
F-box DNA Helicase 1 (FBH1) Contributes to the Destabilization of DNA Damage Repair Machinery in Human Cancers


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
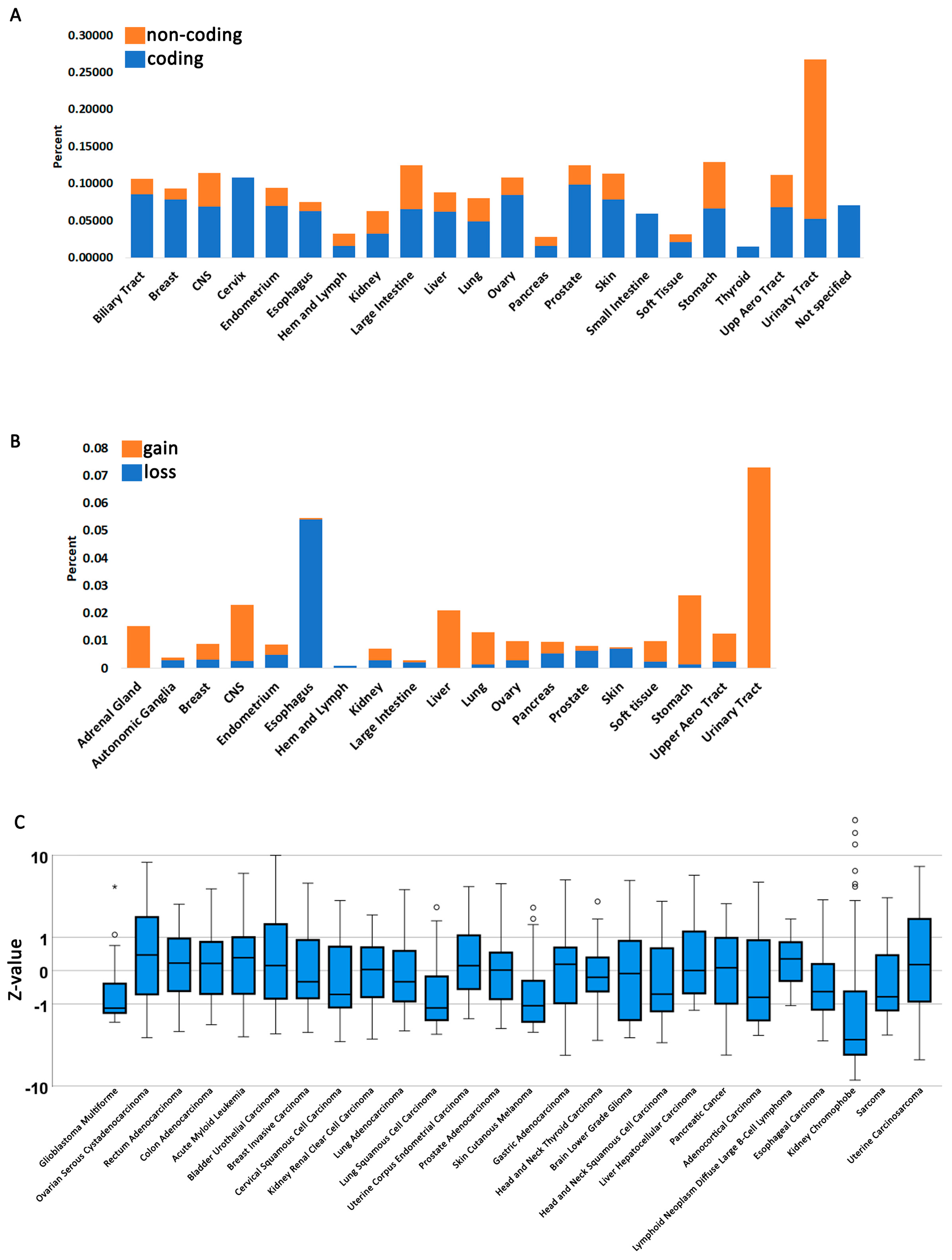
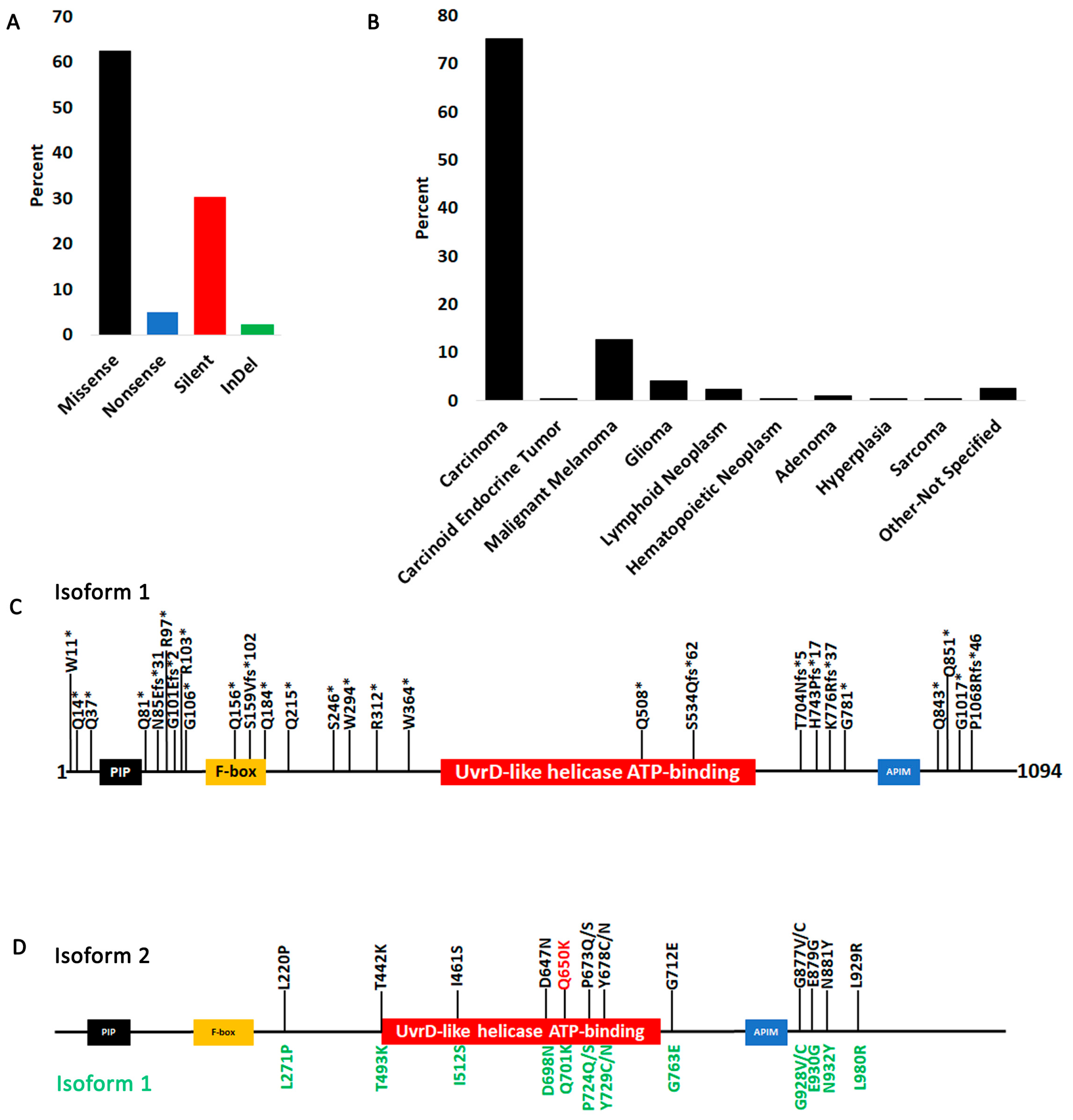
3.1. Fbh1 Gene Alterations and Expression Profiles in Human Cancers
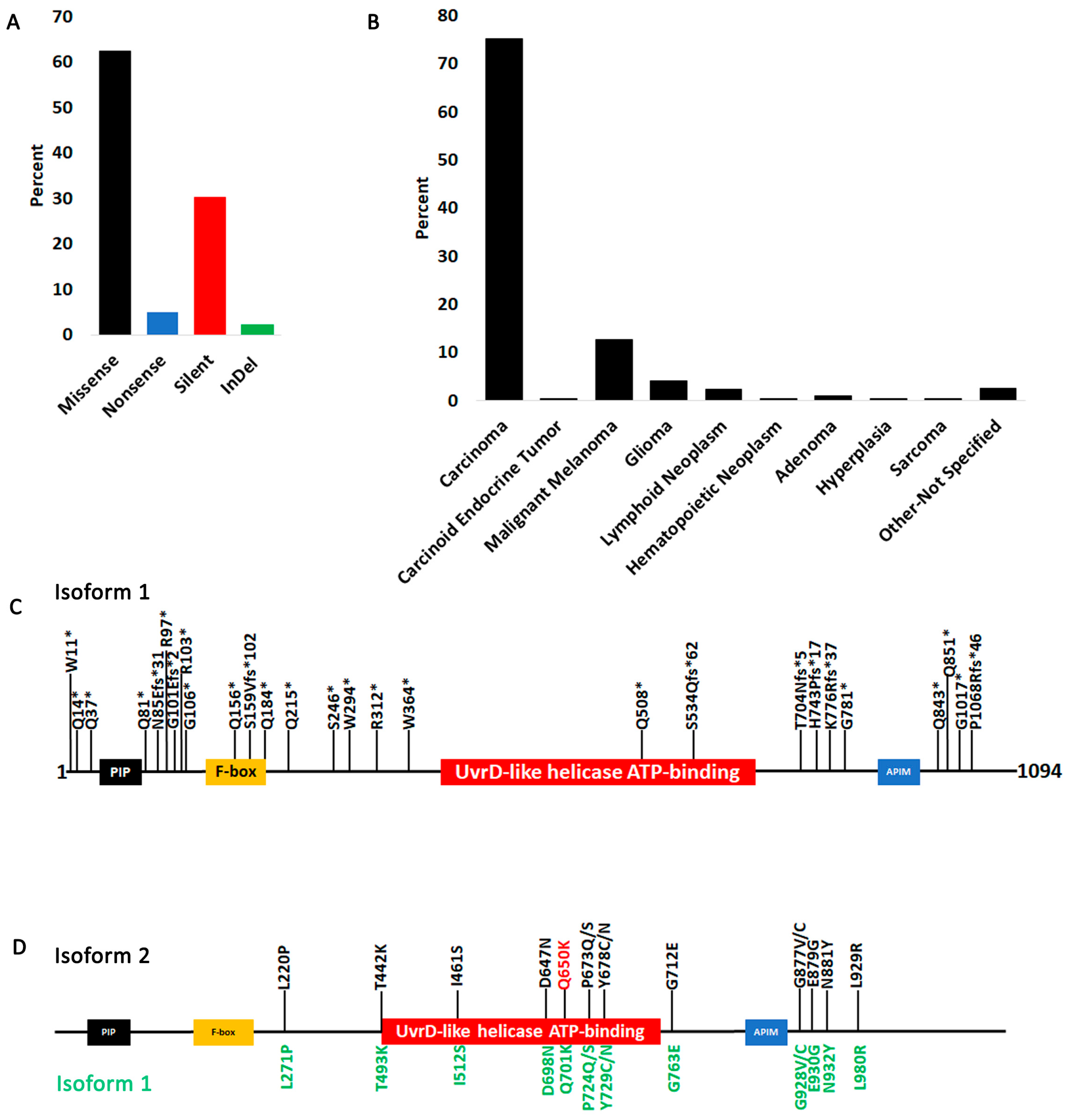
3.2. FBH1 Coding Mutations
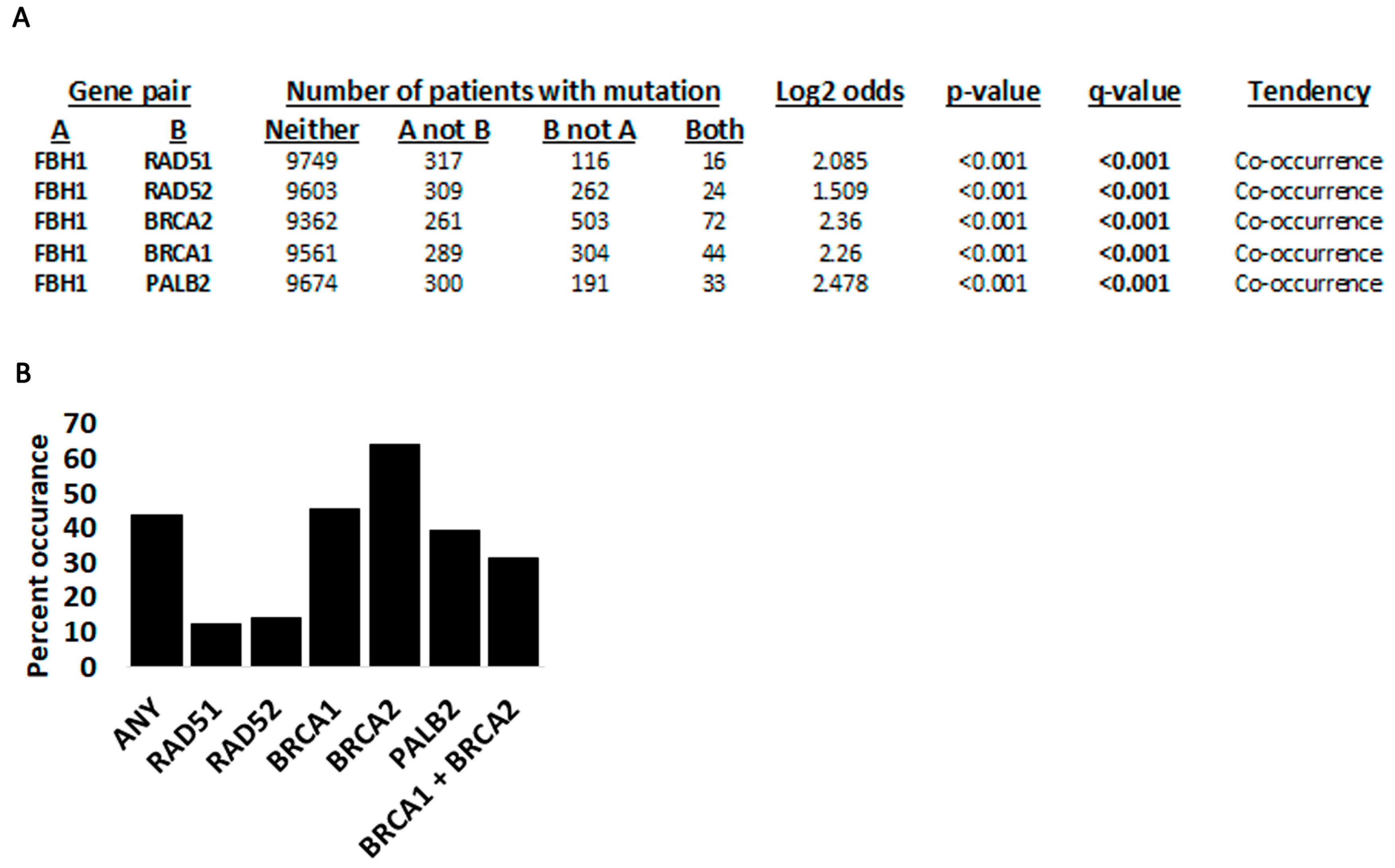
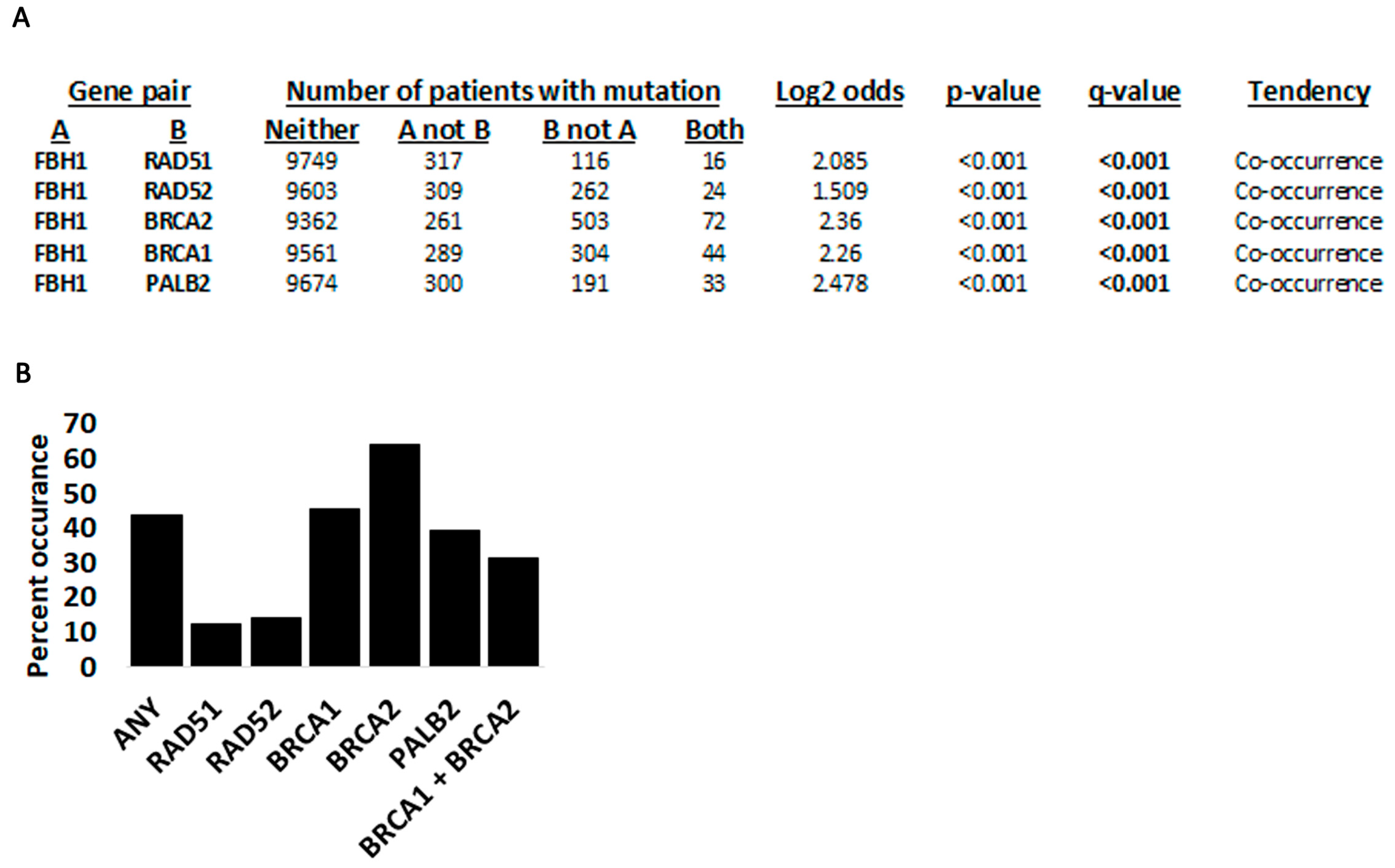
3.3. Fbh1 Co-Occurring Mutations with Other Recombination Genes
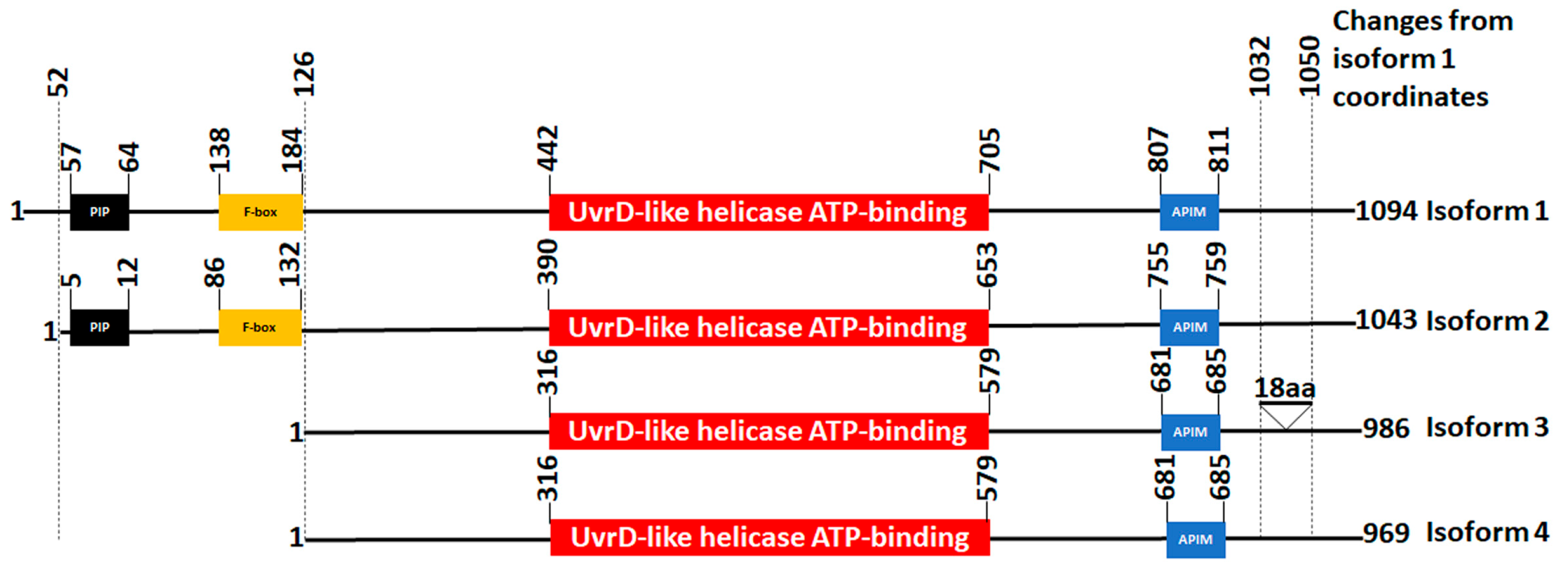
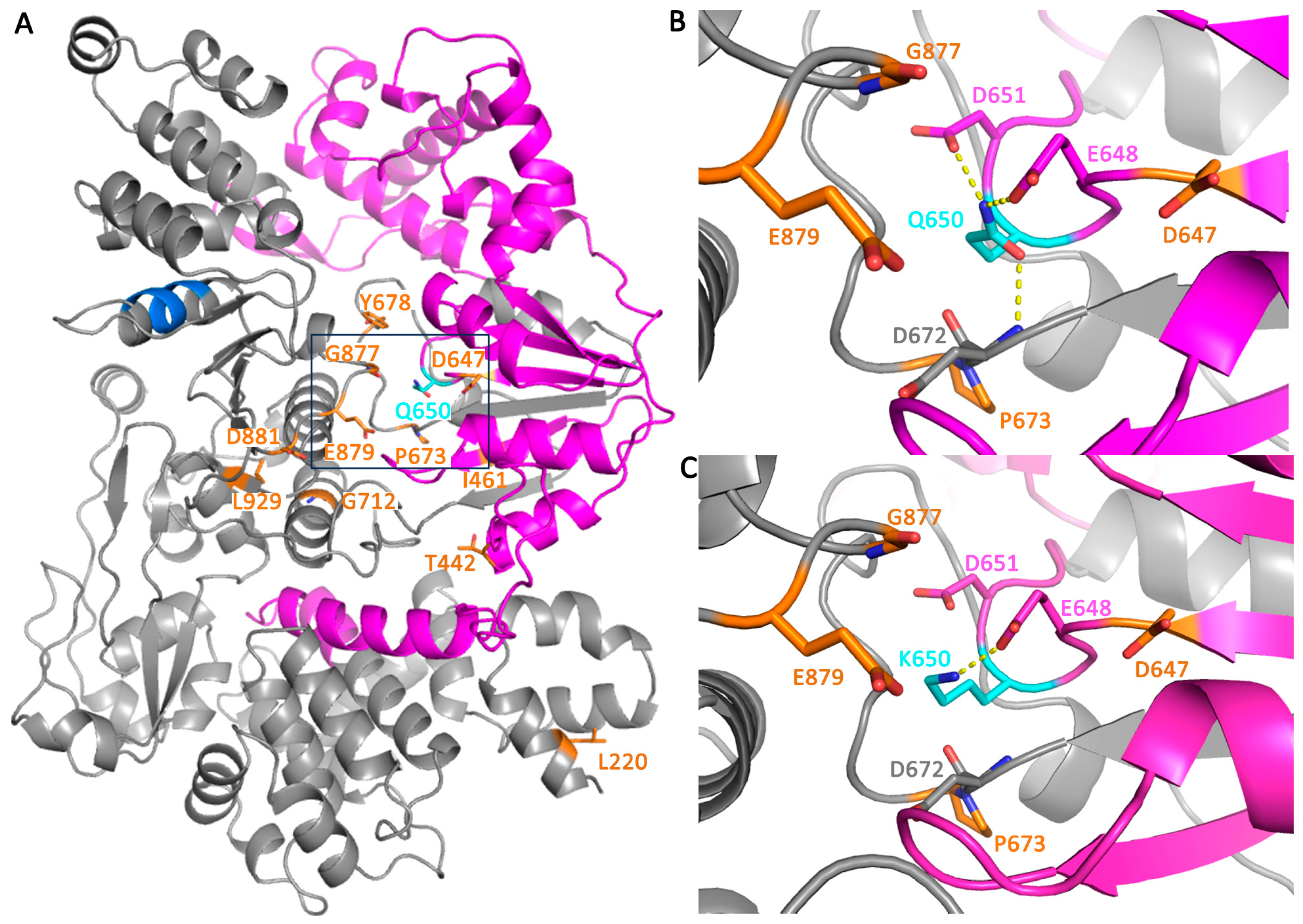
3.4. Structural Analysis of FBH1 Mutations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cavalier-Smith, T. Origins of the machinery of recombination and sex. Heredity 2002, 88, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Amunugama, R.; Fishel, R. Homologous recombination in eukaryotes. Prog. Mol. Biol. Transl. Sci. 2012, 110, 155–206. [Google Scholar] [CrossRef]
- Korolev, S. Advances in structural studies of recombination mediator proteins. Biophys. Chem. 2017, 225, 27–37. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Quinet, A.; Lemacon, D.; Vindigni, A. Replication Fork Reversal: Players and Guardians. Mol. Cell 2017, 68, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef]
- Epum, E.A.; Haber, J.E. DNA replication: The recombination connection. Trends Cell Biol. 2022, 32, 45–57. [Google Scholar] [CrossRef]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Seol, J.H.; Shim, E.Y.; Lee, S.E. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res. 2018, 809, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Shinohara, A. Chromosome architecture and homologous recombination in meiosis. Front. Cell Dev. Biol. 2022, 10, 1097446. [Google Scholar] [CrossRef] [PubMed]
- Nowosielska, A. Bacterial DNA repair genes and their eukaryotic homologues: 5. The role of recombination in DNA repair and genome stability. Acta Biochim. Pol. 2007, 54, 483–494. [Google Scholar] [CrossRef]
- Courcelle, J.; Worley, T.K.; Courcelle, C.T. Recombination Mediator Proteins: Misnomers That Are Key to Understanding the Genomic Instabilities in Cancer. Genes 2022, 13, 437. [Google Scholar] [CrossRef]
- Spies, J.; Polasek-Sedlackova, H.; Lukas, J.; Somyajit, K. Homologous Recombination as a Fundamental Genome Surveillance Mechanism during DNA Replication. Genes 2021, 12, 1960. [Google Scholar] [CrossRef]
- Rahimian, E.; Amini, A.; Alikarami, F.; Pezeshki, S.M.S.; Saki, N.; Safa, M. DNA repair pathways as guardians of the genome: Therapeutic potential and possible prognostic role in hematologic neoplasms. DNA Repair. 2020, 96, 102951. [Google Scholar] [CrossRef]
- Alhegaili, A.S. Role of DNA Repair Deficiency in Cancer Development. Pak. J. Biol. Sci. 2023, 26, 15–22. [Google Scholar] [CrossRef]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef]
- Shao, C.; Wan, J.; Lam, F.C.; Tang, H.; Marley, A.R.; Song, Y.; Miller, C.; Brown, M.; Han, J.; Adeboyeje, G. A comprehensive literature review and meta-analysis of the prevalence of pan-cancer BRCA mutations, homologous recombination repair gene mutations, and homologous recombination deficiencies. Environ. Mol. Mutagen. 2022, 63, 308–316. [Google Scholar] [CrossRef]
- Foo, T.K.; Xia, B. BRCA1-Dependent and Independent Recruitment of PALB2-BRCA2-RAD51 in the DNA Damage Response and Cancer. Cancer Res. 2022, 82, 3191–3197. [Google Scholar] [CrossRef]
- Yoshimura, A.; Imoto, I.; Iwata, H. Functions of Breast Cancer Predisposition Genes: Implications for Clinical Management. Int. J. Mol. Sci. 2022, 23, 7481. [Google Scholar] [CrossRef]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Hanamshet, K.; Mazina, O.M.; Mazin, A.V. Reappearance from Obscurity: Mammalian Rad52 in Homologous Recombination. Genes 2016, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.J.; DiDomenico, S.F.; Patel, M.; Mazin, A.V. RAD52: Paradigm of Synthetic Lethality and New Developments. Front. Genet. 2021, 12, 780293. [Google Scholar] [CrossRef]
- Lok, B.H.; Powell, S.N. Molecular pathways: Understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin. Cancer Res. 2012, 18, 6400–6406. [Google Scholar] [CrossRef]
- Mortensen, U.H.; Lisby, M.; Rothstein, R. Rad52. Curr. Biol. 2009, 19, R676–R677. [Google Scholar] [CrossRef] [PubMed]
- Krogh, B.O.; Symington, L.S. Recombination proteins in yeast. Annu. Rev. Genet. 2004, 38, 233–271. [Google Scholar] [CrossRef] [PubMed]
- Maher, R.L.; Branagan, A.M.; Morrical, S.W. Coordination of DNA replication and recombination activities in the maintenance of genome stability. J. Cell Biochem. 2011, 112, 2672–2682. [Google Scholar] [CrossRef]
- Roy, U.; Greene, E.C. The Role of the Rad55-Rad57 Complex in DNA Repair. Genes 2021, 12, 1390. [Google Scholar] [CrossRef]
- Symington, L.S. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 2002, 66, 630–670. [Google Scholar] [CrossRef]
- Huselid, E.; Bunting, S.F. The Regulation of Homologous Recombination by Helicases. Genes 2020, 11, 498. [Google Scholar] [CrossRef] [PubMed]
- Cejka, P.; Symington, L.S. DNA End Resection: Mechanism and Control. Annu. Rev. Genet. 2021, 55, 285–307. [Google Scholar] [CrossRef] [PubMed]
- Osman, F.; Dixon, J.; Barr, A.R.; Whitby, M.C. The F-Box DNA helicase Fbh1 prevents Rhp51-dependent recombination without mediator proteins. Mol. Cell Biol. 2005, 25, 8084–8096. [Google Scholar] [CrossRef] [PubMed]
- Jalan, M.; Oehler, J.; Morrow, C.A.; Osman, F.; Whitby, M.C. Factors affecting template switch recombination associated with restarted DNA replication. eLife 2019, 8, 41697. [Google Scholar] [CrossRef] [PubMed]
- Fugger, K.; Mistrik, M.; Danielsen, J.R.; Dinant, C.; Falck, J.; Bartek, J.; Lukas, J.; Mailand, N. Human Fbh1 helicase contributes to genome maintenance via pro- and anti-recombinase activities. J. Cell Biol. 2009, 186, 655–663. [Google Scholar] [CrossRef]
- Laulier, C.; Cheng, A.; Huang, N.; Stark, J.M. Mammalian Fbh1 is important to restore normal mitotic progression following decatenation stress. DNA Repair. 2010, 9, 708–717. [Google Scholar] [CrossRef]
- Chu, W.K.; Payne, M.J.; Beli, P.; Hanada, K.; Choudhary, C.; Hickson, I.D. FBH1 influences DNA replication fork stability and homologous recombination through ubiquitylation of RAD51. Nat. Commun. 2015, 6, 5931. [Google Scholar] [CrossRef]
- Liu, G.; Li, J.; He, B.; Yan, J.; Zhao, J.; Wang, X.; Zhao, X.; Xu, J.; Wu, Y.; Zhang, S.; et al. Bre1/RNF20 promotes Rad51-mediated strand exchange and antagonizes the Srs2/FBH1 helicases. Nat. Commun. 2023, 14, 3024. [Google Scholar] [CrossRef]
- Fugger, K.; Mistrik, M.; Neelsen, K.J.; Yao, Q.; Zellweger, R.; Kousholt, A.N.; Haahr, P.; Chu, W.K.; Bartek, J.; Lopes, M.; et al. FBH1 Catalyzes Regression of Stalled Replication Forks. Cell Rep. 2015, 10, 1749–1757. [Google Scholar] [CrossRef]
- Higgs, M.R.; Reynolds, J.J.; Winczura, A.; Blackford, A.N.; Borel, V.; Miller, E.S.; Zlatanou, A.; Nieminuszczy, J.; Ryan, E.L.; Davies, N.J.; et al. BOD1L Is Required to Suppress Deleterious Resection of Stressed Replication Forks. Mol. Cell 2015, 59, 462–477. [Google Scholar] [CrossRef]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef]
- Liu, J.; Chaves-Arquero, B.; Wei, P.; Tencer, A.H.; Ruiz-Albor, A.; Zhang, G.; Blanco, F.J.; Kutateladze, T.G. Molecular insight into the PCNA-binding mode of FBH1. Structure 2023, 31, 511–517.e3. [Google Scholar] [CrossRef]
- Bacquin, A.; Pouvelle, C.; Siaud, N.; Perderiset, M.; Salome-Desnoulez, S.; Tellier-Lebegue, C.; Lopez, B.; Charbonnier, J.B.; Kannouche, P.L. The helicase FBH1 is tightly regulated by PCNA via CRL4(Cdt2)-mediated proteolysis in human cells. Nucleic Acids Res. 2013, 41, 6501–6513. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jimenez, F.; Muinos, F.; Sentis, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, P.; Nordgard, S.H.; Lingjaerde, O.C.; Russnes, H.G.; Rye, I.H.; Sun, W.; Weigman, V.J.; Marynen, P.; Zetterberg, A.; Naume, B.; et al. Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 16910–16915. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Chen, S.; Isik, L.; Tyekucheva, S.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Cancer-specific high-throughput annotation of somatic mutations: Computational prediction of driver missense mutations. Cancer Res. 2009, 69, 6660–6667. [Google Scholar] [CrossRef]
- Wong, W.C.; Kim, D.; Carter, H.; Diekhans, M.; Ryan, M.C.; Karchin, R. CHASM and SNVBox: Toolkit for detecting biologically important single nucleotide mutations in cancer. Bioinformatics 2011, 27, 2147–2148. [Google Scholar] [CrossRef] [PubMed]
- Tokheim, C.; Karchin, R. CHASMplus Reveals the Scope of Somatic Missense Mutations Driving Human Cancers. Cell Syst. 2019, 9, 9–23.e8. [Google Scholar] [CrossRef]
- Cheadle, C.; Vawter, M.P.; Freed, W.J.; Becker, K.G. Analysis of microarray data using Z score transformation. J. Mol. Diagn. 2003, 5, 73–81. [Google Scholar] [CrossRef]
- Guo, Y.; Sheng, Q.; Li, J.; Ye, F.; Samuels, D.C.; Shyr, Y. Large scale comparison of gene expression levels by microarrays and RNAseq using TCGA data. PLoS ONE 2013, 8, e71462. [Google Scholar] [CrossRef]
- Park, J.S.; Choi, E.; Lee, S.H.; Lee, C.; Seo, Y.S. A DNA helicase from Schizosaccharomyces pombe stimulated by single-stranded DNA-binding protein at low ATP concentration. J. Biol. Chem. 1997, 272, 18910–18919. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.H.; Lee, S.H.; Kim, D.H.; Kang, H.Y.; Bae, S.H.; Pan, Z.Q.; Seo, Y.S. The novel human DNA helicase hFBH1 is an F-box protein. J. Biol. Chem. 2002, 277, 24530–24537. [Google Scholar] [CrossRef] [PubMed]
- Pagel, K.A.; Kim, R.; Moad, K.; Busby, B.; Zheng, L.; Tokheim, C.; Ryan, M.; Karchin, R. Integrated Informatics Analysis of Cancer-Related Variants. JCO Clin. Cancer Inform. 2020, 4, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Hamid, A.B.; Frank, L.E.; Bouley, R.A.; Petreaca, R.C. Pan-cancer analysis of co-occurring mutations in RAD52 and the BRCA1-BRCA2-PALB2 axis in human cancers. PLoS ONE 2022, 17, e0273736. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Stelter, M.; Acajjaoui, S.; McSweeney, S.; Timmins, J. Structural and mechanistic insight into DNA unwinding by Deinococcus radiodurans UvrD. PLoS ONE 2013, 8, e77364. [Google Scholar] [CrossRef]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 34, W239–W242. [Google Scholar] [CrossRef]
- Lewis, K.A.; Mullany, S.; Thomas, B.; Chien, J.; Loewen, R.; Shridhar, V.; Cliby, W.A. Heterozygous ATR mutations in mismatch repair-deficient cancer cells have functional significance. Cancer Res. 2005, 65, 7091–7095. [Google Scholar] [CrossRef]
- Vanderschelden, R.K.; Rodriguez-Escriba, M.; Chan, S.H.; Berman, A.J.; Rajkovic, A.; Yatsenko, S.A. Heterozygous TP63 pathogenic variants in isolated primary ovarian insufficiency. J. Assist. Reprod. Genet. 2023, 40, 2211–2218. [Google Scholar] [CrossRef]
- Fero, M.L.; Randel, E.; Gurley, K.E.; Roberts, J.M.; Kemp, C.J. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 1998, 396, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.F.; Nguyen, H.T.; Veitia, R.A. Causes and effects of haploinsufficiency. Biol. Rev. Camb. Philos. Soc. 2019, 94, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 2001, 1, 157–162. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue a | FBH1 | RAD51 | RAD52 | BRCA1 | BRCA2 | PALB2 |
|---|---|---|---|---|---|---|
| CNS | C65Y b | K1551R | H264Y, L1357=, N2189=, C393=, E2635=, P2796T, Y3203H | V213=, G112= | ||
| CNS | I460T | A1438V, E1033*, N383S | K607=, S973L, E1276*, S1753=, V2010=, G2379R, G2596E, K2833N, T3401M | |||
| CNS | N330S R380W | K1551R | H264Y, L1357=, N2189=, C393=, E2635=, P2796T, Y3203H | V213=, G112= | ||
| Endometrium | A546T | A195V | A622V, E733*, L156= | S2835P, Q569H, G25=, P143S, S571F, A1725S, G1761V, A1981S, K2206N, S2216F | R160I | |
| Endometrium | E550K | Z = 2.13 c | R1443Q | S1331Y, H1350N, E1441* | ||
| Endometrium | G928C | A195V | A622V, E733*, L156= | S2835P, Q569H, G25=, P143S, S571F, A1725S, G1761V, A1981S, K2206N, S2216F | R160I | |
| Endometrium | M52L | E1258D, K996Q, E733A | K2316Q | |||
| Endometrium | R975Q | Z = 2.13 | R1443Q | S1331Y, H1350N, E1441* | ||
| Endometrium | V274M | A195V | A622V, E733*, L156= | S571F, G25=, P143S, Q569H, A1725S, G1761V, A1981S, K2206N, S2216F, S2835P | R160I | |
| Endometrium | V593I | E597K | S3144Y | L1092= | ||
| Endometrium | Y729C | M244I | N665S | N1100S, D191N, C738Y, D980N, K1888E | ||
| Large Intestine | A400V | Z = 2.119 | D821Y | S3319Y, F1192C, D479Y, Y2997* | ||
| Large Intestine | G30S | S763F, K1732R, E577* | E2258K, S2052*, L29I, L414=, E1415= | T993=, E554K, R147= | ||
| Large Intestine | H202R | N976S, G401E | A1439=, T1505I | T733A, F440Lfs*12 | ||
| Large Intestine | I512S I989S | K1254T | S3332Y, F701C, L951I, K956T | |||
| Large Intestine | K984T | Z = 2.119 | D821Y | S3319Y, F1192C, D479Y, Y2997* | ||
| Large Intestine | S654N | I1318T, K690R, G57= | E97*, D281Y, S445Y, L613R, R645I, L901I, D1352Y, E2635G | P65= | ||
| Large Intestine | V1093I | S226F | E349V | E3096K, D3410= | R566H | |
| Skin | D64N | G964V | E215G | |||
| Skin | S1067F | Q1800* | A1253=, G1696E | |||
| Stomach | Q473R | K1497R, T1246A, K1104E | T3033Lfs*29, P190H, L413V | |||
| Upper Aerodigestive | E334K T61N | K1183R, P871L | N372H | |||
| Urinary Tract | E668Q | Q1633H |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watson, A.J.; Shaffer, M.L.; Bouley, R.A.; Petreaca, R.C. F-box DNA Helicase 1 (FBH1) Contributes to the Destabilization of DNA Damage Repair Machinery in Human Cancers. Cancers 2023, 15, 4439. https://doi.org/10.3390/cancers15184439
Watson AJ, Shaffer ML, Bouley RA, Petreaca RC. F-box DNA Helicase 1 (FBH1) Contributes to the Destabilization of DNA Damage Repair Machinery in Human Cancers. Cancers. 2023; 15(18):4439. https://doi.org/10.3390/cancers15184439
Chicago/Turabian StyleWatson, Alizhah J., Michaela L. Shaffer, Renee A. Bouley, and Ruben C. Petreaca. 2023. "F-box DNA Helicase 1 (FBH1) Contributes to the Destabilization of DNA Damage Repair Machinery in Human Cancers" Cancers 15, no. 18: 4439. https://doi.org/10.3390/cancers15184439
APA StyleWatson, A. J., Shaffer, M. L., Bouley, R. A., & Petreaca, R. C. (2023). F-box DNA Helicase 1 (FBH1) Contributes to the Destabilization of DNA Damage Repair Machinery in Human Cancers. Cancers, 15(18), 4439. https://doi.org/10.3390/cancers15184439