
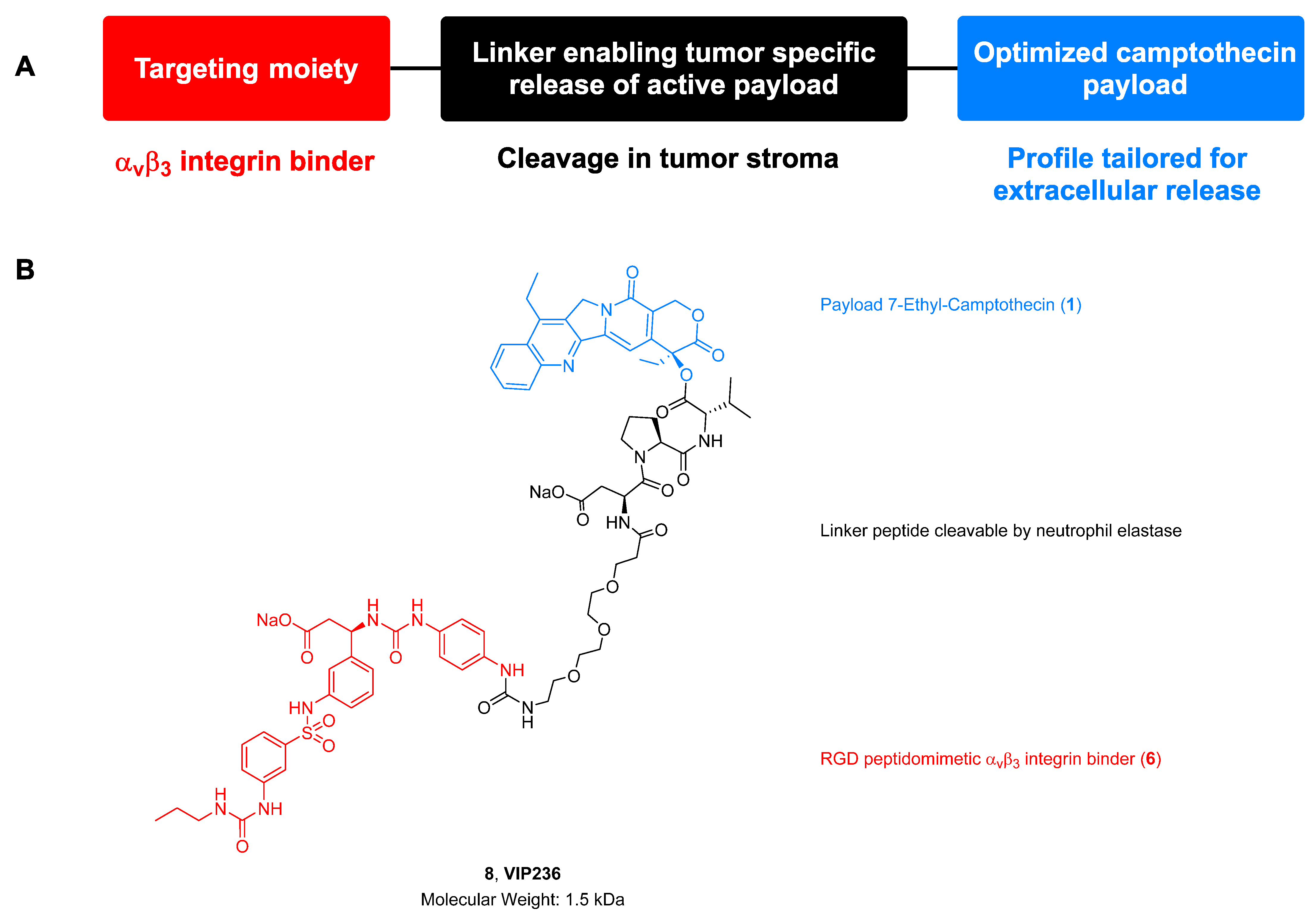
Discovery of VIP236, an αvβ3-Targeted Small-Molecule–Drug Conjugate with Neutrophil Elastase-Mediated Activation of 7-Ethyl Camptothecin Payload for Treatment of Solid Tumors
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
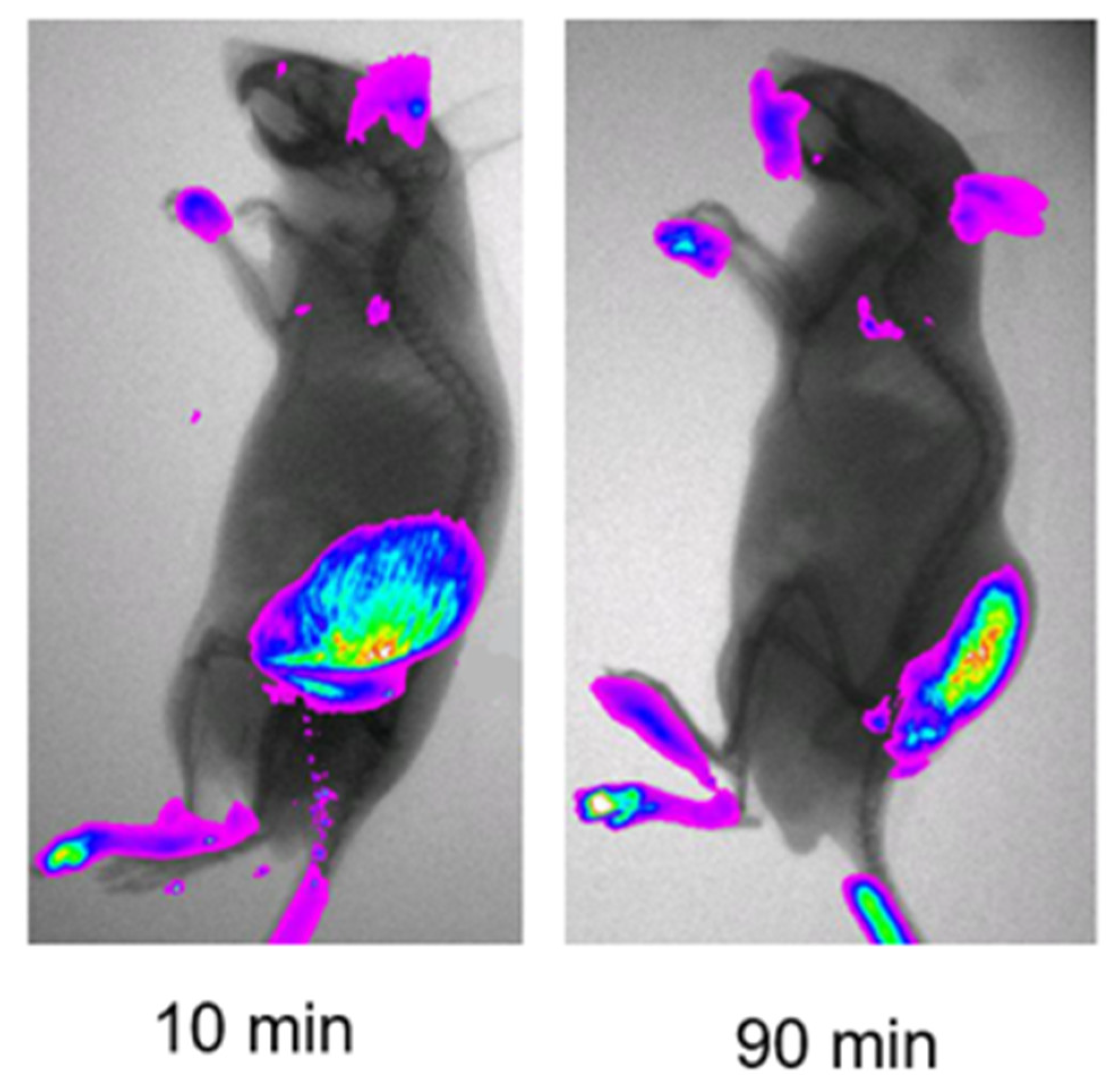
2.1. Imaging Study Showing Elastase-Activity in Tumors
2.2. Compounds
2.3. Stability in Rat Plasma and in Buffer at pH 7.4
2.4. Metabolic Stability in Hepatocytes
2.5. Human and Mouse NE Biochemical Cleavage Assay
2.6. In Vitro Proliferation Assay in the Presence and Absence of NE
2.7. In Vivo Pharmacokinetics in Tumor-Bearing Mice
2.8. VIP236 Bile-Duct-Cannulated Rat Study
2.9. IHC Analysis
2.10. H2AX Staining as a Marker for DNA Damage in SNU16 CDX Mouse Model
2.11. PDX Models
2.12. Orthotopic TNBC Model MA4296 and MA15191
2.13. PCR Analysis
3. Results
3.1. Tumor-Associated NE Cleavage
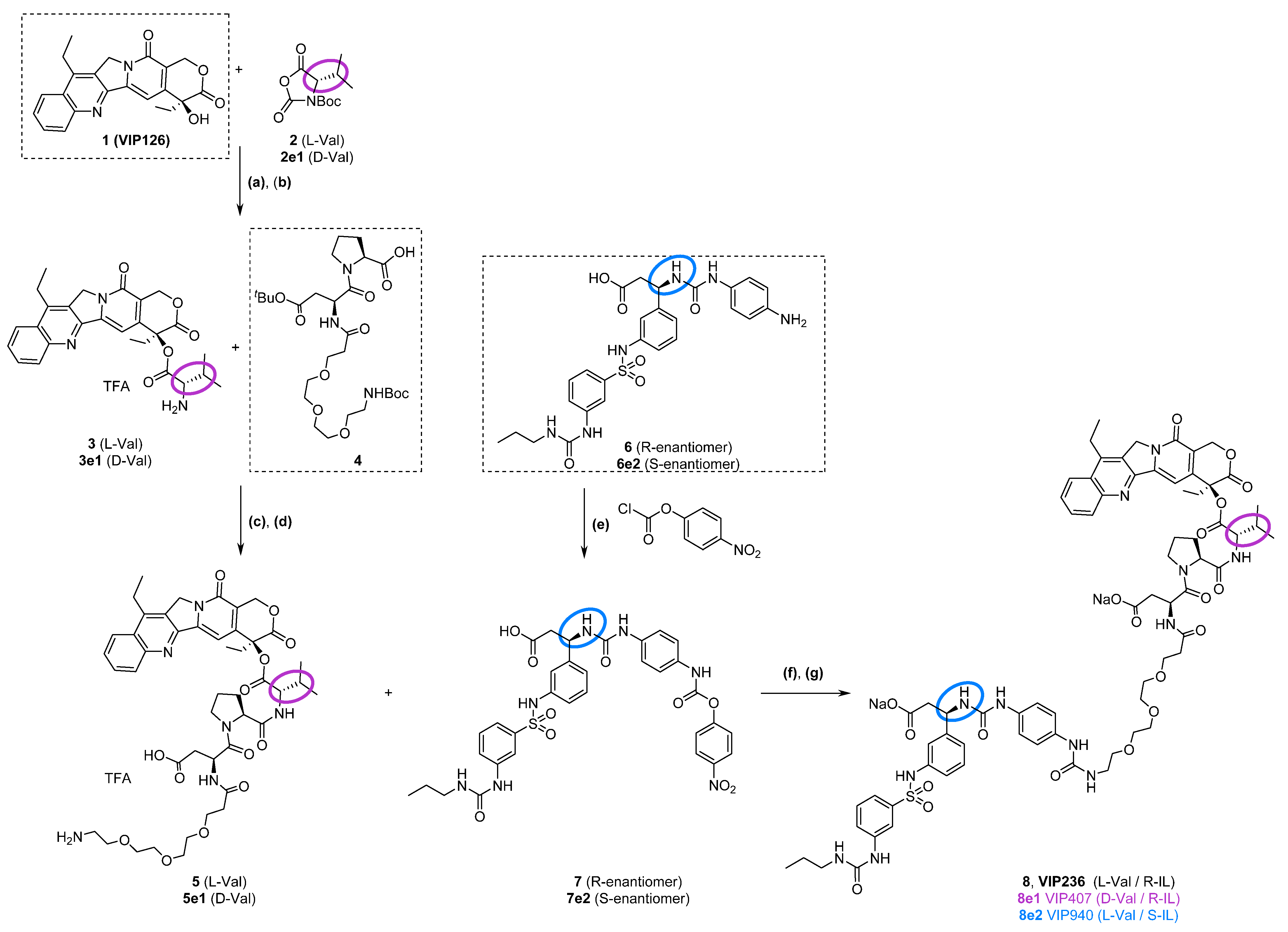
3.2. Synthesis of VIP236 and Epimers
3.3. In Vitro Evaluation: VIP236 Is Highly Stable but Efficiently Cleaved by Mouse and Human NE
- Km = 9.4 and 9.6 μM, respectively;
- kcat = 593 and 691 1/min, respectively.
- Mouse elastase (Figure S4):
- Km = 15.9 and 8.9 μM, respectively;
- kcat = 94 and 80 1/min, respectively.
3.4. In Vivo Pharmacokinetics and Bile Duct Cannulated Rat Study
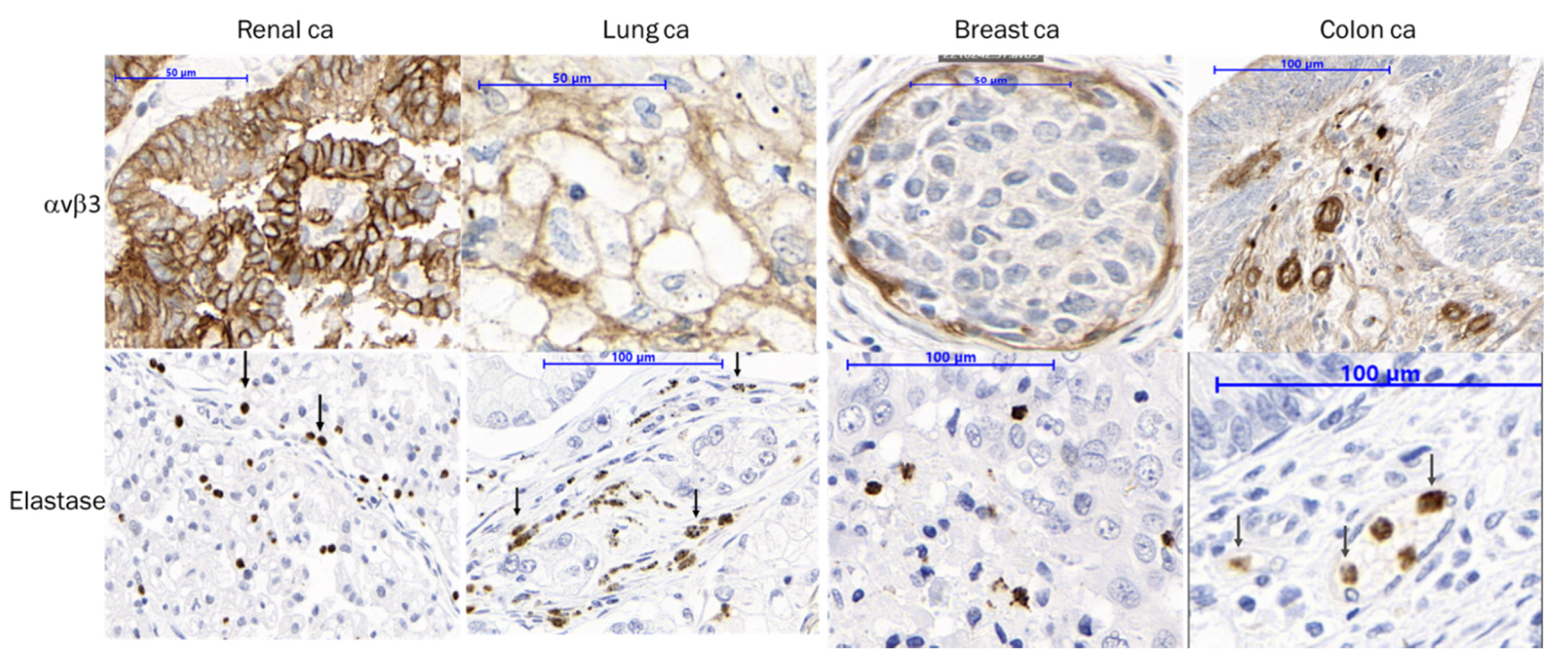
3.5. IHC Staining Confirms αvβ3 and NE Presence in Patient Tumor Samples
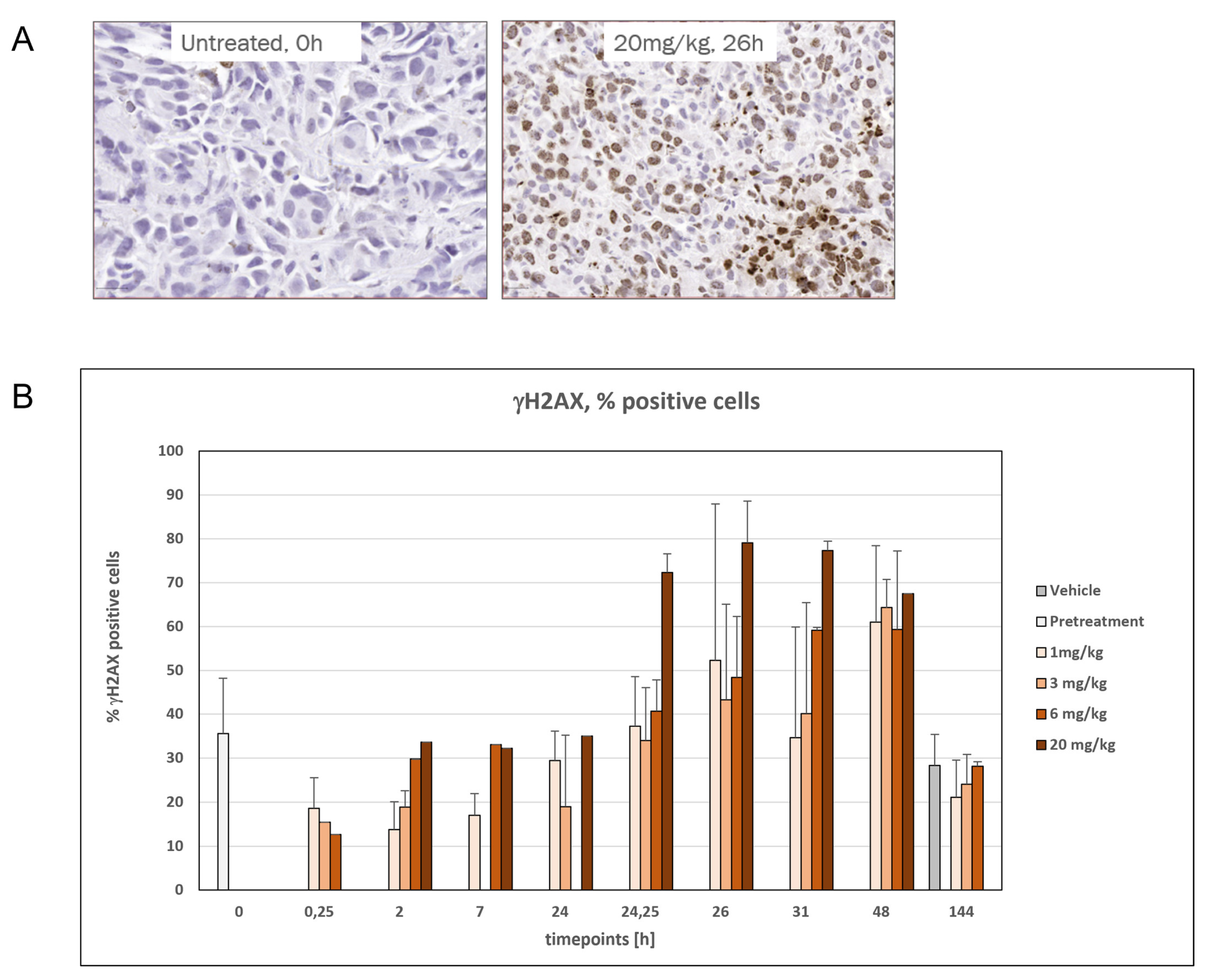
3.6. Time- and Dose-Dependent Induction of DNA Damage by VIP236
3.7. Monotherapy of VIP236 Achieved Potent and Durable Antitumor Activity in PDX Tumor Mouse Models
3.8. Significant Impact of VIP236 on Primary Tumor and Metastases in Orthotopic Metastatic TNBC PDX Mouse Models
3.9. Increase in αvβ3 and NE Staining upon VIP236 Treatment, While TOP1 Expression Is Stable
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwartz, R.S. Paul Ehrlich’s Magic Bullets. N. Engl. J. Med. 2004, 350, 1079–1080. [Google Scholar] [CrossRef]
- Dean, A.Q.; Luo, S.; Twomey, J.D.; Zhang, B. Targeting Cancer with Antibody-Drug Conjugates: Promises and Challenges. mAbs 2021, 13, 1951427. [Google Scholar] [CrossRef] [PubMed]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody–Drug Conjugates—A Tutorial Review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the Potential of Antibody–Drug Conjugates for Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Conilh, L.; Sadilkova, L.; Viricel, W.; Dumontet, C. Payload Diversification: A Key Step in the Development of Antibody–Drug Conjugates. J. Hematol. Oncol. 2023, 16, 3. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Rugo, H.S.; Bardia, A.; Tolaney, S.M.; Arteaga, C.; Cortes, J.; Sohn, J.; Marmé, F.; Hong, Q.; Delaney, R.J.; Hafeez, A.; et al. TROPiCS-02: A Phase III Study Investigating Sacituzumab Govitecan in the Treatment of HR+/HER2− Metastatic Breast Cancer. Future Oncol. 2020, 16, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Colombo, R.; Rich, J.R. The Therapeutic Window of Antibody Drug Conjugates: A Dogma in Need of Revision. Cancer Cell 2022, 40, 1255–1263. [Google Scholar] [CrossRef]
- Zhuang, C.; Guan, X.; Ma, H.; Cong, H.; Zhang, W.; Miao, Z. Small Molecule-Drug Conjugates: A Novel Strategy for Cancer-Targeted Treatment. Eur. J. Med. Chem. 2019, 163, 883–895. [Google Scholar] [CrossRef]
- Cazzamalli, S.; Dal Corso, A.; Widmayer, F.; Neri, D. Chemically-Defined Antibody- and Small Molecule-Drug Conjugates for in Vivo Tumor Targeting Applications: A Comparative Analysis. J. Am. Chem. Soc. 2018, 140, 1617–1621. [Google Scholar] [CrossRef]
- Reddy, J.A.; Dorton, R.; Westrick, E.; Dawson, A.; Smith, T.; Xu, L.-C.; Vetzel, M.; Kleindl, P.; Vlahov, I.R.; Leamon, C.P. Preclinical Evaluation of EC145, a Folate-Vinca Alkaloid Conjugate. Cancer Res. 2007, 67, 4434–4442. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bajjuri, K.M.; Liu, C.; Sinha, S.C. Targeting Cell Surface Alpha(v)Beta(3) Integrins Increases Therapeutic Efficacies of a Legumain Protease-Activated Auristatin Prodrug. Mol. Pharm. 2012, 9, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Alloatti, D.; Giannini, G.; Vesci, L.; Castorina, M.; Pisano, C.; Badaloni, E.; Cabri, W. Camptothecins in Tumor Homing via an RGD Sequence Mimetic. Bioorg. Med. Chem. Lett. 2012, 22, 6509–6512. [Google Scholar] [CrossRef] [PubMed]
- Raposo Moreira Dias, A.; Pina, A.; Dean, A.; Lerchen, H.; Caruso, M.; Gasparri, F.; Fraietta, I.; Troiani, S.; Arosio, D.; Belvisi, L.; et al. Neutrophil Elastase Promotes Linker Cleavage and Paclitaxel Release from an Integrin-Targeted Conjugate. Chem. Weinh. Bergstr. Ger. 2019, 25, 1696–1700. [Google Scholar] [CrossRef]
- Paulus, J.; Sewald, N. Synthesis and Evaluation of a Non-Peptide Small-Molecule Drug Conjugate Targeting Integrin αVβ3. Front. Chem. 2022, 10, 869639. [Google Scholar] [CrossRef]
- Paulus, J.; Nachtigall, B.; Meyer, P.; Sewald, N. RGD Peptidomimetic MMAE-Conjugate Addressing Integrin αVβ3-Expressing Cells with High Targeting Index. Chem. Eur. J. 2023, 29, e202203476. [Google Scholar] [CrossRef]
- Cazzamalli, S.; Figueras, E.; Pethő, L.; Borbély, A.; Steinkühler, C.; Neri, D.; Sewald, N. In Vivo Antitumor Activity of a Novel Acetazolamide–Cryptophycin Conjugate for the Treatment of Renal Cell Carcinomas. ACS Omega 2018, 3, 14726–14731. [Google Scholar] [CrossRef]
- Whalen, K.A.; White, B.H.; Quinn, J.M.; Kriksciukaite, K.; Alargova, R.; Au Yeung, T.P.; Bazinet, P.; Brockman, A.; DuPont, M.M.; Oller, H.; et al. Targeting the Somatostatin Receptor 2 with the Miniaturized Drug Conjugate, PEN-221: A Potent and Novel Therapeutic for the Treatment of Small Cell Lung Cancer. Mol. Cancer Ther. 2019, 18, 1926–1936. [Google Scholar] [CrossRef]
- Millul, J.; Bassi, G.; Mock, J.; Elsayed, A.; Pellegrino, C.; Zana, A.; Dakhel Plaza, S.; Nadal, L.; Gloger, A.; Schmidt, E.; et al. An Ultra-High-Affinity Small Organic Ligand of Fibroblast Activation Protein for Tumor-Targeting Applications. Proc. Natl. Acad. Sci. USA 2021, 118, e2101852118. [Google Scholar] [CrossRef]
- Bennett, G.; Brown, A.; Mudd, G.; Huxley, P.; van Rietschoten, K.; Pavan, S.; Chen, L.; Watcham, S.; Lahdenranta, J.; Keen, N. MMAE Delivery Using the Bicycle Toxin Conjugate BT5528. Mol. Cancer Ther. 2020, 19, 1385–1394. [Google Scholar] [CrossRef]
- Currie, J.-C.; Demeule, M.; Charfi, C.; Zgheib, A.; Larocque, A.; Danalache, B.A.; Ouanouki, A.; Béliveau, R.; Marsolais, C.; Annabi, B. The Peptide-Drug Conjugate TH1902: A New Sortilin Receptor-Mediated Cancer Therapeutic against Ovarian and Endometrial Cancers. Cancers 2022, 14, 1877. [Google Scholar] [CrossRef]
- Mudd, G.E.; Scott, H.; Chen, L.; van Rietschoten, K.; Ivanova-Berndt, G.; Dzionek, K.; Brown, A.; Watcham, S.; White, L.; Park, P.U.; et al. Discovery of BT8009: A Nectin-4 Targeting Bicycle Toxin Conjugate for the Treatment of Cancer. J. Med. Chem. 2022, 65, 14337–14347. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Le Breton, A.; Préat, V. RGD-Based Strategies To Target Alpha(v) Beta(3) Integrin in Cancer Therapy and Diagnosis. Mol. Pharm. 2012, 9, 2961–2973. [Google Scholar] [CrossRef]
- Hieken, T.J.; Farolan, M.; Ronan, S.G.; Shilkaitis, A.; Wild, L.; Gupta, T.K.D. β3 Integrin Expression in Melanoma Predicts Subsequent Metastasis. J. Surg. Res. 1996, 63, 169–173. [Google Scholar] [CrossRef]
- Sun, F.; Wang, J.; Sun, Q.; Li, F.; Gao, H.; Xu, L.; Zhang, J.; Sun, X.; Tian, Y.; Zhao, Q.; et al. Interleukin-8 Promotes Integrin Β3 Upregulation and Cell Invasion through PI3K/Akt Pathway in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 449. [Google Scholar] [CrossRef]
- Wu, C.-M.; Li, T.-M.; Tan, T.-W.; Fong, Y.-C.; Tang, C.-H. Berberine Reduces the Metastasis of Chondrosarcoma by Modulating the αvβ3 Integrin and the PKCδ, c-Src, and AP-1 Signaling Pathways. Evid. Based Complement. Alternat. Med. 2013, 2013, e423164. [Google Scholar] [CrossRef]
- Korkmaz, B.; Moreau, T.; Gauthier, F. Neutrophil Elastase, Proteinase 3 and Cathepsin G: Physicochemical Properties, Activity and Physiopathological Functions. Biochimie 2008, 90, 227–242. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Sato, T.; Takahashi, S.; Mizumoto, T.; Harao, M.; Akizuki, M.; Takasugi, M.; Fukutomi, T.; Yamashita, J. Neutrophil Elastase and Cancer. Surg. Oncol. 2006, 15, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Mohamed Amar, I.A.; Huvelle, S.; Douez, E.; Letast, S.; Henrion, S.; Viaud-Massuard, M.-C.; Aubrey, N.; Allard-Vannier, E.; Joubert, N.; Denevault-Sabourin, C. Dual Intra- and Extracellular Release of Monomethyl Auristatin E from a Neutrophil Elastase-Sensitive Antibody-Drug Conjugate. Eur. J. Med. Chem. 2022, 229, 114063. [Google Scholar] [CrossRef]
- Dornan, D.; Kudirka, R.; Safina, B.; Zhou, M. Elastase-Substrate, Peptide Linker Immunoconjugates, and Uses Thereof. WO2021226440A1, 7 May 2021. [Google Scholar]
- Lerchen, H.-G.; Stelte-Ludwig, B.; Kopitz, C.; Heroult, M.; Zubov, D.; Willuda, J.; Schlange, T.; Kahnert, A.; Wong, H.; Izumi, R.; et al. A Small Molecule–Drug Conjugate (SMDC) Consisting of a Modified Camptothecin Payload Linked to an αVß3 Binder for the Treatment of Multiple Cancer Types. Cancers 2022, 14, 391. [Google Scholar] [CrossRef] [PubMed]
- Perrier, D.; Gibaldi, M. General Derivation of the Equation for Time to Reach a Certain Fraction of Steady State. J. Pharm. Sci. 1982, 71, 474–475. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Nitsche, A.; Neumann, C.; Aumann, J.; Junghahn, I.; Fichtner, I. Sensitive PCR Method for the Detection and Real-Time Quantification of Human Cells in Xenotransplantation Systems. Br. J. Cancer 2002, 87, 1328–1335. [Google Scholar] [CrossRef]
- Sawada, S.; Matsuoka, S.; Nokata, K.; Nagata, H.; Furuta, T.; Yokokura, T.; Miyasaka, T. Synthesis and Antiumor Activity of 20(S)-Camptothecin Derivatives: A-Ring Modified and 7, 10-Disubstituted Camptothecins. Chem. Pharm. Bull. 1991, 39, 3183–3188. [Google Scholar] [CrossRef]
- Kunimoto, T.; Nitta, K.; Tanaka, T.; Uehara, N.; Baba, H.; Takeuchi, M.; Yokokura, T.; Sawada, S.; Miyasaka, T.; Mutai, M. Antitumor Activity of 7-Ethyl-10-[4-(1-Piperidino)-1-Piperidino]Carbonyloxycamptothecin, a Novel Water-Soluble Derivative of Camptothecin, against Murine Tumors1. Cancer Res. 1987, 47, 5944–5947. [Google Scholar]
- Ramesh, M.; Ahlawat, P.; Srinivas, N.R. Irinotecan and Its Active Metabolite, SN-38: Review of Bioanalytical Methods and Recent Update from Clinical Pharmacology Perspectives. Biomed. Chromatogr. 2010, 24, 104–123. [Google Scholar] [CrossRef]
- Lerchen, H.-G.; Stelte-Ludwig, B.; Kopitz, C.; Keldenich, J. Cytostatic Conjugates with Integrin Ligands. WO/2020/094471, 14 May 2020. [Google Scholar]
- Davies, B.; Morris, T. Physiological Parameters in Laboratory Animals and Humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Vonlaufen, A.; Wiedle, G.; Borisch, B.; Birrer, S.; Luder, P.; Imhof, B.A. Integrin αvβ3 Expression in Colon Carcinoma Correlates with Survival. Mod. Pathol. 2001, 14, 1126–1132. [Google Scholar] [CrossRef]
- Ha, S.Y.; Shin, J.; Kim, J.H.; Kang, M.S.; Yoo, H.-Y.; Kim, H.-H.; Um, S.-H.; Kim, S.-H. Overexpression of Integrin αv Correlates with Poor Prognosis in Colorectal Cancer. J. Clin. Pathol. 2014, 67, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Barnes, L.A.; Shields, D.J.; Huang, M.; Lau, S.K.; Prévost, N.; Tarin, D.; Shattil, S.J.; Cheresh, D.A. Integrin αvβ3/c-Src “Oncogenic Unit” Promotes Anchorage-Independence and Tumor Progression. Nat. Med. 2009, 15, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Alday-Parejo, B.; Stupp, R.; Rüegg, C. Are Integrins Still Practicable Targets for Anti-Cancer Therapy? Cancers 2019, 11, 978. [Google Scholar] [CrossRef] [PubMed]
- Hatley, R.J.D.; Macdonald, S.J.F.; Slack, R.J.; Le, J.; Ludbrook, S.B.; Lukey, P.T. An αv-RGD Integrin Inhibitor Toolbox: Drug Discovery Insight, Challenges and Opportunities. Angew. Chem. Int. Ed. 2018, 57, 3298–3321. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, B.S.; Kessler, H.; Kossatz, S.; Reuning, U. RGD-Binding Integrins Revisited: How Recently Discovered Functions and Novel Synthetic Ligands (Re-)Shape an Ever-Evolving Field. Cancers 2021, 13, 1711. [Google Scholar] [CrossRef]
- Achilles, K.; Bednarski, P.J. Quantification of Elastase-Like Activity in 13 Human Cancer Cell Lines and in an Immortalized Human Epithelial Cell Line by RP-HPLC. Biol. Chem. 2003, 384, 817–824. [Google Scholar] [CrossRef]
- Lerchen, H.-G.; von dem Bruch, K. Synthesis of 20-O-Linked 20(S)-Camptothecin Glycoconjugates: Impact of the Side Chain of the Ester-Linked Amino Acid on Epimerization During the Acylation Reaction and on Hydrolytic Stability of the Final Glycoconjugates. J. Prakt. Chem. 2000, 342, 753–760. [Google Scholar] [CrossRef]
- Lerchen, H.-G.; Baumgarten, J.; von dem Bruch, K.; Lehmann, T.E.; Sperzel, M.; Kempka, G.; Fiebig, H.-H. Design and Optimization of 20-O-Linked Camptothecin Glycoconjugates as Anticancer Agents. J. Med. Chem. 2001, 44, 4186–4195. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “Biological Missile” for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Tsumura, R.; Manabe, S.; Takashima, H.; Koga, Y.; Yasunaga, M.; Matsumura, Y. Influence of the Dissociation Rate Constant on the Intra-Tumor Distribution of Antibody-Drug Conjugate against Tissue Factor. J. Control. Release 2018, 284, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Ricciuti, B.; Pradhan, S.M.; Tolaney, S.M. Optimizing the Safety of Antibody–Drug Conjugates for Patients with Solid Tumours. Nat. Rev. Clin. Oncol. 2023, 20, 558–576. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Reichert, J.M.; Senter, P.D.; Lambert, J.M.; Beck, A. Antibody–Drug Conjugates Come of Age in Oncology. Nat. Rev. Drug Discov. 2023, 22, 641–661. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SMDC | NCI H292 IC50 (nM) | NCI H292 + hNE IC50 (nM) | LoVo IC50 (nM) | LoVo + hNE IC50 (nM) |
|---|---|---|---|---|
| VIP236 | 209 | 1.78 | 98 | 3.0 |
| 8e1 | 5000 | 317 | 5520 | 412 |
| 8e2 | 127 | 1.54 | 68 | 3.2 |
| 1 | 1.54 | 1.35 | 1.9 | 1.85 |
| Compound Dosed | VIP236 4 mg/kg IV | 8e2 4 mg/kg IV | 8e1 4 mg/kg IV | Payload 1 (VIP126) 1 mg/kg IV |
|---|---|---|---|---|
| Compound measured | VIP126 | VIP126 | VIP126 | VIP126 |
| AUCtumor [µg·h/L] | 318 | 50.4 | <LLOQ | 185 |
| AUCplasma [µg·h/L] | 52.1 | 43.3 | 2.47 | 301 |
| AUCtumor/AUCplasma | 6.1 | 1.17 | <LLOQ | 0.616 |
| Matrix | Animal | % of Dose Recovered in 48-h | Mean Recovery % |
|---|---|---|---|
| Bile | Rat 1 | 89.7 | 108 ± 17.2 * |
| Rat 2 | 109 | ||
| Rat 3 | 124 | ||
| Urine | Rat 1 | 2.8 | 2.4 ± 0.29 |
| Rat 2 | 2.3 | ||
| Rat 3 | 2.3 |
| Group | n | Treatment | Route | Treatment Days | Dose (mg/kg) | Opt. T/C (%) (day 46) |
|---|---|---|---|---|---|---|
| A | 9 | PBS | i.v. | d20, 21, 27, 28, 34, 35, 41, 42 | - | |
| B | 9 | 8 (VIP-236) | i.v. | d20, 21, 27, 28, 34, 35, 41, 42 | 40 | 4 ** |
| C | 9 | 8 (VIP-236) | i.v. | d20, 27, 34, 41 | 60 | 36 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lerchen, H.-G.; Stelte-Ludwig, B.; Heroult, M.; Zubov, D.; Gericke, K.M.; Wong, H.; Frigault, M.M.; Johnson, A.J.; Izumi, R.; Hamdy, A. Discovery of VIP236, an αvβ3-Targeted Small-Molecule–Drug Conjugate with Neutrophil Elastase-Mediated Activation of 7-Ethyl Camptothecin Payload for Treatment of Solid Tumors. Cancers 2023, 15, 4381. https://doi.org/10.3390/cancers15174381
Lerchen H-G, Stelte-Ludwig B, Heroult M, Zubov D, Gericke KM, Wong H, Frigault MM, Johnson AJ, Izumi R, Hamdy A. Discovery of VIP236, an αvβ3-Targeted Small-Molecule–Drug Conjugate with Neutrophil Elastase-Mediated Activation of 7-Ethyl Camptothecin Payload for Treatment of Solid Tumors. Cancers. 2023; 15(17):4381. https://doi.org/10.3390/cancers15174381
Chicago/Turabian StyleLerchen, Hans-Georg, Beatrix Stelte-Ludwig, Melanie Heroult, Dmitry Zubov, Kersten Matthias Gericke, Harvey Wong, Melanie M. Frigault, Amy J. Johnson, Raquel Izumi, and Ahmed Hamdy. 2023. "Discovery of VIP236, an αvβ3-Targeted Small-Molecule–Drug Conjugate with Neutrophil Elastase-Mediated Activation of 7-Ethyl Camptothecin Payload for Treatment of Solid Tumors" Cancers 15, no. 17: 4381. https://doi.org/10.3390/cancers15174381
APA StyleLerchen, H.-G., Stelte-Ludwig, B., Heroult, M., Zubov, D., Gericke, K. M., Wong, H., Frigault, M. M., Johnson, A. J., Izumi, R., & Hamdy, A. (2023). Discovery of VIP236, an αvβ3-Targeted Small-Molecule–Drug Conjugate with Neutrophil Elastase-Mediated Activation of 7-Ethyl Camptothecin Payload for Treatment of Solid Tumors. Cancers, 15(17), 4381. https://doi.org/10.3390/cancers15174381