
Inhibition of Dipeptidyl Peptidase-4 Activates Autophagy to Promote Survival of Breast Cancer Cells via the mTOR/HIF-1α Pathway
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture and Treatment
2.3. Transfection Experiments
2.4. Western Blot Analysis
2.5. Mouse Breast Cancer Models
2.6. Immunohistochemistry (IHC)
2.7. Fluorescence-Activated Cell Sorting (FACS)
2.8. BrdU Cell Proliferation Assay
2.9. ATP/ADP Ratio Assay
2.10. Statistical Analysis
3. Results
3.1. Breast Cancer Cells Were Highly Sensitive to DPP-4 Suppression-Induced Autophagy
3.2. CXCR4 Played an Essential Role in DPP-4 Inhibition-Induced Autophagy in Breast Cancer Cells
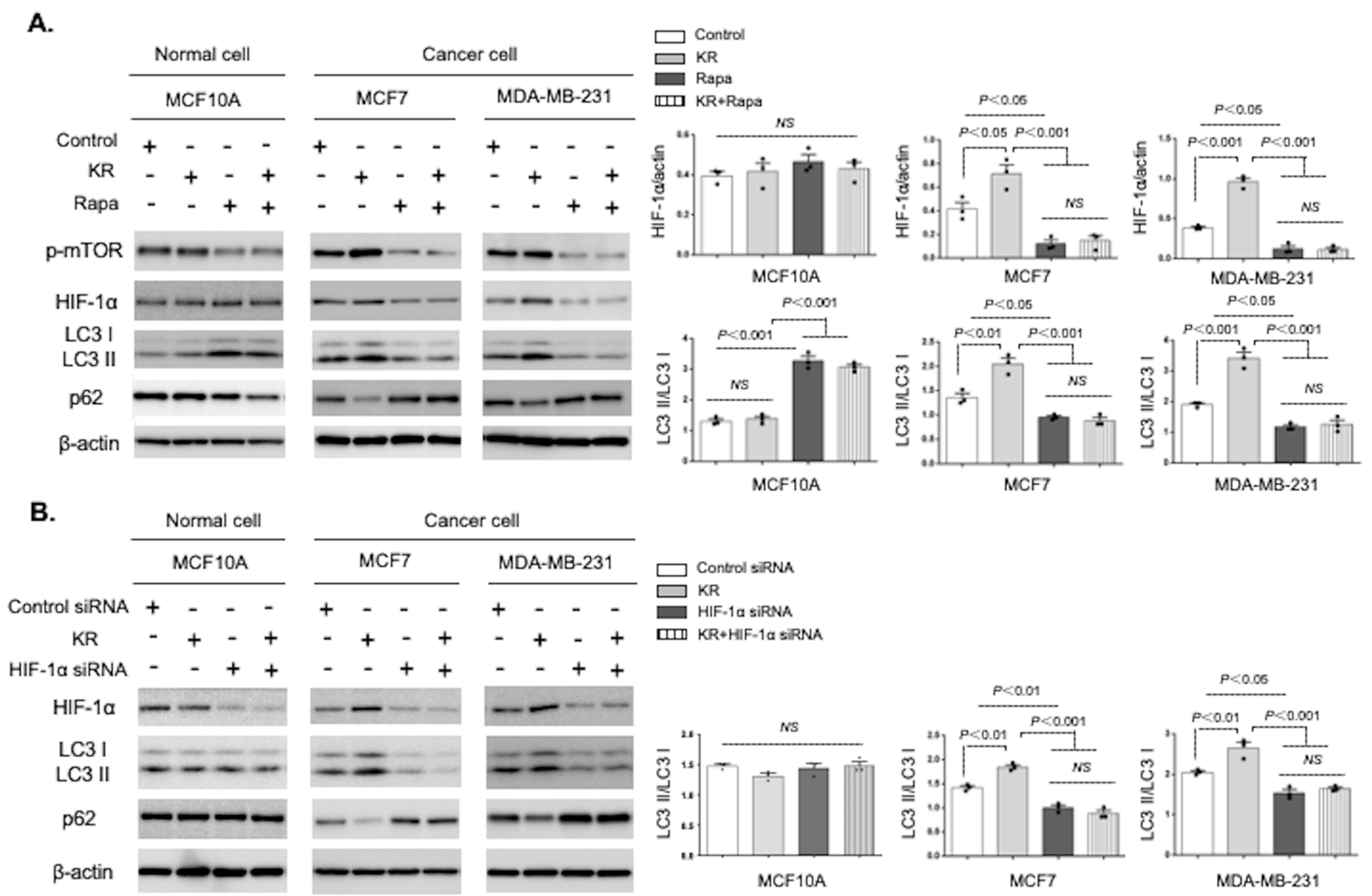
3.3. Autophagic Induction by DPP-4 Inhibition Was Dependent on the mTOR/HIF-1α Axis
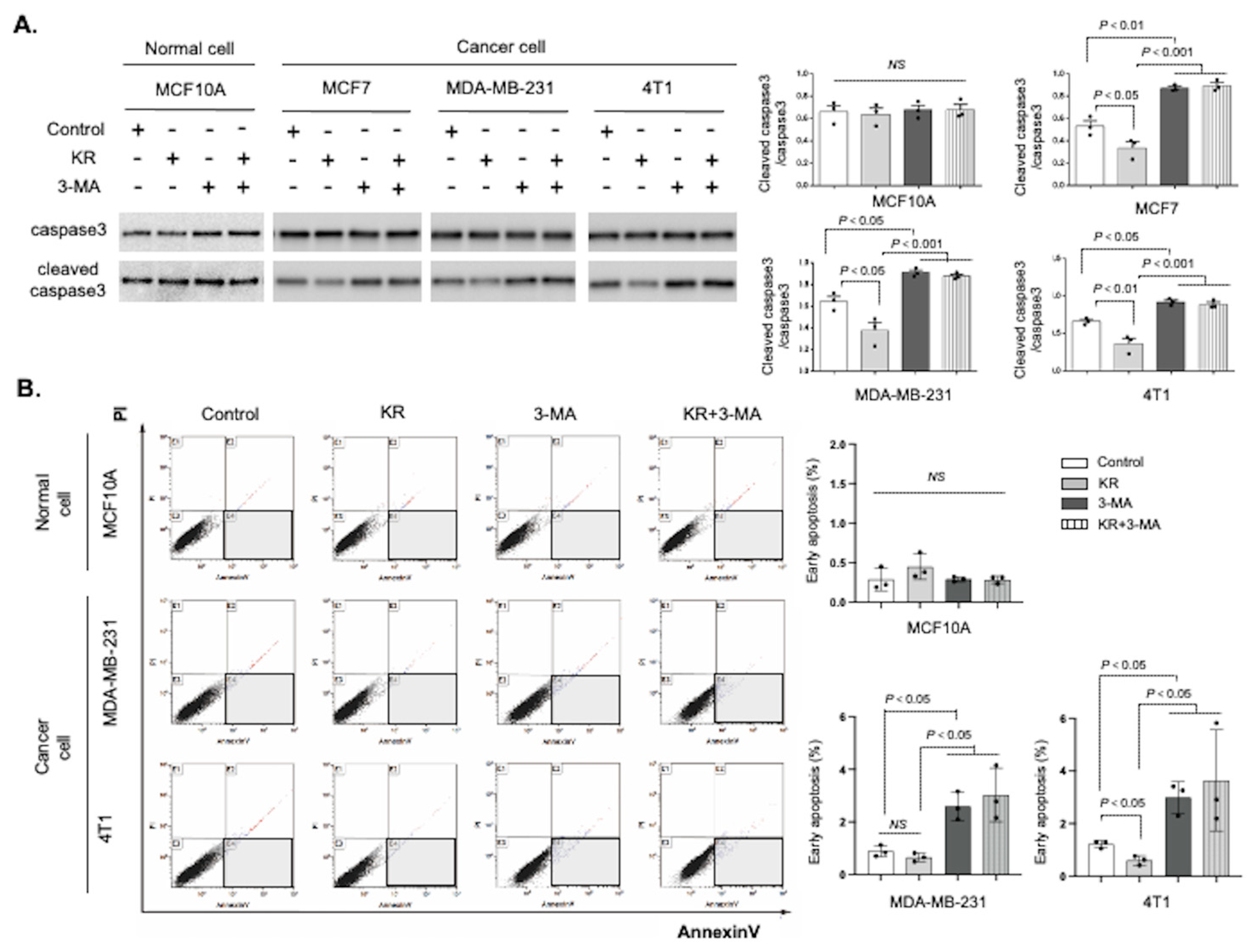
3.4. DPP-4 Inhibition Promoted Breast Cancer Cell Survival via Autophagy
3.5. Metformin Reversed DPP-4 Deficiency-Induced Autophagy and Survival via Suppression of the mTOR/HIF-1α Axis in Mammary Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Rucker, E.B., 3rd; Zhou, B.P. Autophagy regulation in the development and treatment of breast cancer. Acta Biochim. Biophys. Sin. 2016, 48, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.; Song, J.; Lu, J.; et al. Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 728. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. mTORC1 drives HIF-1alpha and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 2015, 34, 2239–2250. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Tan, J.; Zhang, Q. Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol. Int. 2015, 39, 891–898. [Google Scholar] [CrossRef]
- Woo, Y.M.; Shin, Y.; Lee, E.J.; Lee, S.; Jeong, S.H.; Kong, H.K.; Park, E.Y.; Kim, H.K.; Han, J.; Chang, M.; et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1alpha Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PLoS ONE 2015, 10, e0132285. [Google Scholar] [CrossRef]
- de Heer, E.C.; Jalving, M.; Harris, A.L. HIFs, angiogenesis, and metabolism: Elusive enemies in breast cancer. J. Clin. Investig. 2020, 130, 5074–5087. [Google Scholar] [CrossRef]
- Scheen, A.J. A review of gliptins for 2014. Expert Opin. Pharmacother. 2015, 16, 43–62. [Google Scholar] [CrossRef]
- Kanasaki, K.; Kawakita, E.; Koya, D. Relevance of Autophagy Induction by Gastrointestinal Hormones: Focus on the Incretin-Based Drug Target and Glucagon. Front. Pharmacol. 2019, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Kawakita, E.; Koya, D.; Kanasaki, K. CD26/DPP-4: Type 2 Diabetes Drug Target with Potential Influence on Cancer Biology. Cancers 2021, 13, 2191. [Google Scholar] [CrossRef]
- Kajiyama, H.; Kikkawa, F.; Suzuki, T.; Shibata, K.; Ino, K.; Mizutani, S. Prolonged survival and decreased invasive activity attributable to dipeptidyl peptidase IV overexpression in ovarian carcinoma. Cancer Res. 2002, 62, 2753–2757. [Google Scholar]
- Wang, H.; Liu, X.; Long, M.; Huang, Y.; Zhang, L.; Zhang, R.; Zheng, Y.; Liao, X.; Wang, Y.; Liao, Q.; et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 2016, 8, 334ra51. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fan, Y.; Kumagai, A.; Kawakita, E.; Kitada, M.; Kanasaki, K.; Koya, D. Deficiency in Dipeptidyl Peptidase-4 Promotes Chemoresistance through the CXCL12/CXCR4/mTOR/TGFbeta Signaling Pathway in Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 805. [Google Scholar] [CrossRef] [PubMed]
- Abrahami, D.; Dourous, A.; Yin, H.; Yu, O.H.; Fallie, J.L.; Montastruc, F.; Platt, R.W.; Bouganim, N.; Azoulay, L. Incretin based drugs and risk of cholangiocarcinoma among patients with type 2 diabetes: Population based cohort study. BMJ 2018, 363, k4880. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Takagaki, Y.; Yoshitomi, Y.; Ikeda, T.; Li, J.; Kitada, M.; Kumagai, A.; Kawakita, E.; Shi, S.; Kanasaki, K.; et al. Inhibition of Dipeptidyl Peptidase-4 Accelerates Epithelial-Mesenchymal Transition and Breast Cancer Metastasis via the CXCL12/CXCR4/mTOR Axis. Cancer Res. 2019, 79, 735–746. [Google Scholar] [CrossRef]
- Dai, X.; Zeng, J.; Yan, X.; Lin, Q.; Wang, K.; Chen, J.; Shen, F.; Gu, X.; Wang, Y.; Chen, J.; et al. Sitagliptin-mediated preservation of endothelial progenitor cell function via augmenting autophagy enhances ischaemic angiogenesis in diabetes. J. Cell Mol. Med. 2018, 22, 89–100. [Google Scholar] [CrossRef]
- Nathan, D.M.; Buse, J.B.; Davidson, M.B.; Ferrannini, E.; Holman, R.R.; Sherwin, R.; Zinman, B.; American Diabetes Association; European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: A consensus algorithm for the initiation and adjustment of therapy: A consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009, 32, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Landman, G.W.; Kleefstra, N.; van Hateren, K.J.; Groenier, K.H.; Gans, R.O.; Bilo, H.J. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care 2010, 33, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Mallik, R.; Chowdhury, T.A. Metformin in cancer. Diabetes Res. Clin. Pract. 2018, 143, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Yang, Y.; Liu, S. Association between Metformin Therapy and Breast Cancer Incidence and Mortality: Evidence from a Meta-Analysis. J. Breast Cancer 2015, 18, 264–270. [Google Scholar] [CrossRef]
- Tang, G.H.; Satkunam, M.; Pond, G.R.; Steinberg, G.R.; Blandino, G.; Schunemann, H.J.; Muti, P. Association of Metformin with Breast Cancer Incidence and Mortality in Patients with Type II Diabetes: A GRADE-Assessed Systematic Review and Meta-analysis. Cancer Epidemiol. Biomarkers Prev. 2018, 27, 627–635. [Google Scholar] [CrossRef]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Gimarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef]
- Sun, C.; Li, X.; Guo, E.; Li, N.; Zhou, B.; Lu, H.; Huang, J.; Xia, M.; Shan, W.; Wang, B.; et al. MCP-1/CCR-2 axis in adipocytes and cancer cell respectively facilitates ovarian cancer peritoneal metastasis. Oncogene 2020, 39, 1681–1695. [Google Scholar] [CrossRef]
- Shao, S.; Zhao, L.; An, G.; Zhang, L.; Jing, X.; Luo, M.; Li, W.; Meng, D.; Ning, Q.; Zhao, X.; et al. Metformin suppresses HIF-1alpha expression in cancer-associated fibroblasts to prevent tumor-stromal cross talk in breast cancer. FASEB J. 2020, 34, 10860–10870. [Google Scholar] [CrossRef]
- Kawakita, E.; Yang, F.; Kumagai, A.; Takagaki, Y.; Kitada, M.; Yoshitomi, Y.; Ikeda, T.; Nakamura, Y.; Ishigaki, Y.; Kanasaki, K.; et al. Metformin Mitigates DPP-4 Inhibitor-Induced Breast Cancer Metastasis via Suppression of mTOR Signaling. Mol. Cancer Res. 2021, 19, 61–73. [Google Scholar] [CrossRef]
- Saito, A.; Kitayama, J.; Horie, H.; Koinuma, K.; Kawashima, R.; Ohzawa, H.; Yamaguchi, H.; Kawahira, H.; Mimura, T.; Lefor, A.K.; et al. Dipeptidyl Peptidase (DPP)-4 Inhibitor Impairs the Outcomes of Patients with Type 2 Diabetes Mellitus After Curative Resection for Colorectal Cancer. Cancer Res. Commun. 2021, 1, 106–114. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sincrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef] [PubMed]
- Gavilan, E.; Sanchez-Aguayo, I.; Daza, P.; Ruano, D. GSK-3beta signaling determines autophagy activation in the breast tumor cell line MCF7 and inclusion formation in the non-tumor cell line MCF10A in response to proteasome inhibition. Cell Death Dis. 2013, 4, e572. [Google Scholar] [CrossRef][Green Version]
- Claude-Taupin, A.; Fonderflick, L.; Gauthier, T.; Mansi, L.; Pallandre, J.R.; Borg, C.; Perez, V.; Monnien, F.; Algros, M.P.; Vigneron, M.; et al. ATG9A Is Overexpressed in Triple Negative Breast Cancer and Its In Vitro Extinction Leads to the Inhibition of Pro-Cancer Phenotypes. Cells 2018, 7, 248. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, M.; Zhao, J.; Wang, J.; Zhang, Y.; Zhang, Q. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med. Oncol. 2013, 30, 475. [Google Scholar] [CrossRef] [PubMed]
- Murase, H.; Kuno, A.; Miki, T.; Tanno, M.; Yano, T.; Kouzu, H.; Ishikawa, S.; Tobisawa, T.; Ogasawara, M.; Nishizawa, K. Inhibition of DPP-4 reduces acute mortality after myocardial infarction with restoration of autophagic response in type 2 diabetic rats. Cardiovasc. Diabetol. 2015, 14, 103. [Google Scholar] [CrossRef]
- Arab, H.H.; Gad, A.M.; Reda, E.; Yahia, R.; Eid, A.H. Activation of autophagy by sitagliptin attenuates cadmium-induced testicular impairment in rats: Targeting AMPK/mTOR and Nrf2/HO-1 pathways. Life Sci. 2021, 269, 119031. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arezana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Xu, C.; Zhao, H.; Chen, H.; Yao, Q. CXCR4 in breast cancer: Oncogenic role and therapeutic targeting. Drug Des. Dev. Ther. 2015, 9, 4953–4964. [Google Scholar]
- Correia, A.L.; Guimaraes, J.C.; Auf der Maur, P.; De Silva, D.; Trefny, M.P.; Okamoto, R.; Bruno, S.; Schmidt, A.; Mertz, K.; Volkmann, K.; et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature 2021, 594, 566–571. [Google Scholar] [CrossRef]
- Zhang, Z.; Ni, C.; Chen, W.; Wu, P.; Wang, Z.; Yin, J.; Huang, J.; Qiu, F. Expression of CXCR4 and breast cancer prognosis: A systematic review and meta-analysis. BMC Cancer 2014, 14, 49. [Google Scholar] [CrossRef]
- Wang, X.; Cao, Y.; Zhang, S.; Chen, Z.; Fan, L.; Shen, X.; Zhou, S.; Chen, D. Stem cell autocrine CXCL12/CXCR4 stimulates invasion and metastasis of esophageal cancer. Oncotarget 2017, 8, 36149–36160. [Google Scholar] [CrossRef]
- Hu, X.; Mei, S.; Meng, W.; Xue, S.; Jiang, L.; Yang, Y.; Hui, L.; Chen, Y.; Guan, M.X. CXCR4-mediated signaling regulates autophagy and influences acute myeloid leukemia cell survival and drug resistance. Cancer Lett. 2018, 425, 1–12. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, B.; Bao, L.; Jin, L.; Yang, M.; Peng, Y.; Kumar, A.; Wang, J.E.; Wang, C.; Zou, X.; et al. ZMYND8 acetylation mediates HIF-dependent breast cancer progression and metastasis. J. Clin. Investig. 2018, 128, 1937–1955. [Google Scholar] [CrossRef]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Land, S.C.; Tee, A.R. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Currie, C.J.; Poole, C.D.; Jenkins-Jones, S.; Gale, E.A.; Johnson, J.A.; Morgan, C.L. Mortality after incident cancer in people with and without type 2 diabetes: Impact of metformin on survival. Diabetes Care 2012, 35, 299–304. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Esteva, F.J.; Ensor, J.; Hortobagyi, G.N.; Lee, M.H.; Yeung, S.C.J. Metformin and thiazolidinediones are associated with improved breast cancer-specific survival of diabetic women with HER2+ breast cancer. Ann. Oncol. 2012, 23, 1771–1780. [Google Scholar] [CrossRef]
- Jacob, L.; Kostev, K.; Rathmann, W.; Kalder, M. Impact of metformin on metastases in patients with breast cancer and type 2 diabetes. J. Diabetes Complicat. 2016, 30, 1056–1059. [Google Scholar] [CrossRef]
- Mohammed, A.; Janakiram, N.B.; Brewer, M.; Ritchie, R.L.; Marya, A.; Lighfoot, S.; Steele, V.E.; Rao, C.V. Antidiabetic Drug Metformin Prevents Progression of Pancreatic Cancer by Targeting in Part Cancer Stem Cells and mTOR Signaling. Transl. Oncol. 2013, 6, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, I.; Lohmann, A.E.; Ennis, M.; Dowling, R.J.O.; Cescon, D.; Elser, C.; Potvin, K.R.; Haq, R.; Hammm, C.; Chang, M.C.; et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast 2019, 48, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Nanni, O.; Amadori, D.; De Censi, A.; Rocca, A.; Freschi, A.; Bologna, A.; Gianni, L.; Rosetti, F.; Amaducci, L.; Cavanna, L.; et al. Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial. Breast Cancer Res. Treat. 2019, 174, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Qu, B.; Huang, X.; Song, Y.; Gao, P.; Shi, J.; Zhou, C.; Wang, Z. The potential adjunctive benefit of adding metformin to standard treatment in inoperable cancer patients: A meta-analysis of randomized controlled trials. Ann. Transl. Med. 2020, 8, 1404. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawakita, E.; Yang, F.; Shi, S.; Takagaki, Y.; Koya, D.; Kanasaki, K. Inhibition of Dipeptidyl Peptidase-4 Activates Autophagy to Promote Survival of Breast Cancer Cells via the mTOR/HIF-1α Pathway. Cancers 2023, 15, 4529. https://doi.org/10.3390/cancers15184529
Kawakita E, Yang F, Shi S, Takagaki Y, Koya D, Kanasaki K. Inhibition of Dipeptidyl Peptidase-4 Activates Autophagy to Promote Survival of Breast Cancer Cells via the mTOR/HIF-1α Pathway. Cancers. 2023; 15(18):4529. https://doi.org/10.3390/cancers15184529
Chicago/Turabian StyleKawakita, Emi, Fan Yang, Sen Shi, Yuta Takagaki, Daisuke Koya, and Keizo Kanasaki. 2023. "Inhibition of Dipeptidyl Peptidase-4 Activates Autophagy to Promote Survival of Breast Cancer Cells via the mTOR/HIF-1α Pathway" Cancers 15, no. 18: 4529. https://doi.org/10.3390/cancers15184529
APA StyleKawakita, E., Yang, F., Shi, S., Takagaki, Y., Koya, D., & Kanasaki, K. (2023). Inhibition of Dipeptidyl Peptidase-4 Activates Autophagy to Promote Survival of Breast Cancer Cells via the mTOR/HIF-1α Pathway. Cancers, 15(18), 4529. https://doi.org/10.3390/cancers15184529