
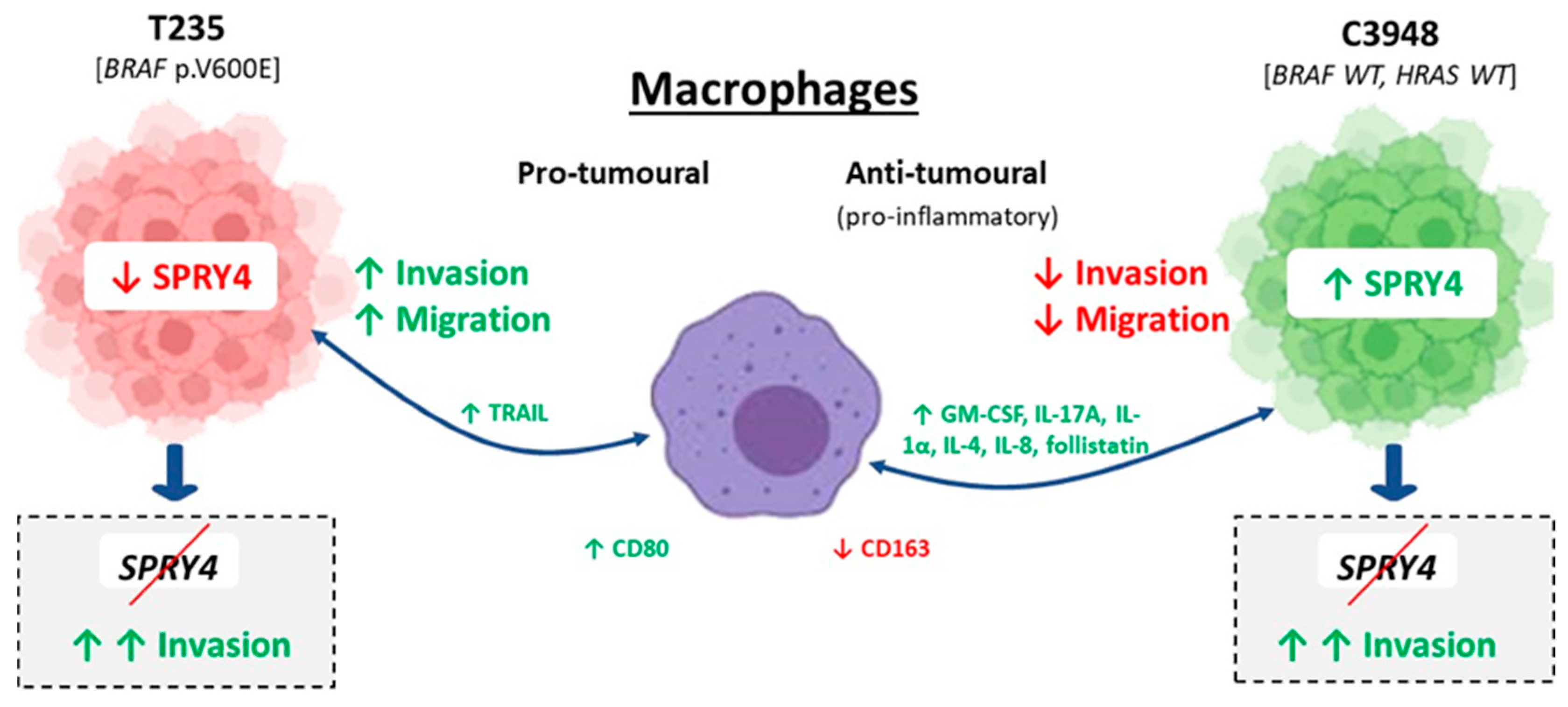
SPRY4 as a Potential Mediator of the Anti-Tumoral Role of Macrophages in Anaplastic Thyroid Cancer Cells

 , , ,
, , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
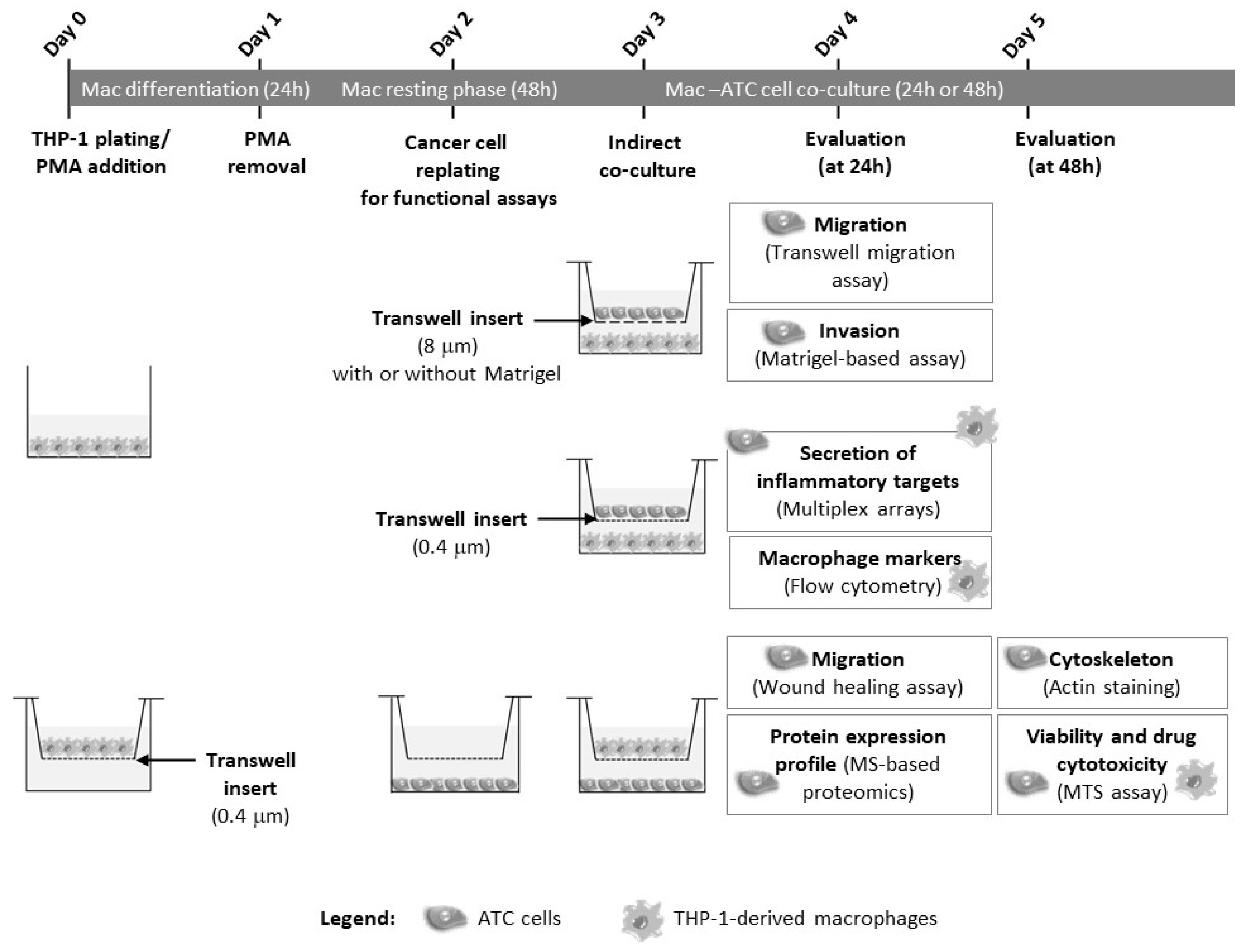
2. Materials and Methods
2.1. Cell Lines
2.2. Macrophages
2.3. Macrophage–ATC Co-Cultures
2.4. Transient Inhibition of SPRY4 Using siRNA-Mediated Silencing
2.5. Metabolic Cell Viability and Cytotoxic Drug Assay
2.6. Migration and Invasion Assays
2.7. Actin Filament Staining
2.8. Flow Cytometry
2.9. Conditioned Medium Analysis
2.10. MS-Based Proteomics
2.11. Western Blot Validation
2.12. Statistical Analysis
3. Results
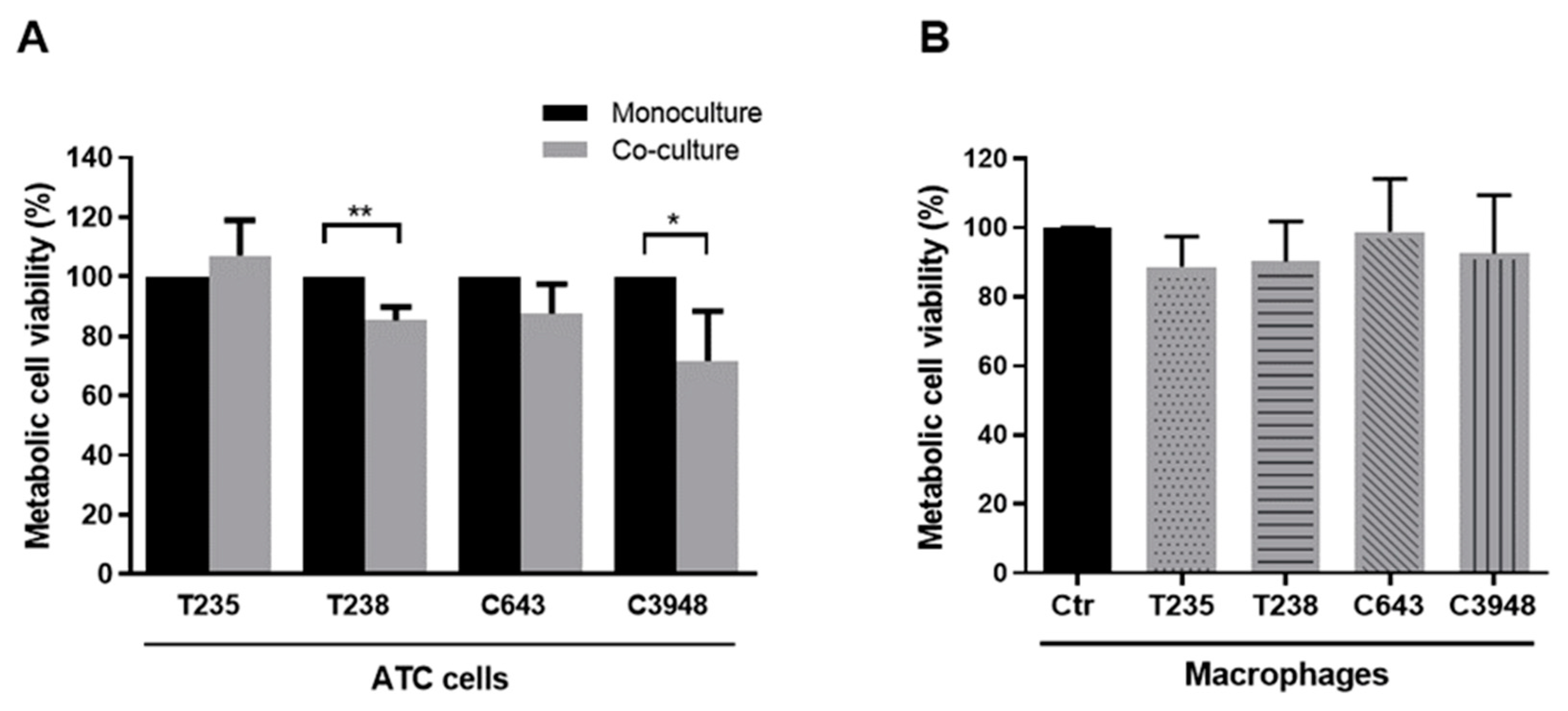
3.1. Macrophages Decrease the Viability of T238 and C3948 ATC Cell Lines, While Maintaining the Intrinsic Chemoresistance Profile of Cancer Cells
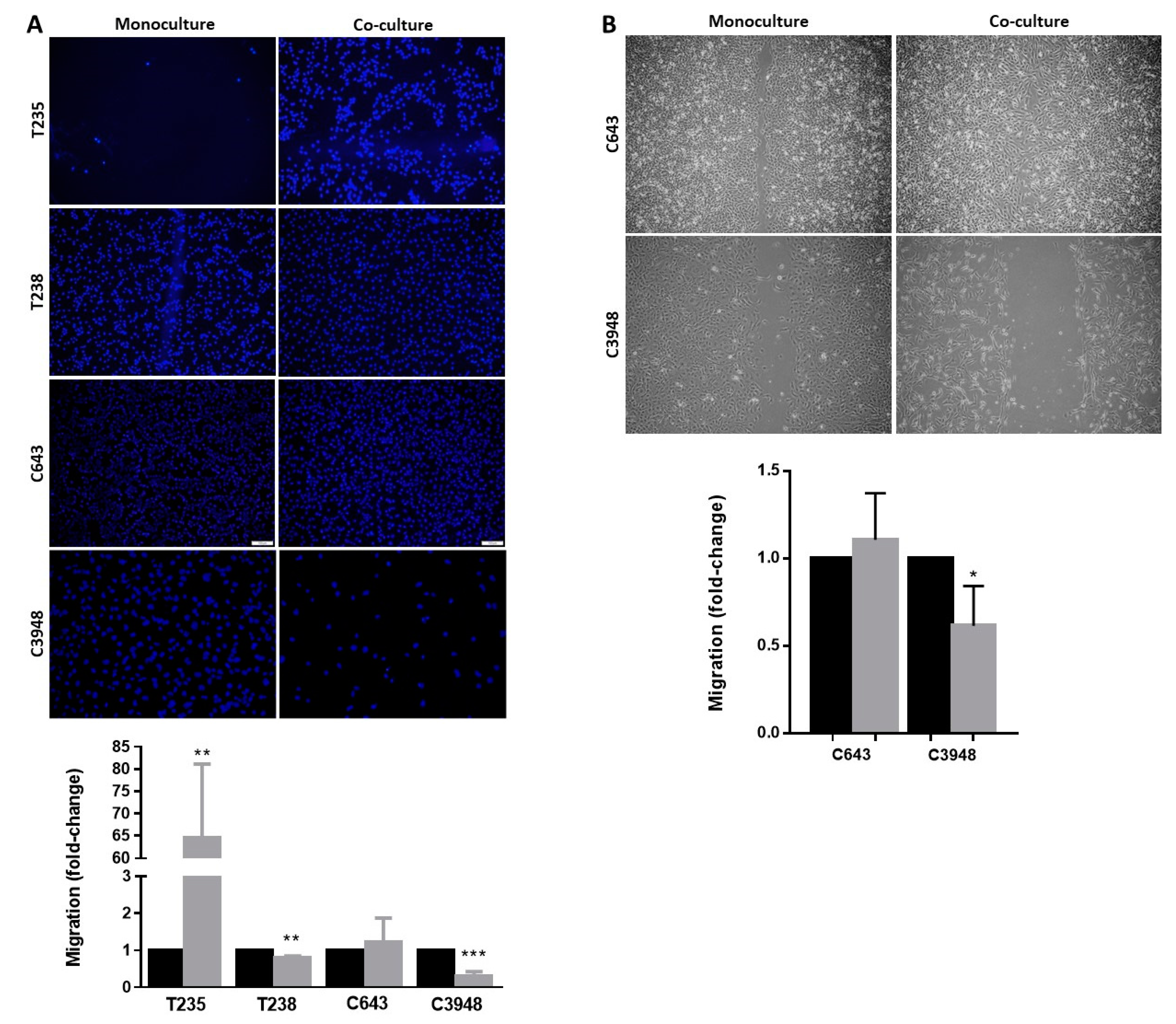
3.2. Macrophages Increase the Migration Ability of T235 ATC Cell Lines, While Decreasing That of T238 and C3948
3.3. Macrophages Increase the Invasion Ability of T235 and C643 ATC Cell Lines, While Decreasing That of C3948
3.4. Co-Culture with Macrophages Affects Differently ATC Cell Cytoskeleton
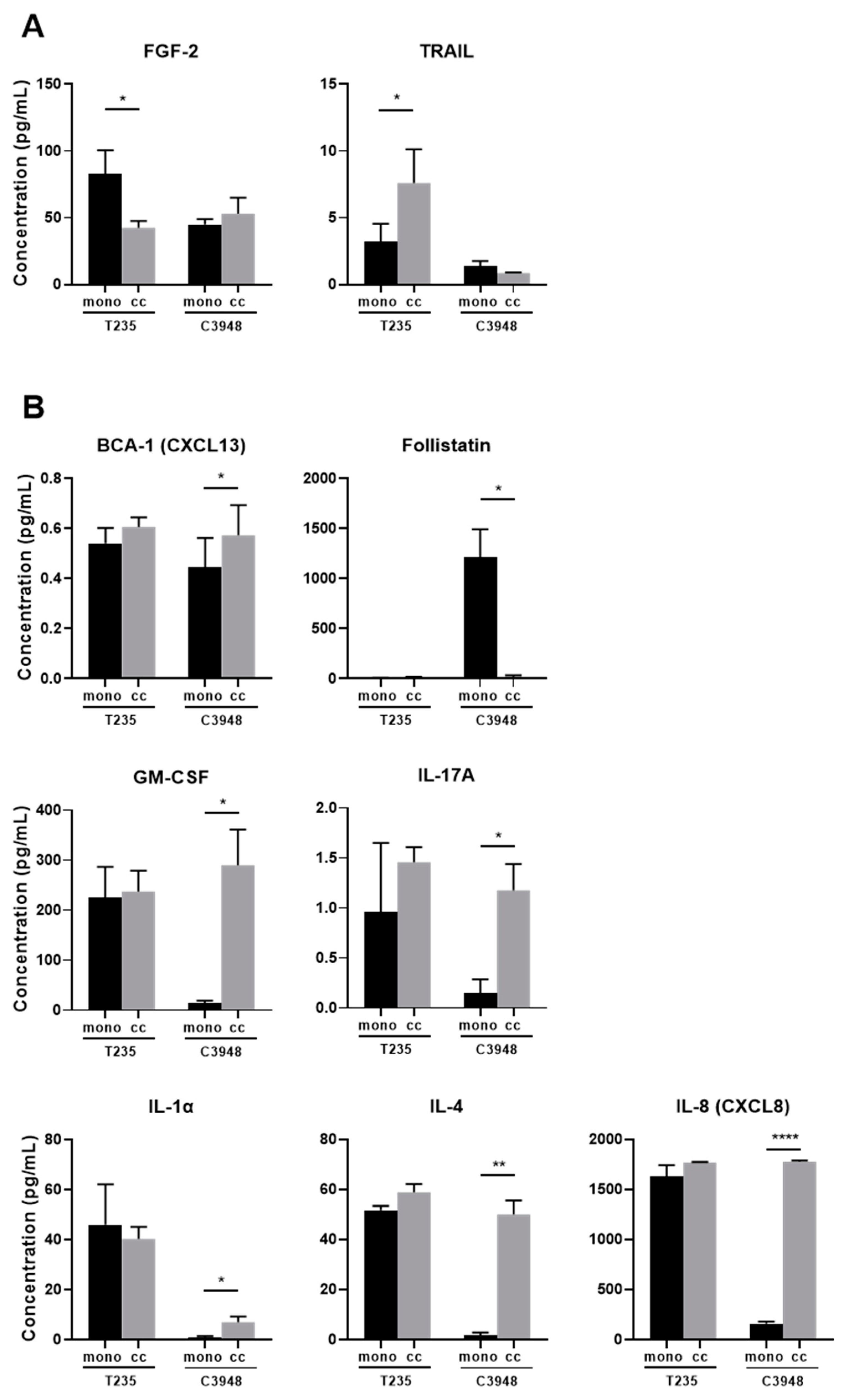
3.5. Co-Culture of Macrophages with C3948 or T235 Exhibit a Different Cytokine Profile
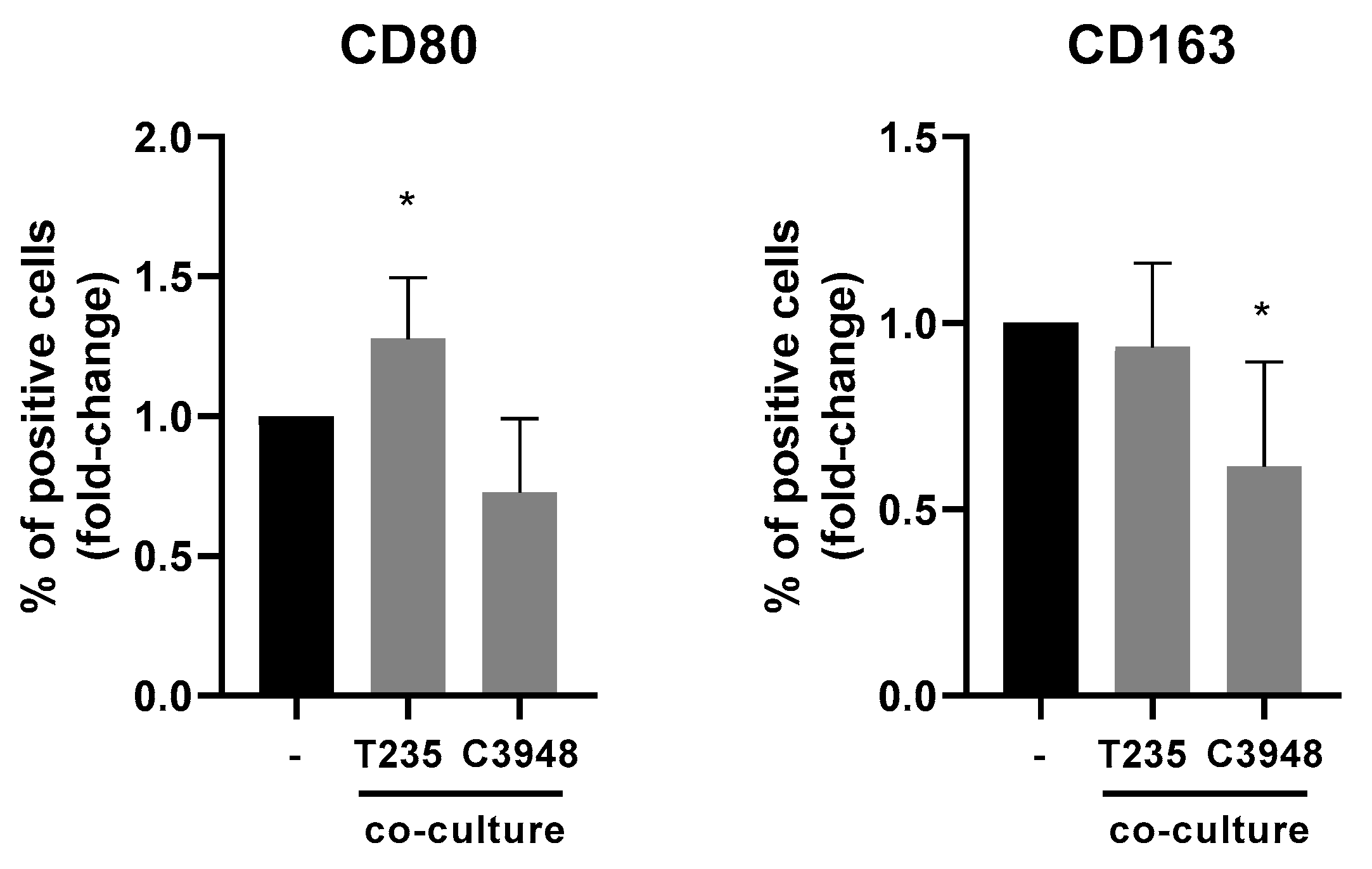
3.6. The Inflammatory Profile of Macrophages Is Different in Co-Culture with C3948 or T235 Cells
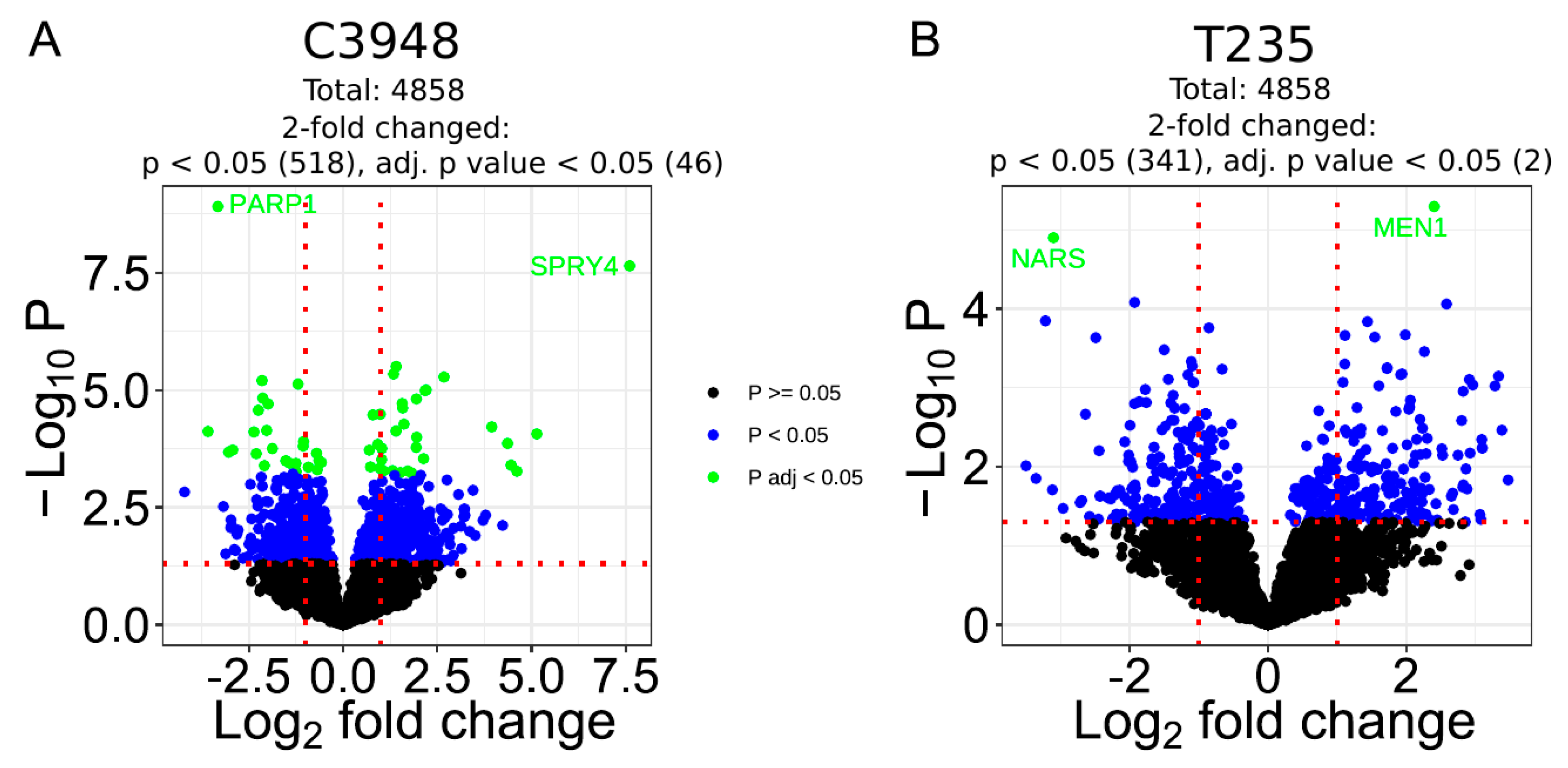
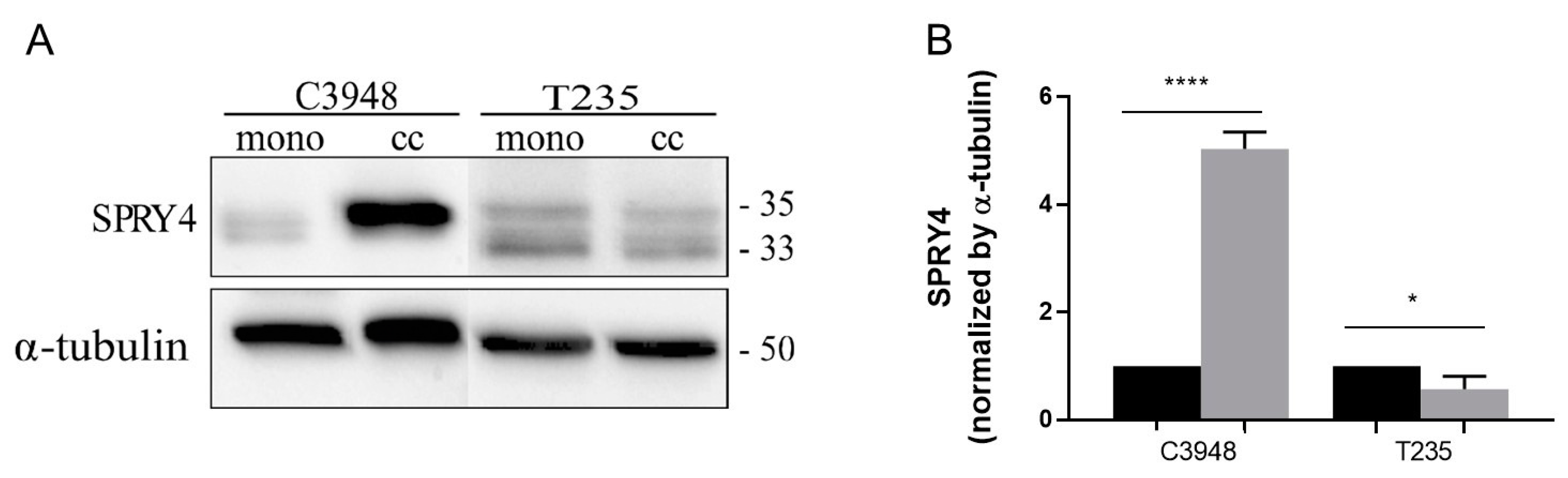
3.7. Co-Culture with Macrophages Differentially Deregulates the Proteome Profile of C3948 and T235 Cells Unveiling SPRY4 as a Differentially Expressed Protein
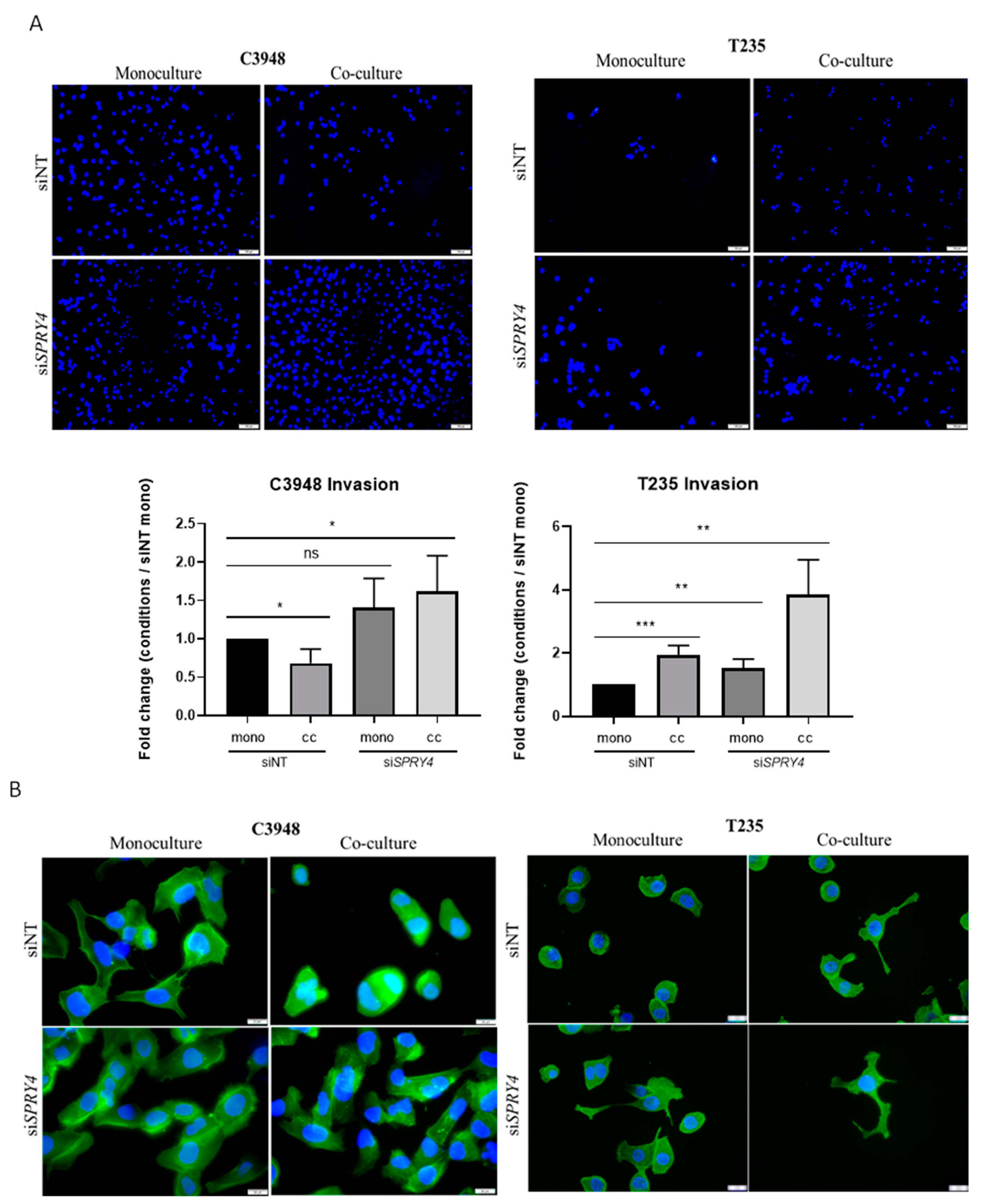
3.8. SPRY4 Silencing Reverts the Macrophage-Induced Reduction of C3948 Cell’s Invasion and Promotes Cytoskeleton Alterations in ATC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ragazzi, M.; Ciarrocchi, A.; Sancisi, V.; Gandolfi, G.; Bisagni, A.; Piana, S. Update on anaplastic thyroid carcinoma: Morphological, molecular, and genetic features of the most aggressive thyroid cancer. Int. J. Endocrinol. 2014, 2014, 790834. [Google Scholar] [CrossRef]
- Abe, I.; Lam, A.K. Anaplastic thyroid carcinoma: Updates on WHO classification, clinicopathological features and staging. Histol. Histopathol. 2020, 36, 239–248. [Google Scholar]
- Xu, B.; Fuchs, T.L.; Dogan, S.; Landa, I.; Katabi, N.; Fagin, J.A.; Tuttle, R.M.; Sherman, E.J.; Gill, A.J.; Ghossein, R. Dissecting Anaplastic Thyroid Carcinoma: A Comprehensive Clinical, Histologic, Immunophenotypic, and Molecular Study of 360 Cases. Thyroid 2020, 30, 1505–1517. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.-C.; et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef]
- Simões-Pereira, J.; Moura, M.M.; Marques, I.J.; Rito, M.; Cabrera, R.A.; Leite, V.; Cavaco, B.M. The role of EIF1AX in thyroid cancer tumourigenesis and progression. J. Endocrinol. Investig. 2019, 42, 313–318. [Google Scholar] [CrossRef]
- Lopes-Ventura, S.; Pojo, M.; Matias, A.T.; Moura, M.M.; Marques, I.J.; Leite, V.; Cavaco, B.M. The efficacy of HRAS and CDK4/6 inhibitors in anaplastic thyroid cancer cell lines. J. Endocrinol. Investig. 2019, 42, 527–540. [Google Scholar] [CrossRef]
- Pita, J.M.; Figueiredo, I.F.; Moura, M.M.; Leite, V.; Cavaco, B.M. Cell cycle deregulation and TP53 and RAS mutations are major events in poorly differentiated and undifferentiated thyroid carcinomas. J. Clin. Endocrinol. Metab. 2014, 99, E497–E507. [Google Scholar] [CrossRef]
- Neff, R.L.; Farrar, W.B.; Kloos, R.T.; Burman, K.D. Anaplastic thyroid cancer. Endocrinol. Metab. Clin. N. Am. 2008, 37, 525–538.xi. [Google Scholar] [CrossRef]
- Smallridge, R.C.; Ain, K.B.; Asa, S.L.; Bible, K.C.; Brierley, J.D.; Burman, K.D.; Kebebew, E.; Lee, N.Y.; Nikiforov, Y.E.; Rosenthal, M.S.; et al. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid 2012, 22, 1104–1139. [Google Scholar] [CrossRef]
- Simões-Pereira, J.; Capitão, R.; Limbert, E.; Leite, V. Anaplastic Thyroid Cancer: Clinical Picture of the Last Two Decades at a Single Oncology Referral Centre and Novel Therapeutic Options. Cancers 2019, 11, 1188. [Google Scholar] [CrossRef] [PubMed]
- Prasongsook, N.; Kumar, A.; Chintakuntlawar, A.V.; Foote, R.L.; Kasperbauer, J.; Molina, J.; Garces, Y.; Ma, D.; Wittich, M.A.N.; Rubin, J.; et al. Survival in Response to Multimodal Therapy in Anaplastic Thyroid Cancer. J. Clin. Endocrinol. Metab. 2017, 102, 4506–4514. [Google Scholar] [CrossRef]
- Bible, K.C.; Kebebew, E.; Brierley, J.; Brito, J.P.; Cabanillas, M.E.; Clark, T.J., Jr.; Di Cristofano, A.; Foote, R.; Giordano, T.; Kasperbauer, J.; et al. 2021 American Thyroid Association Guidelines for Management of Patients with Anaplastic Thyroid Cancer. Thyroid 2021, 31, 337–386. [Google Scholar] [CrossRef] [PubMed]
- Abe, I.; Lam, A.K. Anaplastic Thyroid Carcinoma: Current Issues in Genomics and Therapeutics. Curr. Oncol. Rep. 2021, 23, 31. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.; Afonso, R.A.; Gaspar, M.M.; Reis, C.P. Anaplastic thyroid cancer: How far can we go? EXCLI J. 2020, 19, 800–812. [Google Scholar]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and Trametinib Treatment in Patients with Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef]
- da Silva, T.N.; Rodrigues, R.; Saramago, A.; Pires, C.; Rito, M.; Horta, M.; Martins, C.; Leite, V.; Cavaco, B.M. Target therapy for BRAF mutated anaplastic thyroid cancer: A clinical and molecular study. Eur. J. Endocrinol. 2023, 188, 31–38. [Google Scholar] [CrossRef]
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res. 2021, 81, 1201–1208. [Google Scholar] [CrossRef]
- Stempin, C.C.; Geysels, R.C.; Park, S.; Palacios, L.M.; Volpini, X.; Motran, C.C.; Rodríguez, E.V.A.; Nicola, J.P.; Cheng, S.-Y.; Pellizas, C.G.; et al. Secreted Factors by Anaplastic Thyroid Cancer Cells Induce Tumor-Promoting M2-like Macrophage Polarization through a TIM3-Dependent Mechanism. Cancers 2021, 13, 4821. [Google Scholar] [CrossRef]
- Caillou, B.; Talbot, M.; Weyemi, U.; Pioche-Durieu, C.; Al Ghuzlan, A.; Bidart, J.M.; Chouaib, S.; Schlumberger, M.; Dupuy, C. Tumor-associated macrophages (TAMs) form an interconnected cellular supportive network in anaplastic thyroid carcinoma. PLoS ONE 2011, 6, e22567. [Google Scholar] [CrossRef]
- Ryder, M.; Ghossein, R.A.; Ricarte-Filho, J.C.; Knauf, J.A.; Fagin, J.A. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr. Relat. Cancer 2008, 15, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.W.; Kim, W.W.; Lee, Y.M.; Jeon, M.J.; Kim, W.G.; Song, D.E.; Park, Y.; Chung, K.-W.; Hong, S.J.; Sung, T.-Y. Impact of tumor-associated macrophages and BRAF. Head Neck 2019, 41, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Locati, M. Tumor-associated macrophages as a paradigm of macrophage plasticity, diversity, and polarization: Lessons and open questions. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1478–1483. [Google Scholar] [CrossRef]
- Cunha, L.L.; Ward, L.S. Translating the immune microenvironment of thyroid cancer into clinical practice. Endocr. Relat. Cancer 2022, 29, R67–R83. [Google Scholar] [CrossRef]
- Menicali, E.; Guzzetti, M.; Morelli, S.; Moretti, S.; Puxeddu, E. Immune Landscape of Thyroid Cancers: New Insights. Front. Endocrinol. 2020, 11, 637826. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Liu, C.; Chen, F.-K.; Feng, Z.-P.; Jia, L.; Liu, P.-J.; Yang, Z.-X.; Hou, F.; Deng, Z.-Y. M2-like tumour-associated macrophage-secreted IGF promotes thyroid cancer stemness and metastasis by activating the PI3K/AKT/mTOR pathway. Mol. Med. Rep. 2021, 24, 604. [Google Scholar] [CrossRef] [PubMed]
- Rogers, T.L.; Holen, I. Tumour macrophages as potential targets of bisphosphonates. J. Transl. Med. 2011, 9, 177. [Google Scholar] [CrossRef]
- Ryder, M.; Gild, M.; Hohl, T.M.; Pamer, E.; Knauf, J.; Ghossein, R.; Joyce, J.A.; Fagin, J.A. Genetic and pharmacological targeting of CSF-1/CSF-1R inhibits tumor-associated macrophages and impairs BRAF-induced thyroid cancer progression. PLoS ONE 2013, 8, e54302. [Google Scholar] [CrossRef]
- Pinto, A.T.; Pojo, M.; Simões-Pereira, J.; Roque, R.; Saramago, A.; Roque, L.; Martins, C.; André, S.; Cabeçadas, J.; Leite, V.; et al. Establishment and characterization of a new patient-derived anaplastic thyroid cancer cell line (C3948), obtained through fine-needle aspiration cytology. Endocrine 2019, 66, 288–300. [Google Scholar] [CrossRef]
- Schweppe, R.E.; Klopper, J.P.; Korch, C.; Pugazhenthi, U.; Benezra, M.; Knauf, J.A.; Fagin, J.A.; Marlow, L.A.; Copland, J.A.; Smallridge, R.C.; et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J. Clin. Endocrinol. Metab. 2008, 93, 4331–4341. [Google Scholar] [CrossRef]
- Carvalho, A.S.; Molina, H.; Matthiesen, R. New insights into functional regulation in MS-based drug profiling. Sci. Rep. 2016, 6, 18826. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- El Motiam, A.; Vidal, S.; De La Cruz-Herrera, C.F.; Da Silva-Álvarez, S.; Baz-Martínez, M.; Seoane, R.; Vidal, A.; Rodríguez, M.S.; Xirodimas, D.P.; Carvalho, A.S.; et al. Interplay between SUMOylation and NEDDylation regulates RPL11 localization and function. FASEB J. 2019, 33, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.S.; Ribeiro, H.; Voabil, P.; Penque, D.; Jensen, O.N.; Molina, H.; Matthiesen, R. Global mass spectrometry and transcriptomics array based drug profiling provides novel insight into glucosamine induced endoplasmic reticulum stress. Mol. Cell. Proteom. 2014, 13, 3294–3307. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Matthiesen, R.; Prieto, G.; Amorim, A.; Aloria, K.; Fullaondo, A.; Carvalho, A.S.; Arizmendi, J.M. SIR: Deterministic protein inference from peptides assigned to MS data. J. Proteom. 2012, 75, 4176–4183. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, Article 3. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Oh, Y.T.; Sun, S.Y. Regulation of Cancer Metastasis by TRAIL/Death Receptor Signaling. Biomolecules 2021, 11, 499. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Na, Y.R.; Hong, J.H.; Lee, M.Y.; Jung, J.H.; Jung, D.; Kim, Y.W.; Son, D.; Choi, M.; Kim, K.P.; Seok, S.H. Proteomic Analysis Reveals Distinct Metabolic Differences Between Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF) and Macrophage Colony Stimulating Factor (M-CSF) Grown Macrophages Derived from Murine Bone Marrow Cells. Mol. Cell. Proteom. 2015, 14, 2722–2732. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Panagiotou, G.; Ghaly, W.; Upadhyay, J.; Pazaitou-Panayiotou, K.; Mantzoros, C.S. Serum Follistatin Is Increased in Thyroid Cancer and Is Associated with Adverse Tumor Characteristics in Humans. J. Clin. Endocrinol. Metab. 2021, 106, e2137–e2150. [Google Scholar] [CrossRef] [PubMed]
- Angell, T.E.; Lechner, M.G.; Jang, J.K.; Correa, A.J.; LoPresti, J.S.; Epstein, A.L. BRAFV600E in papillary thyroid carcinoma is associated with increased programmed death ligand 1 expression and suppressive immune cell infiltration. Thyroid 2014, 24, 1385–1393. [Google Scholar] [CrossRef]
- Marques, I.J.; Gomes, I.; Pojo, M.; Pires, C.; Moura, M.M.; Cabrera, R.; Santos, C.; van Ijcken, W.F.J.; Teixeira, M.R.; Ramalho, J.S.; et al. Identification of SPRY4 as a Novel Candidate Susceptibility Gene for Familial Nonmedullary Thyroid Cancer. Thyroid 2021, 31, 1366–1375. [Google Scholar] [CrossRef]
- Cabrita, M.A.; Christofori, G. Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis 2008, 11, 53–62. [Google Scholar] [CrossRef]
- Masoumi-Moghaddam, S.; Amini, A.; Morris, D.L. The developing story of Sprouty and cancer. Cancer Metastasis Rev. 2014, 33, 695–720. [Google Scholar] [CrossRef]
- Tennis, M.A.; Van Scoyk, M.M.; Freeman, S.V.; Vandervest, K.M.; Nemenoff, R.A.; Winn, R.A. Sprouty-4 inhibits transformed cell growth, migration and invasion, and epithelial-mesenchymal transition, and is regulated by Wnt7A through PPARgamma in non-small cell lung cancer. Mol. Cancer Res. 2010, 8, 833–843. [Google Scholar] [CrossRef]
- Hébrant, A.; Dom, G.; Dewaele, M.; Andry, G.; Trésallet, C.; Leteurtre, E.; Dumont, J.E.; Maenhaut, C. mRNA expression in papillary and anaplastic thyroid carcinoma: Molecular anatomy of a killing switch. PLoS ONE 2012, 7, e37807. [Google Scholar] [CrossRef]
- Das, M.K.; Furu, K.; Evensen, H.F.; Haugen, Ø.; Haugen, T.B. Knockdown of SPRY4 and SPRY4-IT1 inhibits cell growth and phosphorylation of Akt in human testicular germ cell tumours. Sci. Rep. 2018, 8, 2462. [Google Scholar] [CrossRef]
- So, W.-K.; Cheng, J.-C.; Liu, Y.; Xu, C.; Zhao, J.; Chang, V.T.W.; Leung, P.C.K. Sprouty4 mediates amphiregulin-induced down-regulation of E-cadherin and cell invasion in human ovarian cancer cells. Tumour Biol. 2016, 37, 9197–9207. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Pozdeyev, N.; Korch, C.; Marlow, L.A.; Smallridge, R.C.; Copland, J.A.; Henderson, Y.C.; Lai, S.Y.; Clayman, G.L.; Onoda, N.; et al. Comprehensive Genetic Characterization of Human Thyroid Cancer Cell Lines: A Validated Panel for Preclinical Studies. Clin. Cancer Res. 2019, 25, 3141–3151. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; García-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, A.T.; Pojo, M.; Rodrigues, R.; Sousa, D.P.; Matthiesen, R.; Carvalho, A.S.; Beck, H.C.; Pires, C.; Eduardo, R.; Pereira, J.S.; et al. SPRY4 as a Potential Mediator of the Anti-Tumoral Role of Macrophages in Anaplastic Thyroid Cancer Cells. Cancers 2023, 15, 4387. https://doi.org/10.3390/cancers15174387
Pinto AT, Pojo M, Rodrigues R, Sousa DP, Matthiesen R, Carvalho AS, Beck HC, Pires C, Eduardo R, Pereira JS, et al. SPRY4 as a Potential Mediator of the Anti-Tumoral Role of Macrophages in Anaplastic Thyroid Cancer Cells. Cancers. 2023; 15(17):4387. https://doi.org/10.3390/cancers15174387
Chicago/Turabian StylePinto, Ana Teresa, Marta Pojo, Ricardo Rodrigues, Diana Pacheco Sousa, Rune Matthiesen, Ana Sofia Carvalho, Hans C. Beck, Carolina Pires, Rodrigo Eduardo, Joana Simões Pereira, and et al. 2023. "SPRY4 as a Potential Mediator of the Anti-Tumoral Role of Macrophages in Anaplastic Thyroid Cancer Cells" Cancers 15, no. 17: 4387. https://doi.org/10.3390/cancers15174387
APA StylePinto, A. T., Pojo, M., Rodrigues, R., Sousa, D. P., Matthiesen, R., Carvalho, A. S., Beck, H. C., Pires, C., Eduardo, R., Pereira, J. S., Leite, V., & Cavaco, B. M. (2023). SPRY4 as a Potential Mediator of the Anti-Tumoral Role of Macrophages in Anaplastic Thyroid Cancer Cells. Cancers, 15(17), 4387. https://doi.org/10.3390/cancers15174387