2002, 59, 1317–1326. [Google Scholar] [CrossRef]
- Köhler, K.; Sanchez-Pulido, L.; Höfer, V.; Marko, A.; Ponting, C.P.; Snijders, A.P.; Feederle, R.; Schepers, A.; Boos, D. The Cdk8/19-cyclin C transcription regulator functions in genome replication through metazoan Sld7. PLoS Biol. 2019, 17, e2006767. [Google Scholar] [CrossRef]
- Koff, A.; Cross, F.; Fisher, A.; Schumacher, J.; Leguellec, K.; Philippe, M.; Roberts, J.M. Human cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell 1991, 66, 1217–1228. [Google Scholar] [CrossRef]
- Handa, K.; Yamakawa, M.; Takeda, H.; Kimura, S.; Takahashi, T. Expression of cell cycle markers in colorectal carcinoma: Superiority of cyclin A as an indicator of poor prognosis. Int. J. Cancer 1999, 84, 225–233. [Google Scholar] [CrossRef]
- Bahnassy, A.A.; Zekri, A.R.; El-Houssini, S.; El-Shehaby, A.M.; Mahmoud, M.R.; Abdallah, S.; El-Serafi, M. Cyclin A and cyclin D1 as significant prognostic markers in colorectal cancer patients. BMC Gastroenterol. 2004, 4, 22. [Google Scholar] [CrossRef]
- Edler, D.; Hallström, M.; Ragnhammar, P.; Blomgren, H. Thymidylate synthase expression in rectal cancer and proliferation, assessed by cyclin A and Ki-67 expression. Anticancer Res. 2002, 22, 3113–3116. [Google Scholar]
- Nozoe, T.; Inutsuka, S.; Honda, M.; Ezaki, T.; Korenaga, D. Clinicopathologic significance of cyclin A expression in colorectal carcinoma. J. Exp. Clin. Cancer Res. 2004, 23, 127–133. [Google Scholar]
- Zhao, D.B.; Chandler, I.; Chen, Z.M.; Pan, H.C.; Popat, S.; Shao, Y.F.; Houlston, R.S. Mismatch repair, minichromosome maintenance complex component 2, cyclin A, and transforming growth factor β receptor type II as prognostic factors for colorectal cancer: Results of a 10-year prospective study using tissue microarray analysis. Chin. Med. J. 2011, 124, 483–490. [Google Scholar] [PubMed]
- Li, J.Q.; Kubo, A.; Wu, F.; Usuki, H.; Fujita, J.; Bandoh, S.; Masaki, T.; Saoo, K.; Takeuchi, H.; Kobayashi, S.; et al. Cyclin B1, unlike cyclin G1, increases significantly during colorectal carcinogenesis and during later metastasis to lymph nodes. Int. J. Oncol. 2003, 22, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Grabsch, H.; Lickvers, K.; Hansen, O.; Takeno, S.; Willers, R.; Stock, W.; Gabbert, H.E.; Mueller, W. Prognostic value of cyclin B1 protein expression in colorectal cancer. Am. J. Clin. Pathol. 2004, 122, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Liang, X.; Jiang, W.; Li, J.; Xu, J.; Cai, X. Cyclin b1 suppresses colorectal cancer invasion and metastasis by regulating e-cadherin. PLoS ONE 2015, 10, e0126875. [Google Scholar] [CrossRef]
- Ye, C.; Wang, J.; Wu, P.; Li, X.; Chai, Y. Prognostic role of cyclin B1 in solid tumors: A meta-analysis. Oncotarget 2017, 8, 2224–2232. [Google Scholar] [CrossRef]
- Firestein, R.; Shima, K.; Nosho, K.; Irahara, N.; Baba, Y.; Bojarski, E.; Giovannucci, E.L.; Hahn, W.C.; Fuchs, C.S.; Ogino, S. CDK8 expression in 470 colorectal cancers in relation to beta-catenin activation, other molecular alterations and patient survival. Int. J. Cancer 2010, 126, 2863–2873. [Google Scholar] [CrossRef]
- Palaiologos, P.; Chrysikos, D.; Theocharis, S.; Kouraklis, G. The Prognostic Value of G1 Cyclins, p21 and Rb Protein in Patients with Colon Cancer. Anticancer Res. 2019, 39, 6291–6297. [Google Scholar] [CrossRef]
- Li, Y.; Wei, J.; Xu, C.; Zhao, Z.; You, T. Prognostic significance of cyclin D1 expression in colorectal cancer: A meta-analysis of observational studies. PLoS ONE 2014, 9, e94508. [Google Scholar] [CrossRef]
- Jun, S.Y.; Kim, J.; Yoon, N.; Maeng, L.S.; Byun, J.H. Prognostic Potential of Cyclin D1 Expression in Colorectal Cancer. J. Clin. Med. 2023, 12, 572. [Google Scholar] [CrossRef]
- Maeda, K.; Chung, Y.; Kang, S.; Ogawa, M.; Onoda, N.; Nishiguchi, Y.; Ikehara, T.; Nakata, B.; Okuno, M.; Sowa, M. Cyclin D1 overexpression and prognosis in colorectal adenocarcinoma. Oncology 1998, 55, 145–151. [Google Scholar] [CrossRef]
- Sarkar, R.; Hunter, I.A.; Rajaganeshan, R.; Perry, S.L.; Guillou, P.; Jayne, D.G. Expression of cyclin D2 is an independent predictor of the development of hepatic metastasis in colorectal cancer. Color. Dis. 2010, 12, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Li, Z.; Lou, C.; Zhang, Y. Expression of phosphorylated Stat5 predicts expression of cyclin D1 and correlates with poor prognosis of colonic adenocarcinoma. Int. J. Color. Dis. 2011, 26, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.X.; Chen, Y.; Li, Y.T.; Tang, J.; Ding, J.; Du, Y.; Wu, Q.; Liu, J.Y. Selected proliferation markers correlated with dynamics of growth in colorectal cancer. Eur. J. Cancer Prev. 2019, 28, 181–187. [Google Scholar] [CrossRef]
- Babic, A.; Miladinovic, N.; Milin Lazovic, J.; Milenkovic, S. Decreased ERβ expression and high cyclin D1 expression may predict early CRC recurrence in high-risk Duk’s B and Duke’s C stage. J. BUON 2021, 26, 536–543. [Google Scholar] [PubMed]
- Palmqvist, R.; Stenling, R.; Oberg, A.; Landberg, G. Expression of cyclin D1 and retinoblastoma protein in colorectal cancer. Eur. J. Cancer 1998, 34, 1575–1581. [Google Scholar] [CrossRef] [PubMed]
- Schwandner, O.; Bruch, H.P.; Broll, R. p21, p27, cyclin D1, and p53 in rectal cancer: Immunohistology with prognostic significance? Int. J. Color. Dis. 2002, 17, 11–19. [Google Scholar] [CrossRef]
- Ioachim, E. Expression patterns of cyclins D1, E and cyclin-dependent kinase inhibitors p21waf1/cip1, p27kip1 in colorectal carcinoma: Correlation with other cell cycle regulators (pRb, p53 and Ki-67 and PCNA) and clinicopathological features. Int. J. Clin. Pract. 2008, 62, 1736–1743. [Google Scholar] [CrossRef]
- Al-Maghrabi, J.; Mufti, S.; Gomaa, W.; Buhmeida, A.; Al-Qahtani, M.; Al-Ahwal, M. Immunoexpression of cyclin D1 in colorectal carcinomas is not correlated with survival outcome. J. Microsc. Ultrastruct. 2015, 3, 62–67. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Irahara, N.; Kure, S.; Shima, K.; Baba, Y.; Toyoda, S.; Chen, L.; Giovannucci, E.L.; Meyerhardt, J.A.; et al. A cohort study of cyclin D1 expression and prognosis in 602 colon cancer cases. Clin. Cancer Res. 2009, 15, 4431–4438. [Google Scholar] [CrossRef]
- Hilska, M.; Collan, Y.U.; O Laine, V.J.; Kössi, J.; Hirsimäki, P.; Laato, M.; Roberts, P.J. The significance of tumor markers for proliferation and apoptosis in predicting survival in colorectal cancer. Dis. Colon Rectum 2005, 48, 2197–2208. [Google Scholar] [CrossRef] [PubMed]
- Holland, T.A.; Elder, J.; McCloud, J.M.; Hall, C.; Deakin, M.; Fryer, A.A.; Elder, J.B.; Hoban, P.R. Subcellular localisation of cyclin D1 protein in colorectal tumours is associated with p21(WAF1/CIP1) expression and correlates with patient survival. Int. J. Cancer 2001, 95, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Jiang, F.; Yang, J. Association of β-Catenin, APC, SMAD3/4, Tp53, and Cyclin D1 Genes in Colorectal Cancer: A Systematic Review and Meta-Analysis. Genet. Res. 2022, 2022, 5338956. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, G.; Wang, H.L.; Wang, L. Analysis of expression of cyclin E, p27kip1 and Ki67 protein in colorectal cancer tissues and its value for diagnosis, treatment and prognosis of disease. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4874–4879. [Google Scholar] [PubMed]
- Melincovici, C.S.; Mihu, C.M.; Mărginean, M.; Boşca, A.B.; Coneac, A.; Moldovan, I.; Crişan, M. The prognostic significance of p53, Bax, Bcl-2 and cyclin E protein overexpression in colon cancer—An immunohistochemical study using the tissue microarray technique. Rom. J. Morphol. Embryol. 2016, 7, 81–89. [Google Scholar]
- Boehm, E.M.; Gildenberg, M.S.; Washington, M.T. The Many Roles of PCNA in Eukaryotic DNA Replication. Enzymes 2016, 39, 231–254. [Google Scholar] [CrossRef]
- Lavezzi, A.M.; Ottaviani, G.; De Ruberto, F.; Fichera, G.; Matturri, L. Prognostic significance of different biomarkers (DNA content, PCNA, karyotype) in colorectal adenomas. Anticancer Res. 2002, 22, 2077–2081. [Google Scholar]
- Al-Sheneber, I.F.; Shibata, H.R.; Sampalis, J.; Jothy, S. Prognostic significance of proliferating cell nuclear antigen expression in colorectal cancer. Cancer 1993, 71, 1954–1959. [Google Scholar] [CrossRef]
- Choi, H.J.; Jung, I.K.; Kim, S.S.; Hong, S.H. Proliferating cell nuclear antigen expression and its relationship to malignancy potential in invasive colorectal carcinomas. Dis. Colon Rectum 1997, 40, 51–59. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Oates, G.D.; Newbold, K.M.; Robson, A.M.; McConkey, C.; Powell, J. Cyclin/proliferation cell nuclear antigen immunohistochemistry does not improve the prognostic power of Dukes’ or Jass’ classifications for colorectal cancer. Br. J. Surg. 1995, 82, 184–187. [Google Scholar] [CrossRef]
- Mayer, A.; Takimoto, M.; Fritz, E.; Schellander, G.; Kofler, K.; Ludwig, H. The prognostic significance of proliferating cell nuclear antigen, epidermal growth factor receptor, and mdr gene expression in colorectal cancer. Cancer 1993, 71, 2454–2460. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.R.; Tanaka, S.; Haruma, K.; Yoshihara, M.; Sumii, K.; Kajiyama, G. Proliferating cell nuclear antigen expression at the invasive tumor margin predicts malignant potential of colorectal carcinomas. Cancer 1994, 73, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Tabuchi, Y.; Nakae, S.; Ohno, M.; Saitoh, Y. Serum carcinoembryonic antigen levels and proliferating cell nuclear antigen labeling index for patients with colorectal carcinoma. Correlation with tumor progression and survival. Cancer 1996, 77, 1741–1746. [Google Scholar] [CrossRef]
- Sandler, R.S.; Baron, J.A.; Tosteson, T.D.; Mandel, J.S.; Haile, R.W. Rectal mucosal proliferation and risk of colorectal adenomas: Results from a randomized controlled trial. Cancer Epidemiol. Biomark. Prev. 2000, 9, 653–656. [Google Scholar]
- Kunihiro, M.; Tanaka, S.; Haruma, K.; Yoshihara, M.; Sumii, K.; Kajiyama, G.; Shimamoto, F. Combined expression of HLA-DR antigen and proliferating cell nuclear antigen correlate with colorectal cancer prognosis. Oncology 1998, 55, 326–333. [Google Scholar] [CrossRef]
- Guzińska-Ustymowicz, K.; Pryczynicz, A.; Kemona, A.; Czyzewska, J. Correlation between proliferation markers: PCNA, Ki-67, MCM-2 and antiapoptotic protein Bcl-2 in colorectal cancer. Anticancer Res. 2009, 29, 3049–3052. [Google Scholar]
- Zhou, H.; Huang, T.; Xiong, Y.; Peng, L.; Wang, R.; Zhang, G.J. The prognostic value of proliferating cell nuclear antigen expression in colorectal cancer: A meta-analysis. Medicine 2018, 97, e13752. [Google Scholar] [CrossRef]
- Gerdes, J.; Schwab, U.; Lemke, H.; Stein, H. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int. J. Cancer 1983, 31, 13–20. [Google Scholar] [CrossRef]
- Schlüter, C.; Duchrow, M.; Wohlenberg, C.; Becker, M.H.; Key, G.; Flad, H.D.; Gerdes, J. The cell proliferation-associated antigen of antibody Ki-67: A very large, ubiquitous nuclear protein with numerous repeated elements, representing a new kind of cell cycle-maintaining proteins. J. Cell Biol. 1993, 123, 513–522. [Google Scholar] [CrossRef]
- Gerdes, J.; Li, L.; Schlueter, C.; Duchrow, M.; Wohlenberg, C.; Gerlach, C.; Stahmer, I.; Kloth, S.; Brandt, E.; Flad, H.D. Immunobiochemical and molecular biologic characterization of the cell proliferation-associated nuclear antigen that is defined by monoclonal antibody Ki-67. Am. J. Pathol. 1991, 138, 867–873. [Google Scholar]
- Brown, D.C.; Gatter, K.C. Monoclonal antibody Ki-67: Its use in histopathology. Histopathology 1990, 17, 489–503. [Google Scholar] [CrossRef]
- Chierico, L.; Rizzello, L.; Guan, L.; Joseph, A.S.; Lewis, A.; Battaglia, G. The role of the two splice variants and extranuclear pathway on Ki-67 regulation in non-cancer and cancer cells. PLoS ONE 2017, 12, e0171815. [Google Scholar] [CrossRef]
- Menon, S.S.; Guruvayoorappan, C.; Sakthivel, K.M.; Rasmi, R.R. Ki-67 protein as a tumour proliferation marker. Clin. Chim. Acta 2019, 491, 39–45. [Google Scholar] [CrossRef]
- Remnant, L.; Kochanova, N.Y.; Reid, C.; Cisneros-Soberanis, F.; Earnshaw, W.C. The intrinsically disorderly story of Ki-67. Open Biol. 2021, 11, 210120. [Google Scholar] [CrossRef]
- Andrés-Sánchez, N.; Fisher, D.; Krasinska, L. Physiological functions and roles in cancer of the proliferation marker Ki-67. J. Cell Sci. 2022, 135, jcs258932. [Google Scholar] [CrossRef]
- Luo, Z.W.; Zhu, M.G.; Zhang, Z.Q.; Ye, F.J.; Huang, W.H.; Luo, X.Z. Increased expression of Ki-67 is a poor prognostic marker for colorectal cancer patients: A meta analysis. BMC Cancer 2019, 19, 123. [Google Scholar] [CrossRef]
- Cheutin, T.; O’Donohue, M.F.; Beorchia, A.; Klein, C.; Kaplan, H.; Ploton, D. Three-dimensional organization of pKi-67: A comparative fluorescence and electron tomography study using FluoroNanogold. J. Histochem. Cytochem. 2003, 51, 1411–1423. [Google Scholar] [CrossRef]
- Bullwinkel, J.; Baron-Lühr, B.; Lüdemann, A.; Wohlenberg, C.; Gerdes, J.; Scholzen, T. Ki-67 protein is associated with ribosomal RNA transcription in quiescent and proliferating cells. J. Cell. Physiol. 2006, 206, 624–635. [Google Scholar] [CrossRef]
- Miller, I.; Min, M.; Yang, C.; Tian, C.; Gookin, S.; Carter, D.; Spencer, S.L. Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep. 2018, 24, 1105–1112.e5. [Google Scholar] [CrossRef]
- Stamatiou, K.; Vagnarelli, P. Chromosome clustering in mitosis by the nuclear protein Ki-67. Biochem. Soc. Trans. 2021, 49, 2767–2776. [Google Scholar] [CrossRef]
- Cuylen, S.; Blaukopf, C.; Politi, A.Z.; Müller-Reichert, T.; Neumann, B.; Poser, I.; Ellenberg, J.; Hyman, A.A.; Gerlich, D.W. Ki-67 acts as a biological surfactant to disperse mitotic chromosomes. Nature 2016, 535, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Cuylen-Haering, S.; Petrovic, M.; Hernandez-Armendariz, A.; Schneider, M.W.G.; Samwer, M.; Blaukopf, C.; Holt, L.J.; Gerlich, D.W. Chromosome clustering by Ki-67 excludes cytoplasm during nuclear assembly. Nature 2020, 587, 285–290. [Google Scholar] [CrossRef]
- Booth, D.G.; Takagi, M.; Sanchez-Pulido, L.; Petfalski, E.; Vargiu, G.; Samejima, K.; Imamoto, N.; Ponting, C.P.; Tollervey, D.; Earnshaw, W.C.; et al. Ki-67 is a PP1-interacting protein that organises the mitotic chromosome periphery. Elife 2014, 27, e01641. [Google Scholar] [CrossRef] [PubMed]
- Sobecki, M.; Mrouj, K.; Camasses, A.; Parisis, N.; Nicolas, E.; Llères, D.; Gerbe, F.; Prieto, S.; Krasinska, L.; David, A.; et al. The cell proliferation antigen Ki-67 organises heterochromatin. Elife 2016, 5, e13722. [Google Scholar] [CrossRef] [PubMed]
- Sales Gil, R.; Vagnarelli, P. Ki-67: More Hidden behind a ‘Classic Proliferation Marker’. Trends Biochem. Sci. 2018, 43, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Nayak, J.; Mohanty, P.; Lenka, A.; Sahoo, N.; Agrawala, S.; Panigrahi, S.K. Histopathological and Immunohistochemical Evaluation of CDX2 and Ki67 in Colorectal Lesions with their Expression Pattern in Different Histologic Variants, Grade, and Stage of Colorectal Carcinomas. J. Microsc. Ultrastruct. 2021, 9, 183–189. [Google Scholar] [CrossRef]
- Saleh, H.A.; Jackson, H.; Banerjee, M. Immunohistochemical expression of bcl-2 and p53 oncoproteins: Correlation with Ki67 proliferation index and prognostic histopathologic parameters in colorectal neoplasia. Appl. Immunohistochem. Mol. Morphol. 2000, 8, 175–812. [Google Scholar] [CrossRef]
- Ishida, H.; Sadahiro, S.; Suzuki, T.; Ishikawa, K.; Kamijo, A.; Tajima, T.; Makuuchi, H.; Murayama, C. Proliferative, infiltrative, and metastatic activities in colorectal tumors assessed by MIB-1 antibody. Oncol. Rep. 2003, 10, 1741–1745. [Google Scholar] [CrossRef]
- Woodland, J.G. CDX-2 and MIB-1 expression in the colorectum: Correlation with morphological features of adenomatous lesions. Br. J. Biomed. Sci. 2006, 63, 68–73. [Google Scholar] [CrossRef]
- Kitabatake, T.; Kojima, K.; Fukasawa, M.; Beppu, T.; Futagawa, S. Correlation of thymidine phosphorylase staining and the Ki-67 labeling index to clinicopathologic factors and hepatic metastasis in patients with colorectal cancer. Surg. Today 2002, 32, 322–328. [Google Scholar] [CrossRef]
- Dziegiel, P.; Forgacz, J.; Suder, E.; Surowiak, P.; Kornafel, J.; Zabel, M. Prognostic significance of metallothionein expression in correlation with Ki-67 expression in adenocarcinomas of large intestine. Histol. Histopathol. 2003, 18, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Valera, V.; Yokoyama, N.; Walter, B.; Okamoto, H.; Suda, T.; Hatakeyama, K. Clinical significance of Ki-67 proliferation index in disease progression and prognosis of patients with resected colorectal carcinoma. Br. J. Surg. 2005, 92, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Gurzu, S.; Jung, J.; Mezei, T.; Pávai, Z. The correlation between the immunostains for p53 and Ki67 with bcl-2 expression and classical prognostic factors in colorectal carcinomas. Rom. J. Morphol. Embryol. 2007, 48, 95–99. [Google Scholar]
- Ma, Y.L.; Peng, J.Y.; Zhang, P.; Liu, W.J.; Huang, L.; Qin, H.L. Immunohistochemical analysis revealed CD34 and Ki67 protein expression as significant prognostic factors in colorectal cancer. Med. Oncol. 2010, 27, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.F.; Amorim, R.; Mota, S.C.; Costa, L.; Pardal, F.; Rodrigues, M.; Longatto-Filho, A. Ki-67 Expression in CRC Lymph Node Metastasis Does Not Predict Survival. Biomed Res. Int. 2015, 2015, 131685. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Mitra, S.; Das, R.N.; Dasgupta, S.; Saha, K.; Chatterjee, U.; Mukherjee, K.; Datta, C.; Chattopadhyay, B.K. Expression of CDX-2 and Ki-67 in different grades of colorectal adenocarcinomas. Indian J. Pathol. Microbiol. 2015, 58, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Z.; Fisher, K.W.; Ren, F.; Lv, J.; Davidson, D.D.; Baldridge, L.A.; Du, X.; Cheng, L. Prognostic value of programmed death ligand 1, p53, and Ki-67 in patients with advanced-stage colorectal cancer. Hum. Pathol. 2018, 71, 20–29. [Google Scholar] [CrossRef]
- Tong, G.; Zhang, G.; Liu, J.; Zheng, Z.; Chen, Y.; Niu, P.; Xu, X. Cutoff of 25% for Ki67 expression is a good classification tool for prognosis in colorectal cancer in the AJCC-8 stratification. Oncol. Rep. 2020, 43, 1187–1198. [Google Scholar] [CrossRef]
- Lei, H.T.; Yan, S.; He, Y.H.; Xu, N.; Zhao, M.; Yu, C.J.; Li, H.L.; Kuang, S.; Cui, Z.H.; Fang, J. Ki67 testing in the clinical management of patients with non-metastatic colorectal cancer: Detecting the optimal cut-off value based on the Restricted Cubic Spline model. Oncol. Lett. 2022, 24, 420. [Google Scholar] [CrossRef]
- Lanza, G., Jr.; Cavazzini, L.; Borghi, L.; Ferretti, S.; Buccoliero, F.; Rubbini, M. Immunohistochemical assessment of growth fractions in colorectal adenocarcinomas with monoclonal antibody Ki-67. Relation to clinical and pathological variables. Pathol. Res. Pract. 1990, 186, 608–618. [Google Scholar] [CrossRef]
- Meteoglu, I.; Erdogdu, I.H.; Tuncyurek, P.; Coskun, A.; Culhaci, N.; Erkus, M.; Barutca, S. Nuclear Factor Kappa B, Matrix Metalloproteinase-1, p53, and Ki-67 Expressions in the Primary Tumors and the Lymph Node Metastases of Colorectal Cancer Cases. Gastroenterol. Res. Pract. 2015, 2015, 945392. [Google Scholar] [CrossRef] [PubMed]
- Ofner, D.; Grothaus, A.; Riedmann, B.; Larcher, P.; Maier, H.; Bankfalvi, A.; Schmid, K.W. MIB1 in colorectal carcinomas: Its evaluation by three different methods reveals lack of prognostic significance. Anal. Cell. Pathol. 1996, 12, 61–70. [Google Scholar] [PubMed]
- Jansson, A.; Sun, X.F. Ki-67 expression in relation to clinicopathological variables and prognosis in colorectal adenocarcinomas. APMIS 1997, 105, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.W.; Lee, J.H.; Kim, H.R. Expression of p16, p53, and Ki-67 in colorectal adenocarcinoma: A study of 356 surgically resected cases. Hepatogastroenterology 2010, 57, 734–740. [Google Scholar]
- Duchrow, M.; Ziemann, T.; Windhövel, U.; Bruch, H.P.; Broll, R. Colorectal carcinomas with high MIB-1 labelling indices but low pKi67 mRNA levels correlate with better prognostic outcome. Histopathology 2003, 42, 566–574. [Google Scholar] [CrossRef]
- Melling, N.; Kowitz, C.M.; Simon, R.; Bokemeyer, C.; Terracciano, L.; Sauter, G.; Izbicki, J.R.; Marx, A.H. High Ki67 expression is an independent good prognostic marker in colorectal cancer. J. Clin. Pathol. 2016, 69, 209–214. [Google Scholar] [CrossRef]
- Ivanecz, A.; Kavalar, R.; Palfy, M.; Pivec, V.; Sremec, M.; Horvat, M.; Potrč, S. Can we improve the clinical risk score? The prognostic value of p53, Ki-67 and thymidylate synthase in patients undergoing radical resection of colorectal liver metastases. HPB 2014, 16, 235–242. [Google Scholar] [CrossRef]
- Fernebro, E.; Bendahl, P.O.; Dictor, M.; Persson, A.; Fernö, M.; Nilbert, M. Immunohistochemical patterns in rectal cancer: Application of tissue microarray with prognostic correlations. Int. J. Cancer 2004, 111, 921–928. [Google Scholar] [CrossRef]
- Ishida, H.; Miwa, H.; Tatsuta, M.; Masutani, S.; Imamura, H.; Shimizu, J.; Ezumi, K.; Kato, H.; Kawasaki, T.; Furukawa, H.; et al. Ki-67 and CEA expression as prognostic markers in Dukes’ C colorectal cancer. Cancer Lett. 2004, 207, 109–115. [Google Scholar] [CrossRef]
- Lumachi, F.; Orlando, R.; Marino, F.; Chiara, G.B.; Basso, S.M. Expression of p53 and Ki-67 as prognostic factors for survival of men with colorectal cancer. Anticancer Res. 2012, 32, 3965–3967. [Google Scholar]
- Hur, H.; Tulina, I.; Cho, M.S.; Min, B.S.; Koom, W.S.; Lim, J.S.; Ahn, J.B.; Kim, N.K. Biomarker-Based Scoring System for Prediction of Tumor Response After Preoperative Chemoradiotherapy in Rectal Cancer by Reverse Transcriptase Polymerase Chain Reaction Analysis. Dis. Colon Rectum 2016, 59, 1174–1182. [Google Scholar] [CrossRef]
- Valera, V.A.; Walter, B.A.; Yokoyama, N.; Koyama, Y.; Iiai, T.; Okamoto, H.; Hatakeyama, K. Prognostic groups in colorectal carcinoma patients based on tumor cell proliferation and classification and regression tree (CART) survival analysis. Ann. Surg. Oncol. 2007, 14, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Erstad, D.J.; Tumusiime, G.; Cusack, J.C., Jr. Prognostic and Predictive Biomarkers in Colorectal Cancer: Implications for the Clinical Surgeon. Ann. Surg. Oncol. 2015, 22, 3433–3450. [Google Scholar] [CrossRef] [PubMed]
- Das, V.; Kalita, J.; Pal, M. Predictive and prognostic biomarkers in colorectal cancer: A systematic review of recent advances and challenges. Biomed. Pharmacother. 2017, 87, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Caramaschi, S.; Mangogna, A.; Salviato, T.; Ammendola, S.; Barresi, V.; Manco, G.; Canu, P.G.; Zanelli, G.; Bonetti, L.R. Cytoproliferative activity in colorectal poorly differentiated clusters: Biological significance in tumor setting. Ann. Diagn. Pathol. 2021, 53, 151772. [Google Scholar] [CrossRef]
- Palmqvist, R.; Sellberg, P.; Oberg, A.; Tavelin, B.; Rutegård, J.N.; Stenling, R. Low tumour cell proliferation at the invasive margin is associated with a poor prognosis in Dukes’ stage B colorectal cancers. Br. J. Cancer 1999, 79, 577–581. [Google Scholar] [CrossRef]
- Salminen, E.; Palmu, S.; Vahlberg, T.; Roberts, P.J.; Söderström, K.O. Increased proliferation activity measured by immunoreactive Ki67 is associated with survival improvement in rectal/recto sigmoid cancer. World J. Gastroenterol. 2005, 11, 3245–3249. [Google Scholar] [CrossRef]
- Xi, H.Q.; Zhao, P. Clinicopathological significance and prognostic value of EphA3 and CD133 expression in colorectal carcinoma. J. Clin. Pathol. 2011, 64, 498–503. [Google Scholar] [CrossRef]
- Li, P.; Xiao, Z.T.; Braciak, T.A.; Ou, Q.J.; Chen, G.; Oduncu, F.S. Association between Ki67 Index and Clinicopathological Features in Colorectal Cancer. Oncol. Res. Treat. 2016, 39, 696–702. [Google Scholar] [CrossRef]
- Petrowsky, H.; Sturm, I.; Graubitz, O.; Kooby, D.A.; Staib-Sebler, E.; Gog, C.; Köhne, C.H.; Hillebrand, T.; Daniel, P.T.; Fong, Y.; et al. Relevance of Ki-67 antigen expression and K-ras mutation in colorectal liver metastases. Eur. J. Surg. Oncol. 2001, 27, 80–87. [Google Scholar] [CrossRef]
- Nash, G.M.; Gimbel, M.; Shia, J.; Nathanson, D.R.; Ndubuisi, M.I.; Zeng, Z.S.; Kemeny, N.; Paty, P.B. KRAS mutation correlates with accelerated metastatic progression in patients with colorectal liver metastases. Ann. Surg. Oncol. 2010, 17, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Torén, W.; Ansari, D.; Andersson, R. Immunohistochemical investigation of prognostic biomarkers in resected colorectal liver metastases: A systematic review and meta-analysis. Cancer Cell Int. 2018, 18, 217. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K.; Shimada, M.; Higashijima, J.; Nakao, T.; Nishi, M.; Takasu, C.; Kashihara, H.; Eto, S.; Bando, Y. Ki-67 and Survivin as Predictive Factors for Rectal Cancer Treated with Preoperative Chemoradiotherapy. Anticancer Res. 2018, 38, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Taha, A.; Taha-Mehlitz, S.; Petzold, S.; Achinovich, S.L.; Zinovkin, D.; Enodien, B.; Pranjol, M.Z.I.; Nadyrov, E.A. Prognostic Value of Immunohistochemical Markers for Locally Advanced Rectal Cancer. Molecules 2022, 27, 596. [Google Scholar] [CrossRef]
- Papagiorgis, P.C. Segmental distribution of some common molecular markers for colorectal cancer (CRC): Influencing factors and potential implications. Tumour Biol. 2016, 37, 5727–5734. [Google Scholar] [CrossRef]
- Reimers, M.S.; Zeestraten, E.C.; Kuppen, P.J.; Liefers, G.J.; van de Velde, C.J. Biomarkers in precision therapy in colorectal cancer. Gastroenterol. Rep. 2013, 1, 166–183. [Google Scholar] [CrossRef]
- Allar, B.G.; Messaris, E.; Poylin, V.Y.; Schlechter, B.L.; Cataldo, T.E. Oncotype DX testing does not affect clinical practice in stage IIa colon cancer. Med. Oncol. 2022, 39, 59. [Google Scholar] [CrossRef]
- Duchrow, M.; Häsemeyer, S.; Broll, R.; Bruch, H.P.; Windhövel, U. Assessment of proliferative activity in colorectal carcinomas by quantitative reverse transcriptase-polymerase chain reaction (RT-PCR). Cancer Investig. 2001, 19, 588–596. [Google Scholar] [CrossRef]
- Michael-Robinson, J.M.; Reid, L.E.; Purdie, D.M.; Biemer-Hüttmann, A.E.; Walsh, M.D.; Pandeya, N.; Simms, L.A.; Young, J.P.; Leggett, B.A.; Jass, J.R.; et al. Proliferation, apoptosis, and survival in high-level microsatellite instability sporadic colorectal cancer. Clin. Cancer Res. 2001, 7, 2347–2356. [Google Scholar]
- Evans, C.; Morrison, I.; Heriot, A.G.; Bartlett, J.B.; Finlayson, C.; Dalgleish, A.G.; Kumar, D. The correlation between colorectal cancer rates of proliferation and apoptosis and systemic cytokine levels; plus their influence upon survival. Br. J. Cancer 2006, 94, 1412–1419. [Google Scholar] [CrossRef]
- Kaczmarek, E.; Banasiewicz, T.; Seraszek-Jaros, A.; Krokowicz, P.; Grochowalski, M.; Majewski, P.; Zurawski, J.; Paszkowski, J.; Drews, M. Digital image analysis of inflammation markers in colorectal mucosa by using a spatial visualization method. Pathol. Res. Pract. 2014, 210, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.Q.; Yang, Y.L.; Dou, K.F.; Li, K.Z. Expression of PCNA and CD44mRNA in colorectal cancer with venous invasion and its relationship to liver metastasis. World J. Gastroenterol. 2003, 9, 2863–2865. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Li, J.; Pan, X.; Zhang, C.; Wei, D.; Gao, C. Increased Expression of PCNA-AS1 in Colorectal Cancer and its Clinical Association. Clin. Lab. 2017, 63, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, J.; Ma, S.; Zhang, S. Long Noncoding RNA MALAT-1 Can Predict Metastasis and a Poor Prognosis: A Meta-Analysis. Pathol. Oncol. Res. 2015, 21, 1259–1264. [Google Scholar] [CrossRef]
- Perakis, S.O.; Thomas, J.E.; Pichler, M. Non-coding RNAs Enabling Prognostic Stratification and Prediction of Therapeutic Response in Colorectal Cancer Patients. Adv. Exp. Med. Biol. 2016, 937, 183–204. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Yu, P.; Lin, W.; Okugawa, Y.; Boland, C.R.; Goel, A. A RNA-Sequencing approach for the identification of novel long non-coding RNA biomarkers in colorectal cancer. Sci. Rep. 2018, 8, 575. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, R.N.; Fan, X.; Wang, J.; Xu, C.; An, B.; Wang, Q.; Wang, J.; Leung, E.L.; Sui, X.; et al. Clinical diagnostic value of long non-coding RNAs in Colorectal Cancer: A systematic review and meta-analysis. J. Cancer 2020, 11, 5518–5526. [Google Scholar] [CrossRef]
- Chen, S.; Shen, X. Long noncoding RNAs: Functions and mechanisms in colon cancer. Mol. Cancer 2020, 19, 167. [Google Scholar] [CrossRef]
- Zhang, L.; Li, C.; Su, X. Emerging impact of the long noncoding RNA MIR22HG on proliferation and apoptosis in multiple human cancers. J. Exp. Clin. Cancer Res. 2020, 39, 271. [Google Scholar] [CrossRef]
- Zhang, J.; Li, K.; Zheng, H.; Zhu, Y. Research progress review on long non-coding RNA in colorectal cancer. Neoplasma 2021, 68, 240–252. [Google Scholar] [CrossRef]
- Lulli, M.; Napoli, C.; Landini, I.; Mini, E.; Lapucci, A. Role of Non-Coding RNAs in Colorectal Cancer: Focus on Long Non-Coding RNAs. Int. J. Mol. Sci. 2022, 23, 13431. [Google Scholar] [CrossRef] [PubMed]
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Hombach, S.; Kretz, M. Non-coding RNAs: Classification, Biology and Functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Sawada, G.; Kurashige, J.; Uchi, R.; Matsumura, T.; Ueo, H.; Takano, Y.; Eguchi, H.; Sudo, T.; Sugimachi, K.; et al. Amplification of PVT-1 is involved in poor prognosis via apoptosis inhibition in colorectal cancers. Br. J. Cancer 2014, 110, 164–171. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.; Tong, C.W.; Wu, M.; Cho, W.C. MicroRNAs in the prognosis and therapy of colorectal cancer: From bench to bedside. World J. Gastroenterol. 2018, 24, 2949–2973. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.; Liao, X.; Chen, Z.; Li, L.; Tian, T.; Yu, L.; Li, R. LINC00858 promotes colorectal cancer by sponging miR-4766-5p to regulate PAK2. Cell Biol. Toxicol. 2020, 36, 333–347. [Google Scholar] [CrossRef]
- Zhan, Y.X.; Luo, G.H. DNA methylation detection methods used in colorectal cancer. World J. Clin. Cases 2019, 7, 2916–2929. [Google Scholar] [CrossRef]
- Ragusa, M.; Barbagallo, C.; Statello, L.; Condorelli, A.G.; Battaglia, R.; Tamburello, L.; Barbagallo, D.; Di Pietro, C.; Purrello, M. Non-coding landscapes of colorectal cancer. World J. Gastroenterol. 2015, 21, 11709–11739. [Google Scholar] [CrossRef]
- Wang, J.; Song, Y.X.; Ma, B.; Wang, J.J.; Sun, J.X.; Chen, X.W.; Zhao, J.H.; Yang, Y.C.; Wang, Z.N. Regulatory Roles of Non-Coding RNAs in Colorectal Cancer. Int. J. Mol. Sci. 2015, 16, 19886–19919. [Google Scholar] [CrossRef]
- He, J.; Wu, W. Comprehensive landscape and future perspectives of long noncoding RNAs (lncRNAs) in colorectal cancer (CRC): Based on a bibliometric analysis. Noncoding RNA Res. 2022, 8, 33–52. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Pretorius, A.; Klein, A. Biomarkers for Stratification in Colorectal Cancer: MicroRNAs. Cancer Control 2019, 26, 1073274819862784. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, I.L.; Penna, K.G.B.D.; Dos Santos Carneiro, M.A.; Libera, L.S.D.; Ramos, J.E.P.; Saddi, V.A. Tissue micro-RNAs associated with colorectal cancer prognosis: A systematic review. Mol. Biol. Rep. 2021, 48, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhong, C.; Duan, S. The tumorigenic function of LINC00858 in cancer. Biomed. Pharmacother. 2021, 143, 112235. [Google Scholar] [CrossRef] [PubMed]
- Sha, Q.K.; Chen, L.; Xi, J.Z.; Song, H. Long non-coding RNA LINC00858 promotes cells proliferation, migration and invasion by acting as a ceRNA of miR-22-3p in colorectal cancer. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, F.; Sun, Y.; Fan, Q.; Cui, G. Expression of long non-coding RNA PANDAR and its prognostic value in colorectal cancer patients. Int. J. Biol. Markers 2017, 32, e218–e223. [Google Scholar] [CrossRef]
- Rivandi, M.; Pasdar, A.; Hamzezadeh, L.; Tajbakhsh, A.; Seifi, S.; Moetamani-Ahmadi, M.; Ferns, G.A.; Avan, A. The prognostic and therapeutic values of long noncoding RNA PANDAR in colorectal cancer. J. Cell. Physiol. 2019, 234, 1230–1236. [Google Scholar] [CrossRef]
- Xu, J.; Shao, T.; Song, M.; Xie, Y.; Zhou, J.; Yin, J.; Ding, N.; Zou, H.; Li, Y.; Zhang, J. MIR22HG acts as a tumor suppressor via TGFβ/SMAD signaling and facilitates immunotherapy in colorectal cancer. Mol. Cancer 2020, 19, 51. [Google Scholar] [CrossRef]
- Thorenoor, N.; Faltejskova-Vychytilova, P.; Hombach, S.; Mlcochova, J.; Kretz, M.; Svoboda, M.; Slaby, O. Long non-coding RNA ZFAS1 interacts with CDK1 and is involved in p53-dependent cell cycle control and apoptosis in colorectal cancer. Oncotarget 2016, 7, 622–637. [Google Scholar] [CrossRef]
- Fu, Y.; Huang, R.; Li, J.; Xie, X.; Deng, Y. LncRNA ENSG00000254615 Modulates Proliferation and 5-FU Resistance by Regulating p21 and Cyclin D1 in Colorectal Cancer. Cancer Investig. 2021, 39, 696–710. [Google Scholar] [CrossRef]
- Yang, H.; Wang, S.; Kang, Y.J.; Wang, C.; Xu, Y.; Zhang, Y.; Jiang, Z. Long non-coding RNA SNHG1 predicts a poor prognosis and promotes colon cancer tumorigenesis. Oncol. Rep. 2018, 40, 261–271. [Google Scholar] [CrossRef]
- Liu, K.L.; Wu, J.; Li, W.K.; Li, N.S.; Li, Q.; Lao, Y.Q. LncRNA SNHG7 is an Oncogenic Biomarker Interacting with MicroRNA-193b in Colon Carcinogenesis. Clin. Lab. 2019, 65, 2199–2204. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zhang, G.; Liu, Y. Long non-coding RNA XIST sponges miR-34a to promotes colon cancer progression via Wnt/β-catenin signaling pathway. Gene 2018, 665, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Zhang, N.; Ma, J.; Huang, J.; Chen, J.; Wang, L.; Zhang, J. Long noncoding RNA FAL1 promotes proliferation and inhibits apoptosis of human colon cancer cells. IUBMB Life 2018, 70, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, M.Q.; Kesavan, Y.; Ramalingam, S. Non-Coding RNA as Biomarkers for Survival in Colorectal Cancer Patients. Curr. Aging Sci. 2023. [Google Scholar] [CrossRef]
- Baharudin, R.; Rus Bakarurraini, N.Q.; Ismail, I.; Lee, L.H.; Ab Mutalib, N.S. MicroRNA Methylome Signature and Their Functional Roles in Colorectal Cancer Diagnosis, Prognosis, and Chemoresistance. Int. J. Mol. Sci. 2022, 23, 7281. [Google Scholar] [CrossRef]
- Peng, Q.; Zhang, X.; Min, M.; Zou, L.; Shen, P.; Zhu, Y. The clinical role of microRNA-21 as a promising biomarker in the diagnosis and prognosis of colorectal cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 44893–44909. [Google Scholar] [CrossRef]
- Gu, X.; Jin, R.; Mao, X.; Wang, J.; Yuan, J.; Zhao, G. Prognostic value of miRNA-181a/b in colorectal cancer: A meta-analysis. Biomark. Med. 2018, 12, 299–308. [Google Scholar] [CrossRef]
- Moody, L.; Dvoretskiy, S.; An, R.; Mantha, S.; Pan, Y.X. The Efficacy of miR-20a as a Diagnostic and Prognostic Biomarker for Colorectal Cancer: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 1111. [Google Scholar] [CrossRef]
- Wu, Y.; Hong, Q.; Lu, F.; Zhang, Z.; Li, J.; Nie, Z.; He, B. The Diagnostic and Prognostic Value of miR-155 in Cancers: An Updated Meta-analysis. Mol. Diagn. Ther. 2023, 27, 283–301. [Google Scholar] [CrossRef]
- Sabarimurugan, S.; Madhav, M.R.; Kumarasamy, C.; Gupta, A.; Baxi, S.; Krishnan, S.; Jayaraj, R. Prognostic Value of MicroRNAs in Stage II Colorectal Cancer Patients: A Systematic Review and Meta-Analysis. Mol. Diagn. Ther. 2020, 24, 15–30. [Google Scholar] [CrossRef]
- Gao, S.; Zhao, Z.Y.; Wu, R.; Zhang, Y.; Zhang, Z.Y. Prognostic value of microRNAs in colorectal cancer: A meta-analysis. Cancer Manag. Res. 2018, 10, 907–929. [Google Scholar] [CrossRef] [PubMed]
- Galamb, O.; Barták, B.K.; Kalmár, A.; Nagy, Z.B.; Szigeti, K.A.; Tulassay, Z.; Igaz, P.; Molnár, B. Diagnostic and prognostic potential of tissue and circulating long non-coding RNAs in colorectal tumors. World J. Gastroenterol. 2019, 25, 5026–5048. [Google Scholar] [CrossRef] [PubMed]
- Qi, P.; Xu, M.D.; Ni, S.J.; Huang, D.; Wei, P.; Tan, C.; Zhou, X.Y.; Du, X. Low expression of LOC285194 is associated with poor prognosis in colorectal cancer. J. Transl. Med. 2013, 11, 122. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Zhu, J.H.; Yao, X.Q. Long non-coding RNA PVT1 as a novel potential biomarker for predicting the prognosis of colorectal cancer. Int. J. Biol. Markers 2018, 33, 415–422. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, M.; Liang, L.; Li, J.; Chen, Y.X. Over-expression of lncRNA DANCR is associated with advanced tumor progression and poor prognosis in patients with colorectal cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11480–11484. [Google Scholar]
- Ren, Y.K.; Xiao, Y.; Wan, X.B.; Zhao, Y.Z.; Li, J.; Li, Y.; Han, G.S.; Chen, X.B.; Zou, Q.Y.; Wang, G.C.; et al. Association of long non-coding RNA HOTTIP with progression and prognosis in colorectal cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11458–11463. [Google Scholar]
- Shen, X.; Bai, Y.; Luo, B.; Zhou, X. Upregulation of lncRNA BANCR associated with the lymph node metastasis and poor prognosis in colorectal cancer. Biol. Res. 2017, 50, 32. [Google Scholar] [CrossRef]
- Tan, W.; Song, Z.Z.; Xu, Q.; Qu, X.; Li, Z.; Wang, Y.; Yu, Q.; Wang, S. Up-Regulated Expression of SPRY4-IT1 Predicts Poor Prognosis in Colorectal Cancer. Med. Sci. Monit. 2017, 23, 309–314. [Google Scholar] [CrossRef]
- Ozawa, T.; Matsuyama, T.; Toiyama, Y.; Takahashi, N.; Ishikawa, T.; Uetake, H.; Yamada, Y.; Kusunoki, M.; Calin, G.; Goel, A. CCAT1 and CCAT2 long noncoding RNAs, located within the 8q.24.21 ‘gene desert’, serve as important prognostic biomarkers in colorectal cancer. Ann. Oncol. 2017, 28, 1882–1888. [Google Scholar] [CrossRef]
- Zhang, X.T.; Pan, S.X.; Wang, A.H.; Kong, Q.Y.; Jiang, K.T.; Yu, Z.B. Long Non-Coding RNA (lncRNA) X-Inactive Specific Transcript (XIST) Plays a Critical Role in Predicting Clinical Prognosis and Progression of Colorectal Cancer. Med. Sci. Monit. 2019, 25, 6429–6435. [Google Scholar] [CrossRef]
- Liu, X.; Liu, X.; Qiao, T.; Chen, W. Prognostic and clinicopathological significance of long non-coding RNA UCA1 in colorectal cancer: Results from a meta-analysis. Medicine 2019, 98, e18031. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Lan, Z.; Lai, Q.; Li, A.; Liu, S.; Wang, X. LncRNA SNHG6 plays an oncogenic role in colorectal cancer and can be used as a prognostic biomarker for solid tumors. J. Cell. Physiol. 2020, 235, 7620–7634. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, Y.; Guo, L.; Zhang, C.; Tang, S. Long non-coding RNA FTX predicts a poor prognosis of human cancers: A meta-analysis. Biosci. Rep. 2021, 41, BSR20203995. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.B.; Long, P.; Zhao, Z.; Zhang, Y.R.; Chu, X.D.; Zhao, X.X.; Ding, H.; Huan, S.W.; Pan, Y.L.; Pan, J.H. Long Noncoding RNA KCNQ1OT1 is a Prognostic Biomarker and mediates CD8+ T cell exhaustion by regulating CD155 Expression in Colorectal Cancer. Int. J. Biol. Sci. 2021, 17, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Du, J.; Li, Y.; Ma, J.; Shi, W. Association between small nucleolar RNA host gene expression and survival outcome of colorectal cancer patients: A meta-analysis based on PRISMA and bioinformatics analysis. Front. Oncol. 2023, 13, 1094131. [Google Scholar] [CrossRef]
- Li, C.; Zhou, L.; He, J.; Fang, X.Q.; Zhu, S.W.; Xiong, M.M. Increased long noncoding RNA SNHG20 predicts poor prognosis in colorectal cancer. BMC Cancer 2016, 16, 655. [Google Scholar] [CrossRef]
- Zhuang, C.; Zheng, L.; Wang, P. Prognostic role of long non-coding RNA HNF1A-AS1 in Chinese cancer patients: A meta-analysis. Onco Targets Ther. 2018, 11, 5325–5332. [Google Scholar] [CrossRef]
- Xie, H.; Ma, B.; Gao, Q.; Zhan, H.; Liu, Y.; Chen, Z.; Ye, S.; Li, J.; Yao, L.; Huang, W. Long non-coding RNA CRNDE in cancer prognosis: Review and meta-analysis. Clin. Chim. Acta 2018, 485, 262–271. [Google Scholar] [CrossRef]
- Li, X.; Liang, Q.X.; Lin, J.R.; Peng, J.; Yang, J.H.; Yi, C.; Yu, Y.; Zhang, Q.C.; Zhou, K.R. Epitranscriptomic technologies and analyses. Sci. China Life Sci. 2020, 63, 501–515. [Google Scholar] [CrossRef]
- Fontanges, Q.; De Mendonca, R.; Salmon, I.; Le Mercier, M.; D’Haene, N. Clinical Application of Targeted Next Generation Sequencing for Colorectal Cancers. Int. J. Mol. Sci. 2016, 17, 2117. [Google Scholar] [CrossRef]
- Lee, M.K.C.; Loree, J.M. Current and emerging biomarkers in metastatic colorectal cancer. Curr. Oncol. 2019, 26 (Suppl. S1), S7–S15. [Google Scholar] [CrossRef] [PubMed]
- Koulis, C.; Yap, R.; Engel, R.; Jardé, T.; Wilkins, S.; Solon, G.; Shapiro, J.D.; Abud, H.; McMurrick, P. Personalized Medicine-Current and Emerging Predictive and Prognostic Biomarkers in Colorectal Cancer. Cancers 2020, 12, 812. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.J.; Zhao, Q.; Liu, J.; Zheng, J.B.; Qiu, M.Z.; Ju, H.Q.; Xu, R.H. Novel Genetic and Epigenetic Biomarkers of Prognostic and Predictive Significance in Stage II/III Colorectal Cancer. Mol. Ther. 2021, 29, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Sorich, M.J.; Wiese, M.D.; Rowland, A.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized, controlled trials. Ann. Oncol. 2015, 26, 13–21. [Google Scholar] [CrossRef]
- Park, S.M.; Choi, S.B.; Lee, Y.S.; Lee, I.K. Predictive value of KRAS mutation and excision repair cross-complementing 1 (ERCC1) protein overexpression in patients with colorectal cancer administered FOLFOX regimen. Asian J. Surg. 2021, 44, 715–722. [Google Scholar] [CrossRef]
- Popat, S.; Houlston, R.S. A systematic review and meta-analysis of the relationship between chromosome 18q genotype, DCC status and colorectal cancer prognosis. Eur. J. Cancer 2005, 41, 2060–2070. [Google Scholar] [CrossRef]
- Whitfield, M.L.; George, L.K.; Grant, G.D.; Perou, C.M. Common markers of proliferation. Nat. Rev. Cancer 2006, 6, 99–106. [Google Scholar] [CrossRef]
- Coppedè, F.; Lopomo, A.; Spisni, R.; Migliore, L. Genetic and epigenetic biomarkers for diagnosis, prognosis and treatment of colorectal cancer. World J. Gastroenterol. 2014, 20, 943–956. [Google Scholar] [CrossRef]
- Zhang, H.; Dong, S.; Feng, J. Epigenetic profiling and mRNA expression reveal candidate genes as biomarkers for colorectal cancer. J. Cell. Biochem. 2019, 120, 10767–10776. [Google Scholar] [CrossRef]
- Kong, C.; Fu, T. Value of methylation markers in colorectal cancer (Review). Oncol. Rep. 2021, 46, 177. [Google Scholar] [CrossRef]
- Lee, S.; Cho, N.Y.; Choi, M.; Yoo, E.J.; Kim, J.H.; Kang, G.H. Clinicopathological features of CpG island methylator phenotype-positive colorectal cancer and its adverse prognosis in relation to KRAS/BRAF mutation. Pathol. Int. 2008, 58, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Boughanem, H.; Cabrera-Mulero, A.; Hernández-Alonso, P.; Clemente-Postigo, M.; Casanueva, F.F.; Tinahones, F.J.; Morcillo, S.; Crujeiras, A.B.; Macias-Gonzalez, M. Association between variation of circulating 25-OH vitamin D and methylation of secreted frizzled-related protein 2 in colorectal cancer. Clin. Epigenet. 2020, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Barchitta, M.; Quattrocchi, A.; Maugeri, A.; Vinciguerra, M.; Agodi, A. LINE-1 hypomethylation in blood and tissue samples as an epigenetic marker for cancer risk: A systematic review and meta-analysis. PLoS ONE 2014, 9, e109478. [Google Scholar] [CrossRef] [PubMed]
- Boughanem, H.; Martin-Nuñez, G.M.; Torres, E.; Arranz-Salas, I.; Alcaide, J.; Morcillo, S.; Tinahones, F.J.; Crujeiras, A.B.; Macias-Gonzalez, M. Impact of Tumor LINE-1 Methylation Level and Neoadjuvant Treatment and Its Association with Colorectal Cancer Survival. J. Pers. Med. 2020, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Jiang, D.; Li, Y.; Jin, M.; Chen, K. The role of LINE-1 methylation in predicting survival among colorectal cancer patients: A meta-analysis. Int. J. Clin. Oncol. 2017, 22, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Javierre, B.M.; Rodriguez-Ubreva, J.; Al-Shahrour, F.; Corominas, M.; Graña, O.; Ciudad, L.; Agirre, X.; Pisano, D.G.; Valencia, A.; Roman-Gomez, J.; et al. Long-range epigenetic silencing associates with deregulation of Ikaros targets in colorectal cancer cells. Mol. Cancer Res. 2011, 9, 1139–1151. [Google Scholar] [CrossRef]
- Winter, J.M.; Sheehan-Hennessy, L.; Yao, B.; Pedersen, S.K.; Wassie, M.M.; Eaton, M.; Chong, M.; Young, G.P.; Symonds, E.L. Detection of hypermethylated BCAT1 and IKZF1 DNA in blood and tissues of colorectal, breast and prostate cancer patients. Cancer Biomark. 2022, 34, 493–503. [Google Scholar] [CrossRef]
- Moriichi, K.; Fujiya, M.; Kobayashi, Y.; Murakami, Y.; Iwama, T.; Kunogi, T.; Sasaki, T.; Ijiri, M.; Takahashi, K.; Tanaka, K.; et al. Autofluorescence Imaging Reflects the Nuclear Enlargement of Tumor Cells as well as the Cell Proliferation Ability and Aberrant Status of the p53, Ki-67, and p16 Genes in Colon Neoplasms. Molecules 2019, 24, 1106. [Google Scholar] [CrossRef]
- Zygulska, A.L.; Pierzchalski, P. Novel Diagnostic Biomarkers in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 852. [Google Scholar] [CrossRef]
- Cerrito, M.G.; Grassilli, E. Identifying Novel Actionable Targets in Colon Cancer. Biomedicines 2021, 9, 579. [Google Scholar] [CrossRef]
- Abbes, S.; Baldi, S.; Sellami, H.; Amedei, A.; Keskes, L. Molecular methods for colorectal cancer screening: Progress with next-generation sequencing evolution. World J. Gastrointest. Oncol. 2023, 15, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, F.; Mastroiaco, V.; Di Marco, A.; Compagnoni, C.; Capece, D.; Zazzeroni, F.; Capalbo, C.; Alesse, E.; Tessitore, A. Next-generation sequencing: Recent applications to the analysis of colorectal cancer. J. Transl. Med. 2017, 15, 246. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Z.; Zhang, Z.; Zhang, W.; Zhang, M.; Shen, Z.; Ye, Y.; Jiang, K.; Wang, S. Landscape of cell heterogeneity and evolutionary trajectory in ulcerative colitis-associated colon cancer revealed by single-cell RNA sequencing. Chin. J. Cancer Res. 2021, 33, 271–288. [Google Scholar] [CrossRef]
- Xu, S.; Li, Y.; Huang, H.; Miao, X.; Gu, Y. Identification of KIF21B as a Biomarker for Colorectal Cancer and Associated with Poor Prognosis. J. Oncol. 2022, 2022, 7905787. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, X.; Xu, Q.; Gao, X.; Linghu, E. A Prognostic Model of Colon Cancer Based on the Microenvironment Component Score via Single Cell Sequencing. In Vivo 2022, 36, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Cervantes, A.; Ciardiello, F.; De Luca, A.; Pinto, C. The liquid biopsy in the management of colorectal cancer patients: Current applications and future scenarios. Cancer Treat. Rev. 2018, 70, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kastrisiou, M.; Zarkavelis, G.; Pentheroudakis, G.; Magklara, A. Clinical Application of Next-Generation Sequencing as A Liquid Biopsy Technique in Advanced Colorectal Cancer: A Trick or A Treat? Cancers 2019, 11, 1573. [Google Scholar] [CrossRef]
- Basnet, S.; Zhang, Z.Y.; Liao, W.Q.; Li, S.H.; Li, P.S.; Ge, H.Y. The Prognostic Value of Circulating Cell-Free DNA in Colorectal Cancer: A Meta-Analysis. J. Cancer 2016, 7, 1105–1113. [Google Scholar] [CrossRef]
- Rodríguez-Casanova, A.; Bao-Caamano, A.; Lago-Lestón, R.M.; Brozos-Vázquez, E.; Costa-Fraga, N.; Ferreirós-Vidal, I.; Abdulkader, I.; Vidal-Insua, Y.; Rivera, F.V.; Candamio Folgar, S.; et al. Evaluation of a Targeted Next-Generation Sequencing Panel for the Non-Invasive Detection of Variants in Circulating DNA of Colorectal Cancer. J. Clin. Med. 2021, 10, 4487. [Google Scholar] [CrossRef]
- Lan, Y.T.; Chang, S.C.; Lin, P.C.; Lin, C.H.; Liang, W.Y.; Chen, W.S.; Jiang, J.K.; Yang, S.H.; Lin, J.K. High concordance of mutation patterns in 10 common mutated genes between tumor tissue and cell-free DNA in metastatic colorectal cancer. Am. J. Cancer Res. 2021, 11, 2228–2237. [Google Scholar]
- Güttlein, L.; Luca, M.R.; Esteso, F.; Fresno, C.; Mariani, J.; Otero Pizarro, M.; Brest, E.; Starapoli, S.; Kreimberg, K.; Teves, P.; et al. Liquid biopsy for KRAS, NRAS and BRAF mutation testing in advanced colorectal cancer patients: The Argentinean experience. Future Oncol. 2022, 18, 3277–3287. [Google Scholar] [CrossRef] [PubMed]
- Heuvelings, D.J.I.; Wintjens, A.G.W.E.; Luyten, J.; Wilmink, G.E.W.A.; Moonen, L.; Speel, E.M.; de Hingh, I.H.J.T.; Bouvy, N.D.; Peeters, A. DNA and RNA Alterations Associated with Colorectal Peritoneal Metastases: A Systematic Review. Cancers 2023, 15, 549. [Google Scholar] [CrossRef] [PubMed]
- Koveitypour, Z.; Panahi, F.; Vakilian, M.; Peymani, M.; Seyed Forootan, F.; Nasr Esfahani, M.H.; Ghaedi, K. Signaling pathways involved in colorectal cancer progression. Cell Biosci. 2019, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Cai, P.; Xie, J.; Wei, Y. The diagnostic accuracy of digital PCR, ARMS and NGS for detecting KRAS mutation in cell-free DNA of patients with colorectal cancer: A protocol for systematic review and meta-analysis. Medicine 2020, 99, e20708. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.; Townsend, D. History and future technical innovation in positron emission tomography. J. Med. Imaging 2017, 4, 011013. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Pelosi, E.; Deandreis, D. The role of 18F-fluoro-deoxy-glucose positron emission tomography (FDG-PET) in the management of patients with colorectal cancer. Eur. J. Surg. Oncol. 2007, 33, 1–6. [Google Scholar] [CrossRef]
- Flamen, P.; Stroobants, S.; Van Cutsem, E.; Dupont, P.; Bormans, G.; De Vadder, N.; Penninckx, F.; Van Hoe, L.; Mortelmans, L. Additional value of whole-body positron emission tomography with fluorine-18-2-fluoro-2-deoxy-D-glucose in recurrent colorectal cancer. J. Clin. Oncol. 1999, 17, 894–901. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Kameyama, R.; Izuishi, K.; Takebayashi, R.; Hagiike, M.; Asakura, M.; Haba, R.; Nishiyama, Y. Detection of colorectal cancer using ¹⁸F-FLT PET: Comparison with ¹⁸F-FDG PET. Nucl. Med. Commun. 2009, 30, 841–845. [Google Scholar] [CrossRef]
- Deng, S.M.; Zhang, W.; Zhang, B.; Chen, Y.Y.; Li, J.H.; Wu, Y.W. Correlation between the Uptake of 18F-Fluorodeoxyglucose (18F-FDG) and the Expression of Proliferation-Associated Antigen Ki-67 in Cancer Patients: A Meta-Analysis. PLoS ONE 2015, 10, e0129028. [Google Scholar] [CrossRef]
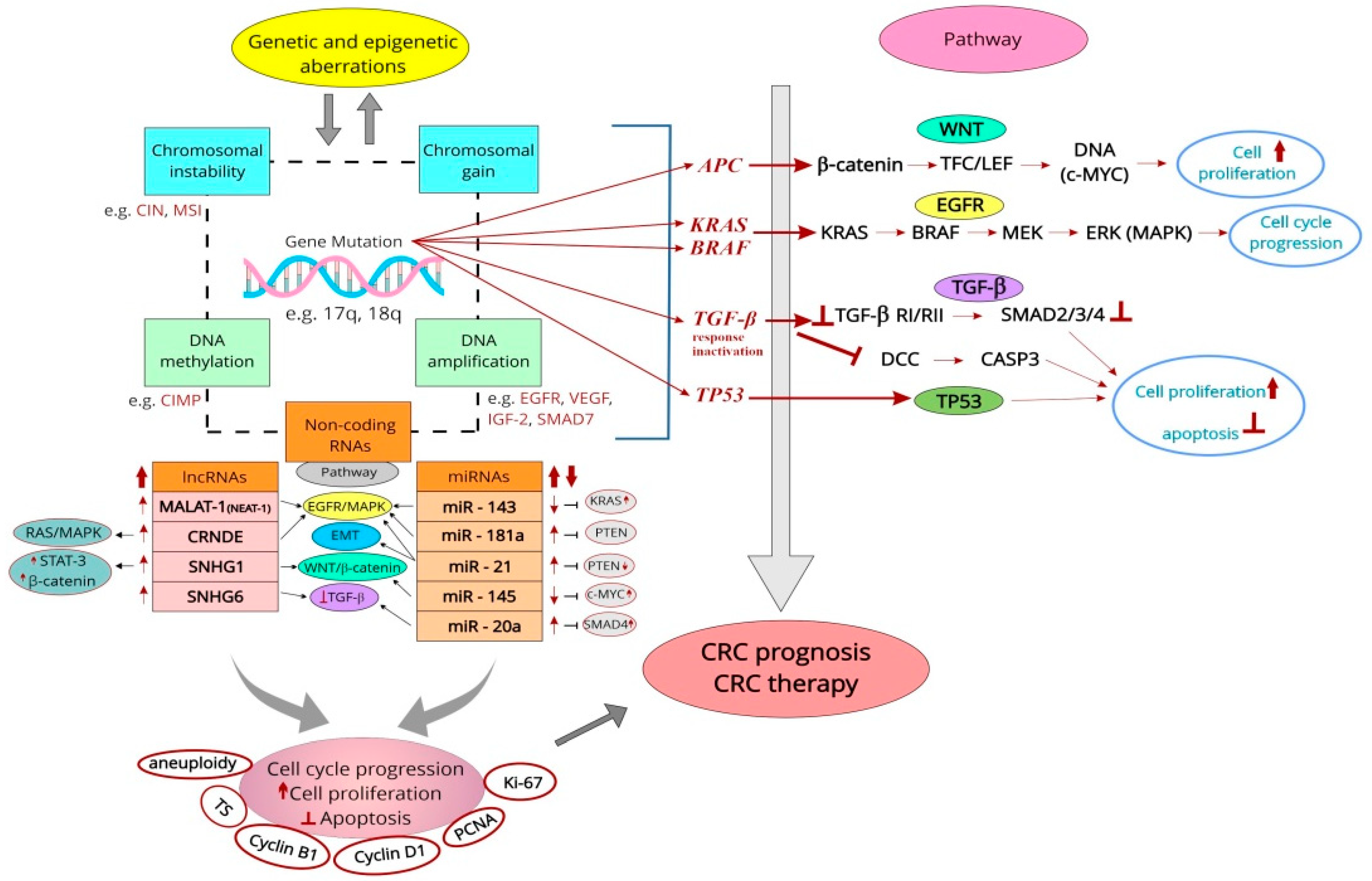

{kind=link}
| Type of Cyclin | Material (No. of Cases) and Method | Findings | Year of Publication | Ref. No. |
|---|---|---|---|---|
| A (A2) | CRC (73); IHC, SI | Mean: 12.26 ± 5.8; SI was correlated with tumor differentiation; ↑expression correlated with ↓OS; ↑expression is an independent negative prognostic factor (HR 7.82, 95% CI, 0.02–60.12) (UA) and (HR 13.89; 95% CI 1.01–190.58) (MA) | 1999 | [159] |
| CRC (60); IHC, SI | ↑Expression associated with ↓OS; independent prognostic factor | 2004 | [160] | |
| CRC (167); IHC | (+) Expression (61.1%); (+) expression correlated with ↓survival; (+) expression, LN meta, and Dukes’ stage were independently associated with unfavorable prognosis | 2004 | [162] | |
| CRC (219); IHC; qRT-PCR | (+) Expression (83%), extra gene copies (6.2%); correlation with stage and differentiation; ↑expression independently associated with improved survival (UA), (HR 0.57, 95% CI 0.33–0.98) (MA) | 2005 | [155] | |
| CRC (790); IHC | Expression above the median predicted an improved patient prognosis (HR 0.71, 95% CI 0.53–0.95); cell proliferation and (+) expression were prognostic indicators of patient outcome | 2011 | [163] | |
| B (B1) | C (22), ADs (62); CAs in ADs (17), pCRC (194), LN meta (21); IHC | ↑B1 expression from C through ADs to pCRC; ↑expression with increasing degree of dysplasia in ADs, from peripheral ADs to central CAs, and from primary to metastatic foci; ↓in pCRC with large size, mucinous type, deep invasion, or short PPS time | 2003 | [164] |
| CRC (342); IHC | ↑Expression (78.7%); no association with histopathologic features; no impact on OS and DFS (UA) | 2004 | [165] | |
| CRC (219); IHC, qRT-PCR | (+) Expression (83%), extra gene copies (9%); no prognostic value | 2005 | [155] | |
| CRC (150); WB; qRT-PCR; IHC | ↑mRNA expression (92.7%); ↑expression negatively related to LN and distant meta, and TNM; ↓expression associated with poor OS | 2015 | [166] | |
| C | CRC (219); IHC; qRT-PCR | ↑Expression (88%), extra gene copies (26.9%); ↑expression correlated with CCNC amplification; protein expression tends to associate with DSS; CCNC amplification related to an unfavorable prognosis, (HR 1.72, 95% CI 1.00–2.94) (MA) | 2005 | [155] |
| D (D1, D3) | CRC (123); IHC | ↑D1 expression correlated with poor OS and DFS; an independent predictor of disease recurrence | 1998 | [172] |
| CRC (90); IHC | Nuclear/cytoplasmic expression of cyclin D1; no prognostic value | 1998 | [177] | |
| CRC (73); IHC, SI | Mean: 6.9 ± 6.3; SI correlated with tumor differentiation; ↑in LN meta vs. those without; ↑in advanced than in early CAs; ↑expression tends to associate with poor prognosis | 1999 | [159] | |
| CRC (126); IHC | (+) Expression (58.7%); cytoplasmic (HR 0.56, 95% CI 0.31–1.0) or nuclear level (HR 0.24, 95% CI 0.07–0.81) related to ↑survival | 2001 | [183] | |
| RC (160); IHC, (+) at the 10% level | (+) D1 expression (48%); no prognostic role of this marker | 2002 | [178] | |
| CRC (60); IHC, SI | ↑SI within deeply invasive tumors and LN meta; ↑expression and D1 amplification associated with ↓OS; independent prognostic factor | 2004 | [160] | |
| CRC (219); IHC; qRT-PCR | (+) D1 expression (11%) and extra gene copies (55%), cyclin D3 (36%) and extra gene copies (20.5%); no prognostic role of these markers | 2005 | [155] | |
| CC and RC (363), Dukes’ A–D, TMA; IHC | (+) Nuclear staining of cyclin D1 reflected better survival | 2005 | [182] | |
| CRC (97); IHC, (+) at >5% cells | ↑Expression (5.9%); ↑levels in mucous differentiation; ↑expression correlated with stage, LN meta; no prognostic value | 2008 | [179] | |
| CC (602), stage I–IV; IHC | ↑Expression (55%) was related to low cancer-specific mortality (HR 0.57, 95% CI 0.39–0.84) (MA), and for low overall mortality (HR 0.74, 95% CI 0.57–0.98); ↑expression related to ↑survival | 2009 | [181] | |
| CRC (84), TMA; cyclin D1, D2, D3; IHC | D2 expression at the margin associated with vascular invasion, LN meta, and CRLM; ↑D2 and D3 associated with vascular invasion, CRLM, and ↓DSS (cyclin D2) | 2010 | [173] | |
| CRC (169); IHC | (+) D1 expression related to shorter survival | 2011 | [174] | |
| CRC (117), TMA; IHC | ↓Nuclear expression associated with negative lymphovascular invasion; no prognostic value of cyclin D1 | 2015 | [180] | |
| CRC with meta (1205); IHC | ↑Expression (46.7%); ↑D1, EGFR expression, late stage after S indicated ↓RFT (UA); no independent factor of prognosis (MA) | 2019 | [175] | |
| CC (102); IHC | (+) Expression of cyclin D1 correlated with a worse 5-yrs survival rate in pts with advanced stage (III, IV) | 2019 | [169] | |
| CRC (101), Dukes’ B and C stages; IHC | ↑Expression more often in DFS ≤24 group vs. ≥48 group and had 5.2 higher odds of having DFS ˂24 mo; ↑expression correlated with early recurrence in high-risk Duke’s B and C stage | 2021 | [176] | |
| E | CRC (219); IHC, qRT-PCR | (+) Expression (25%) and extra gene copies (19.1%); no prognostic value | 2005 | [155] |
| CRC (97); IHC, (+) at >5% cells | ↑Expression (30%); (+) correlation with p21waf1/cip1, PCNA-LI and Ki-67; no prognostic value | 2008 | [179] | |
| CRC (200), benign alterations (200); IHC; RT-PCR | ↑Expression with TNM and decreasing tumor differentiation; (+) expression correlated with shorter PFS and median survival | 2016 | [185] | |
| CRC (31), TMA; IHC | (+) Expression (34.78%); no prognostic role of this marker | 2016 | [186] | |
| CC (102); IHC | (+) Correlation with cyclin D1; no prognostic role | 2019 | [169] |
| S No. | Material (No. of Cases) and Methods | Findings | Prognostic Role | Year of Publication | Ref. No. |
|---|---|---|---|---|---|
| 1. | CRC (40); IHC, PI | ↑PI in both cancer and epithelial cells of adjacent C crypts in those who died vs. survivors | PI is an independent predictor of recurrence and poor survival in both groups | 1993 | [189] |
| 2. | CRC (82) and LN meta (18); IHC, q estimation | Similar to the median and range of the % of (+) cells in primary tumors and LN meta | An inverse relationship between the % of (+) cells and survival times | 1993 | [192] |
| 3. | CRC (60) and ADs (35); IHC; FCM for DNA content | Mean: 38% (ADs); mean: 50.4% (CRC); aneuploid ACs had a tendency to poorer prognosis, especially in Dukes’ C female pts | Can be an indirect indicator of cells in the S phase; is not an independent prognostic factor | 1994 | [123] |
| 4. | CRC (125); IHC, LI, image analysis | LI without significant correlation with clinical characteristics (stage, grade, age, sex, fixity) | No prognostic role for survival | 1994 | [124] |
| 5. | CRC (49); IHC, LI | ↑LI of tumors with venous invasion (mean: 51.7%); with LN meta (mean: 50.5%); with meta to the liver (mean: 55.2%); ↑LI associated with less differentiated tumors | Evaluation of LI at the invasive tumor margin may help identify CRC with ↑malignant potential | 1994 | [193] |
| 6. | CRC biopsies (50); FCM, LI | LI from 38.7% to 53.0%; in diploid tumors (27), the median LI in G0/G1: 71.5%, in S: 10.5%, in G2/M: 17.4% | Is expressed throughout the cell cycle; prognostic role—probable | 1995 | [108] |
| 7. | CC (50) and 40 RC; IHC (79), LI | LI improved the prediction of survival when used with histopathological classification (Dukes’ or Jass’) (MA) | Little prognostic power of LI (UA); not predictive for RC; ↓LI related to the worst prognosis | 1995 | [191] |
| 8. | CRC (57); IHC, LI | ↑Deep invasion, CRLM, and ↑stages with ↑LI (>49.4%) vs. ↓LI; ↑survival curves for pts with (−) CEA and ↓LI vs. pts with (+) CEA and ↑LI; ↑survival curves for pts with (+) CEA and ↓LI vs. pts with (+) CEA and ↑LI | Serum CEA and PCNA LI for cancer pts are useful in the evaluation of tumor progression and prognosis | 1996 | [194] |
| 9. | CRC (86); IHC, LI | ↑LI with stage, histologic differentiation, lymphatic and vascular invasion, LN meta, and CRLM; ↑LI in tumors with DNA aneuploidy | ↑Recurrence rate with LI > than the mean LI; ↓4-yr survival rates for overall and curative pts with LI > than the mean LI | 1996 | [190] |
| 10. | CRC (59); IHC, LI | Lesions combining HLA-DR expression and a relatively ↓LI had the best prognosis | HLA-DR expression with ↓LI is an important outcome predictor | 1998 | [196] |
| 11. | CRC (47); IHC, >60% nuclei (+) | (+) Correlation with Bcl-2, LN meta, and tumor location | May be an indicator of the development of LN meta | 2009 | [197] |
| S No. | Material (No. of Cases) and Methods | Findings | Prognostic Role for Survival | Year of Publication/Country | Ref. No. |
|---|---|---|---|---|---|
| 1. | pAC (139); IHC, mAb Ki-67, LI; S | ↑In mucinous vs. non-mucinous CRC; inverse correlation with grading in non-mucinous AC | No | 1990; Italy | [231] |
| 2. | pCRC (125); IHC, LI; S | No correlations with clinicopathological data | No | 1994; UK | [124] |
| 3. | CRC (106); IHC, MIB-1, 3 methods of estimation; S | No correlation with clinical outcome | No | 1996; Austria | [233] |
| 4. | CRC (70); stages II and III; IHC, MIB-1, LI; S | Relation to disease recurrence, retained in stage II; LI > 45% associated with ↑risk for disease recurrence vs. LI ≤ 45% (MA) | Yes | 1997; USA | [125] |
| 5. | CRC (255); Dukes’ A–D; IHC, MIB-1, weak (<50%), strong (>50%); S | Level > 50% (62%); <50% (38%); no correlations with clinicopathological variables | No | 1997; Sweden | [234] |
| 6. | CRC (56); Dukes’ B; survival analysis (47); IHC, anti-Ki-67, morphometry; S | Mean value in luminal border (27.4%), invasive margin (36.8%); ↓LI at the invasive margin correlated with poorer survival (RR 12.1, 95% CI 1.1–1.33) (UA and MA) | Yes | 1999; Sweden | [247] |
| 7. | CRC (52); AD (56); IHC, MIB-1, LI; S | ↓LI in AD (30.05%) vs. CRC (38.12%); ↑correlated with poor differentiation and Duke’s stage | Nd | 2000; USA | [218] |
| 8. | CRC (30); IHC, LI; S | No correlation with tumor stage and grade | Nd | 2001; France | [127] |
| 9. | CRLM (41); IHC, MIB-1; LI at the hot spot; S | Mean value (38%); LI ≥50% related to shorter survival vs. low scores; ↑score an independent adverse prognostic factor (RR 3.04) (MA) | Yes | 2001; Germany | [251] |
| 10. | CRC (25); pTNM; stages I–IV; IHC, MIB-1, LI, morphometry; RT-PCR; S | Median protein LI (61%), median mRNA LI (0.88 amol); better OS for the group with ↓LI and ↓mRNA level vs. median | Yes | 2001; Germany | [259] |
| 11. | CRC (100); MSI-H (31), MSI-L (29), MSS (40); IHC, PI; S | ↑PI (90.1%) in MSI-H vs. MSI-L (69.5%) and vs. MSS (69.5%); ↑PI showed a trend toward predicting ↑survival only within MSI-H cancers | Probably yes | 2001; Australia | [260] |
| 12. | CC (465); Dukes’ B2 and C; S alone (151) or S + FU-based CTx (314); IHC, LI | No significant association with clinical outcome | No | 2002; USA | [142] |
| 13. | pCRC (74); CRLM (37); IHC, MIB-1, LI; S | LI ≥30% more frequently in lymphatic and venous invasion, LN meta, and CRLM; ↑in primary tumors vs. CRLM (24.3 ± 17.9 vs. 5.0 ± 4.2); LI ≥30% in pCRC correlated with ↑frequency of metachronous CRLM | Nd | 2002; Japan | [221] |
| 14. | CC (706); stages II and III; S alone (275) or S + FU-leucovorin CTx (431); IHC, LI | Tumors with ↑number of (+) cells had improved outcomes vs. tumors with few (+) cells; association with RFS (RR 0.76) and with OS (RR 0.62) | Yes | 2003; USA | [147] |
| 15. | CRC (47); IHC, MIB-1, LI; ISH for mRNA with DIG-labelled cRNA probe, LI; S | Median protein LI (59%), mean mRNA LI (42%); ↑protein but ↓mRNA are likely to proliferate more slowly, which possibly explains the pts’ improved outcome | No | 2003; Germany | [236] |
| 16. | CRC (81); IHC, anti-Ki-67, IRS; S | ↑Expression in the low differentiated tumors; inverse correlation to survival | Yes | 2003; PL | [222] |
| 17. | pCRC (311 including 82 with distant meta); AD and CA in situ (22); IHC, MIB-1; S | ↑Rate in AD with severe atypia and CA in situ; ↓rate in poorly differentiated and mucinous AC vs. well- and moderately differentiated tumors | Nd | 2003; Japan | [219] |
| 18. | CC (144), RC (90); IHC; MIB-1, semiq estimation; S | ↑In LN meta of short-term (505 d) vs. long-term survivors (4150.5 d) | No, but an indicator of survival in Dukes’ C | 2004; Japan | [240] |
| 19. | RC and rectosigmoid AC (146); IHC, MIB-1, high (>40%) and low (≤40%), hot spot areas (>50%); S | Better OS for ↑values vs. those with ↓values; the presence of hot spot areas associated with better survival (MA); hot spot areas one of the prognostic factor | Yes | 2005; Finland | [248] |
| 20. | CRC (106); IHC, MIB-1, PI; S | Mean PI (38.0%); (+) correlation with advanced T status, LN and distant meta, and ↑pTNM stage; an independent prognostic factor for long-term survival; pts with high PI were at greater risk for death (HR 2.1, 95% CI 1.1–4.1) (MA) | Yes | 2005; Japan | [223] |
| 21. | CC (53), RC (33); stages I–IV; IHC, MIB-1, group A (<40%) and B (≥40%); S | Mean LI (0.44 ± 0.16); no correlation with sex, age, and clinical stage; ↑level correlated with ↓survival; an independent predictor of survival (MA) | Yes | 2005; Brazil | [96] |
| 22. | CRC (pCC + pRC) (363), Dukes’ A–D; IHC, LI; S | In RC, pts with a LI ≥5% had a better prognosis than those with a lower index | Yes | 2005; Finland | [182] |
| 23. | CRC (40); IHC, NCL-Ki67p, PI; S | Mean PI (52.39%); pts who developed either local recurrence or meta had a significantly raised PI; PI ≤52.7% with a trend to improved survival | No for OS (MA) | 2006; UK | [261] |
| 24. | CRC (38): mucinous (14), non-mucinous (24); stage B1, B2, C1, C; IHC, anti-Ki-67, hot spot, NIH’s Image I; S | Median (35%); (+) correlation with age, LN meta, and with Dukes’ MAC staging (25% in B1, 60% in C2); ↑with grade | Nd | 2007; Romania | [224] |
| 25. | CRC (47): mucinous (5), non-mucinous (42); pT3, G2; IHC, MIB-1, negative <50%; positive > 50%; S | (+) Correlation with LN meta | Nd | 2009; PL | [197] |
| 26. | CC (40), rectosigmoid or rectal AC (33); CRLM (27); IHC, MIB-1; qRT-PCR; S | pCRC (81.8%) vs. CRLM (36.2%); ↓of the GPS in CRLM and confirmed their ↓proliferative levels by qRT PCR | Nd | 2009; New Zealand | [115] |
| 27. | CRC (152), stages I–IV; IHC, rabbit anti-Ki-67, semiq estimation; S | (+) Correlation with the UICC stage and differentiation; (+) pts had the ↓cumulative survival vs. pts with no expression (MA) | Yes | 2010; China | [225] |
| 28. | CRC (356); IHC; S | No association with clinicopathological variables | Nd | 2010; Korea | [235] |
| 29. | CRLM (188/124 for Ki-67); IHC; S | ↑Expression (62%); ↑expression as an independent predictor of poor survival after colon resection (HR 2.6, 95% CI 1.4–4.8) | Yes | 2010; USA | [252] |
| 30. | CRC (201); stages I–IV; IHC, anti-Ki-67, semiq estimation, (+) (score ≥5); S | (+) Expression (59.7%); (+) correlation with tumor size, grade, invasive depth, LN meta, distant meta, TNM; independent prognostic factor of favorable OS (HR 0.34, 95% CI 0.16–0.72) (MA) | Yes | 2011; China | [249] |
| 31. | CRC (31), men with Dukes’ B AC; IHC, MIB-1, semiq estimation; S | Median (46.9 ± 19.2%); inverse relationship with OS (r = −0.67) | Yes | 2012; Italy | [241] |
| 32. | TMA with CRLM (98); IHC, MIB-1; cut-off value for (+) phenotypes (>50%); S | More (+) pts among the long-term survivors; pts with ↑ expression lived longer (HR 0.82, 95% CI 0.68–0.98) (MA); positive predictor for AS, but not for DFS | Yes | 2014; Slovenia | [238] |
| 33. | RC (111); IHC, MIB-1, LI; SCRT + S | ↑Expression correlated with pTR; in females (+) correlation with pTNM in a long break after SCRT | Nd | 2014; PL | [88] |
| 34. | TMA CRC (672), including CRC with LN meta (210); IHC, anti-Ki67, LI; S | Median in pCRC (68.2%), in LN meta (55%); ↑in pCRC vs. CRC LN meta; (+) correlation with tumor penetration and differentiation | No | 2015; Portugal | [226] |
| 35. | CRC (110) including Dukes’ C; IHC, MIB-1, LI; semiq estimation; S | ↑Expression in LN meta vs. pCRC | No | 2015; Turkey | [232] |
| 36. | CRC (74) including mucinous AC (5); IHC, LI; S | LI of well (14%), moderate (31%), and poorly differentiated AC (43%); (+) correlation with stage and grade | Nd | 2015; India | [227] |
| 37. | CRC (2233), I–IV stage; IHC, MIB-1, low (<50%), high (≥50%); S | Pts in stage III with ↑level had ↑3-yr DFS and OS vs. ↓level pts; improved 3-yr PFS for stage IV pts in the ↑vs. ↓level group | Yes | 2016; Germany/pts of Chinese origin | [250] |
| 38. | TMA CRC (1800); IHC, anti-Ki-67, low (0–10%), moderate (>10–25%), high (>25%); S | ↑Expression associated with low stage and LN status; an independent prognostic factor of favorable survival | Yes | 2016; Germany | [237] |
| 39. | TMA CRC (254), stage II and III; IHC, anti Ki-67, low (<20%) and high (≥20%); S | ↑LI associated with ↑TNM stage; ↓LI related to RFS (UA); ↑LI (HR 2.62, 95% CI 1.12–6.14; an independent predictor of unfavorable prognosis (MA) | Yes | 2018; China | [228] |
| 40. | RC (46), stage II and III; IHC, MIB-1, Image System (Nikon), LI, cut-off value (30%); CRT + S | No difference between ↓ and ↑expression groups in clinicopathological factors; ↑LI correlated with lower 5-yr DFS vs. group with ↓LI (53% and 88%), as was the 5-yr OS (68% and 100%) | Yes | 2018; Japan | [254] |
| 41. | CRC (1090), stage 0-IV; IHC; anti-Ki-67, semiq estimation; cut-off value of 25%; S | (+) Correlation with invasive depth, differentiation, and size, AJCC-8, (+) no. of LN and CTx status; ↑level related to poor prognosis and independently predicts prognosis in the AJCC-8; no differences for DFS and OS in stage IV | Yes | 2020; China | [229] |
| 42. | CRC (38), non-neoplastic polyps (2) and AD (20); IHC, anti-Ki-67, LI; S | CRC: ↑LI in higher grade and stage; AD: ↑intensity and high score similar to CRC; non-neoplastic polyps: ↑LI and medium intensity; ↑LI from non-neoplastic to neoplastic cases | Nd | 2021; India | [217] |
| 43. | CRC (210), stages I–III; IHC, polyclonal Ab, LI, cut-off value 60%; S | LI ≥60% indicated a high-risk ratio for both distant meta (HR 2.56, 95% CI 1.08–6.06) and death (HR 2.64, 95% CI 1.07–6.54) | Yes | 2022; China | [230] |
| 44. | RC (154), RC I–II after RT + S (2–3 d after) (64), RC I–III after S (90); IHC, image analysis application package | ↑Level with a survival rate of less than 3 yrs in both pts after RT and S | Yes | 2022; Switzerland, Germany, UK | [255] |
| Type of lncRNA | Material/Research Model | Expression Level | Findings | Ref. No. |
|---|---|---|---|---|
| LOC285194 | CRC (81); CRC cell lines: CaCO-2, HCT8, LoVo and C (CCC-HIE-2 cells); qRT-PCR | ↓ | A poor DFS; an independent predictor of DFS (MA) | [303] |
| PVT1 | Pairs of CRC and C (164); CRC cell lines: RKO and HCT116; siRNA transfection; cell proliferation and invasion assays; gene expression array; qRT-PCR; array-CGH and copy no. analysis; gene set enrichment analysis; WB | ↑ | Promoted cell proliferation; a poor prognosis; an independent risk factor for OS (UA and MA) | [275] |
| Pairs of CRC and C (210); qRT-PCR | ↑ | A shorter DFS and OS; an independent predictor of poor prognosis (MA) | [304] | |
| DANCR | CRC (104); qRT-PCR | ↑ | A shorter OS and DFS; an independent poor prognostic factor for both OS and DFS (MA) | [305] |
| HOTTIP | CRC (156), C (21); qRT-PCR | ↑ | An unfavorable as well as an independent poor prognostic factor (MA) | [306] |
| SNHG20 | CRC and C (107) | ↑ | An independent poor prognostic factor for OS (MA) | [316] |
| BANCR | CRC (106), C (65), qRT-PCR | ↑ | A shorter OS; an independent poor prognostic factor (HR 2.24, 95% CI 1.22–4.16) | [307] |
| SPRY4-IT1 | CRC (106); qRT-PCR | ↑ | A poor OS; an independent prognostic factor (HR 2.34, 95% CI 1.14–4.82) | [308] |
| CCAT1 and CCAT2 | CRC (280) and C (20); qRT-PCR | ↑ | A poor RFS and OS | [309] |
| XIST | CRC (196); CRC cell lines: LOVO, HT-29, HCT8, HCT116, SW480, and DLD1 and C (HCoEpics cells); qRT-PCR | ↑ | Could predict PFS and OS; could act as independent risk factor for poor prognosis | [310] |
| LINC00858 | Pairs of CRC (115); CRC cell lines: T-29, HT-15, SW837 and SW1463; qRT-PCR; siRNA transfection; cell proliferation and apoptosis assays; colony formation assay; dual luciferase reporter assays; RIP; WB | ↑ | An independent poor prognostic factor | [284] |
| Pairs of CRC (50) and 20 female BALB/c nude mouse; qRT-PCR; ISH; MTT assay; BrdU staining; FCM, wound healing, and Transwell assays; luciferase activity assay and RIP; IHC; WB; HE staining | ↑ | Prognostic factor for OS | [277] | |
| MIR22HG | CRC (79) and C (84); CRC cell lines LoVo and HCT116; bioinformatics screen; qRT-PCR; MTT and Transwell assays; mouse model | ↓ | A poor OS and DFS; promoted cell survival, proliferation and tumor meta in vitro and in vivo | [287] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasprzak, A. Prognostic Biomarkers of Cell Proliferation in Colorectal Cancer (CRC): From Immunohistochemistry to Molecular Biology Techniques. Cancers 2023, 15, 4570. https://doi.org/10.3390/cancers15184570
Kasprzak A. Prognostic Biomarkers of Cell Proliferation in Colorectal Cancer (CRC): From Immunohistochemistry to Molecular Biology Techniques. Cancers. 2023; 15(18):4570. https://doi.org/10.3390/cancers15184570
Chicago/Turabian StyleKasprzak, Aldona. 2023. "Prognostic Biomarkers of Cell Proliferation in Colorectal Cancer (CRC): From Immunohistochemistry to Molecular Biology Techniques" Cancers 15, no. 18: 4570. https://doi.org/10.3390/cancers15184570
APA StyleKasprzak, A. (2023). Prognostic Biomarkers of Cell Proliferation in Colorectal Cancer (CRC): From Immunohistochemistry to Molecular Biology Techniques. Cancers, 15(18), 4570. https://doi.org/10.3390/cancers15184570