


PCR-Based Strategy for Introducing CRISPR/Cas9 Machinery into Hematopoietic Cell Lines

 , , ,
, , ,  ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials Used in This Work
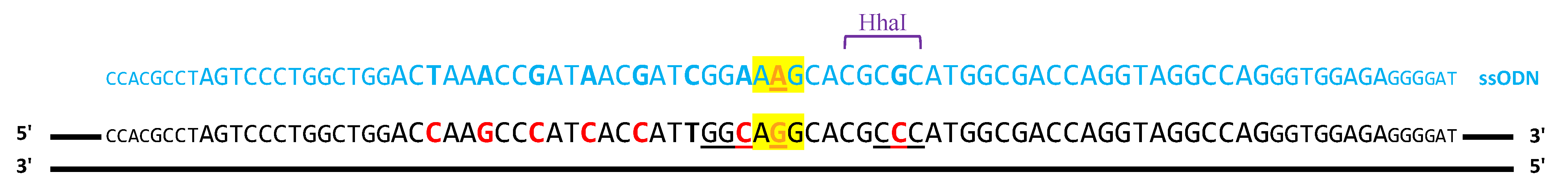
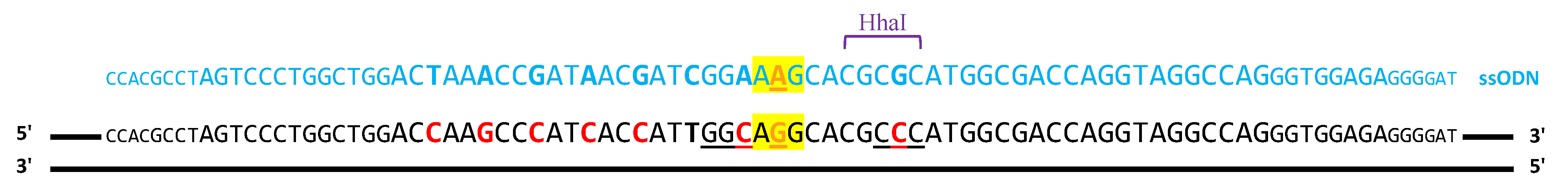
2.1.1. ssODN Template to Induce Homologous Recombination
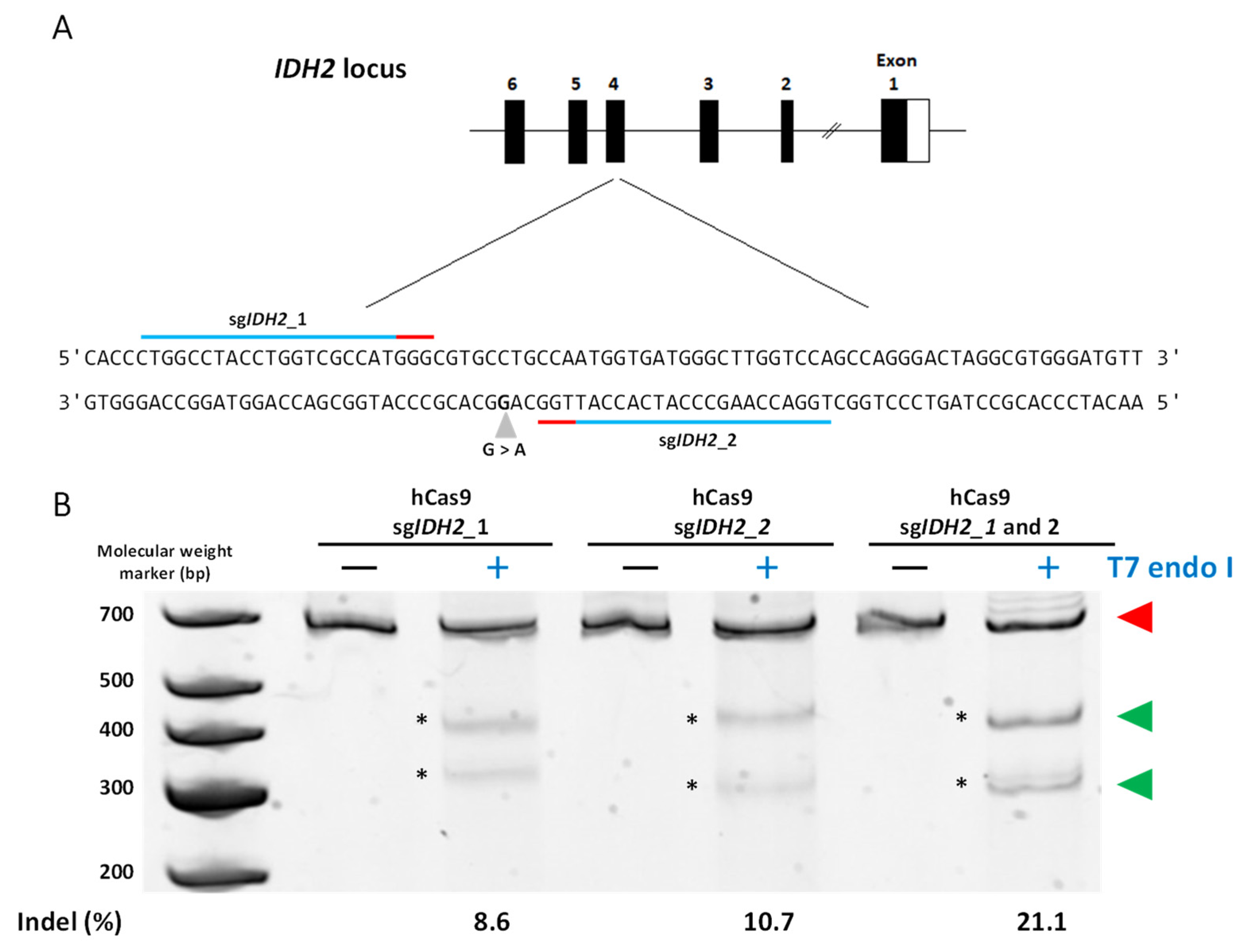
2.1.2. sgRNAs Used to Induce Cuts in DNA
2.1.3. PX458-sgMYBL2 Vector
2.1.4. Cell Lines Used in This Work
2.1.5. Primers used in this work
2.2. Methods
2.2.1. Nucleic Acids Handling and Analysis
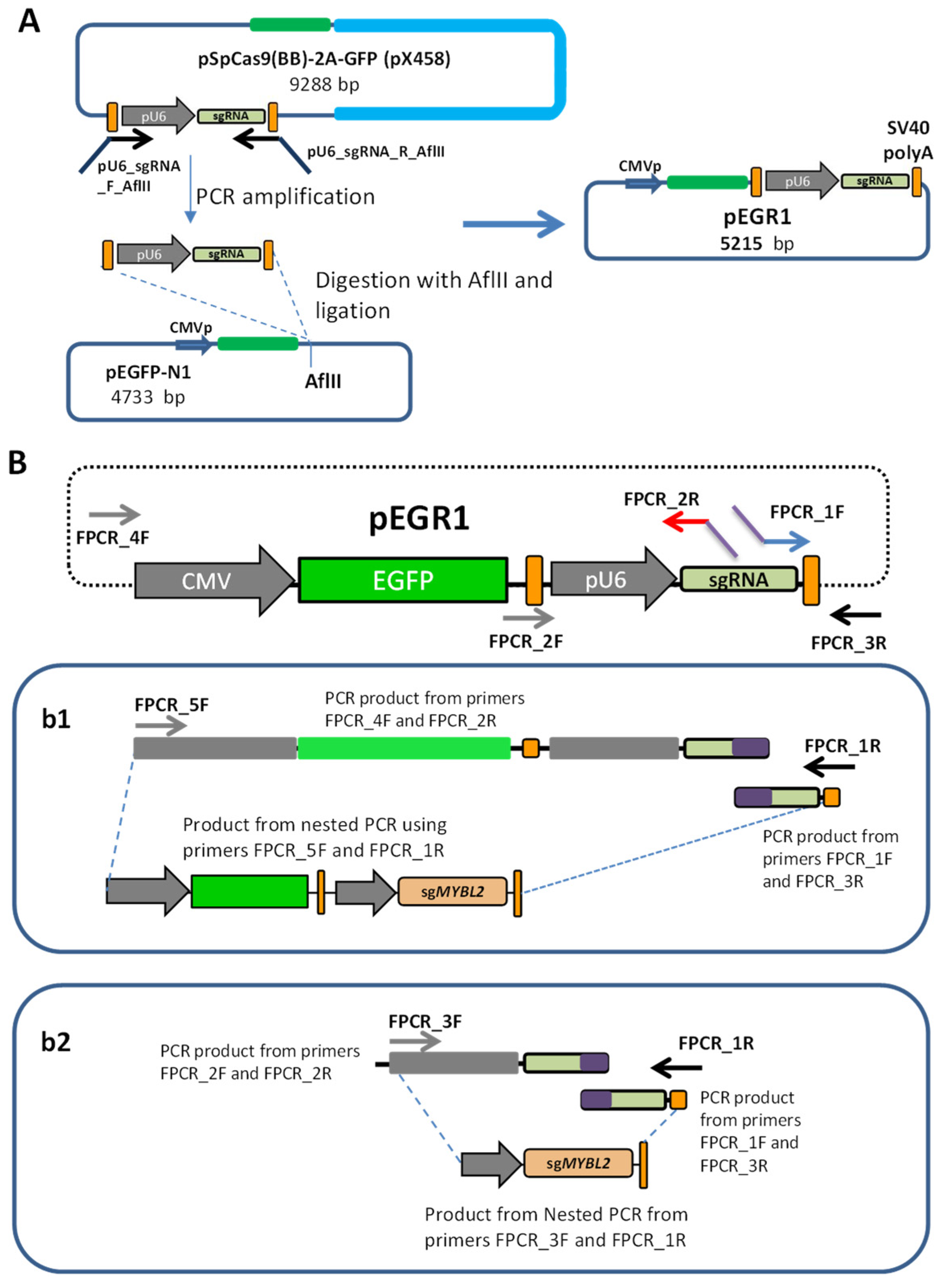
Cloning the sgRNA Cassette to Create pEGR1 Vector
Creating the sgRNA Constructs by Fusion PCR
PCR-Amplification of the Cas9 Cassette
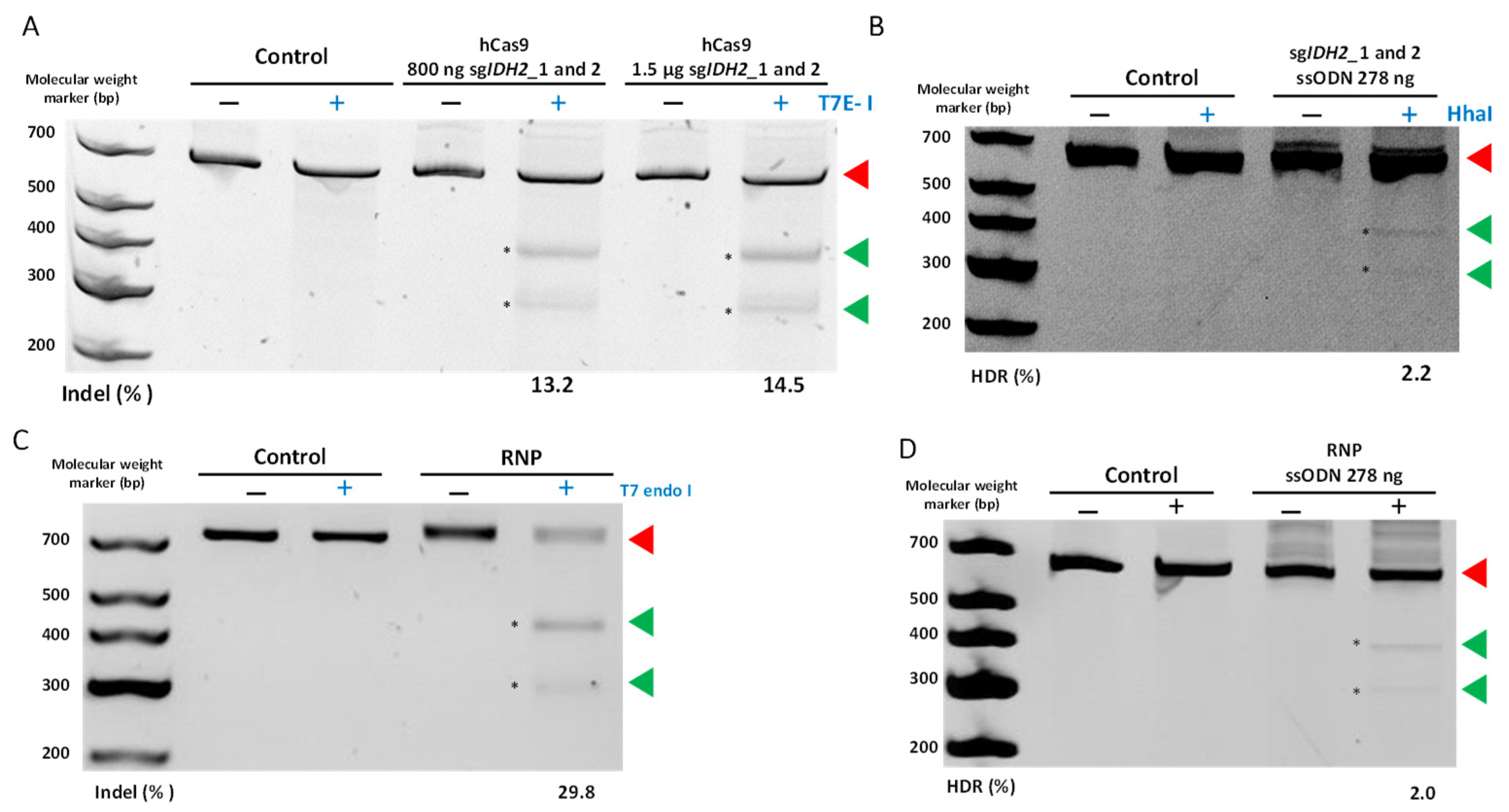
Analysis of DNA Editing Efficiencies
Inference of CRISPR Edits (ICE) Analysis
On- and Off-Target Analysis Using Next-Generation Sequencing
2.2.2. Cell Culture
Culture of Commercial Cell Lines
Assembling Ribonucleoprotein Complexes
Nucleofection of Cell Lines
Transfection of CRISPR Constructs
Generation of the NB4 and HL60 Cells Constitutively Expressing Cas9
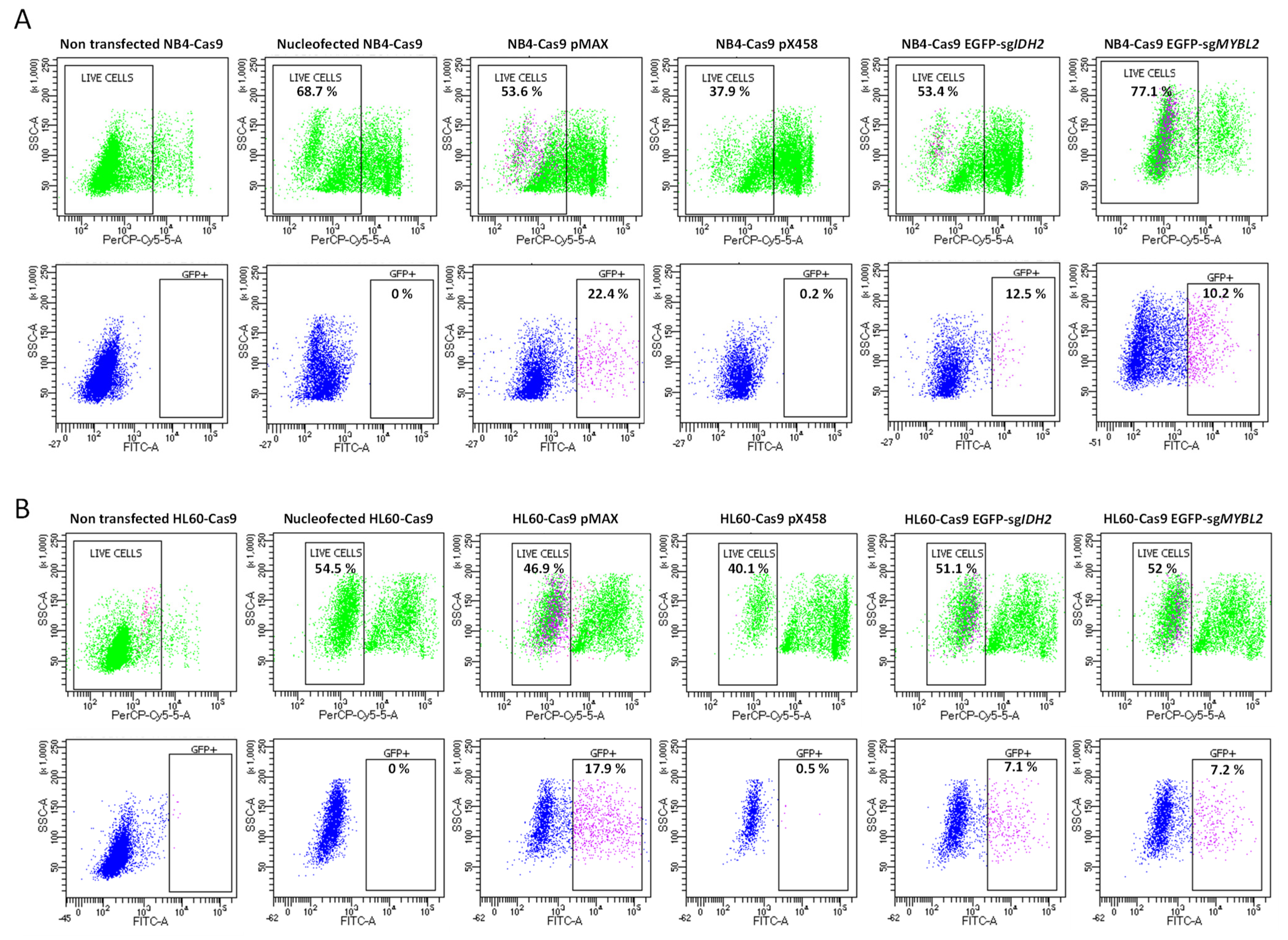
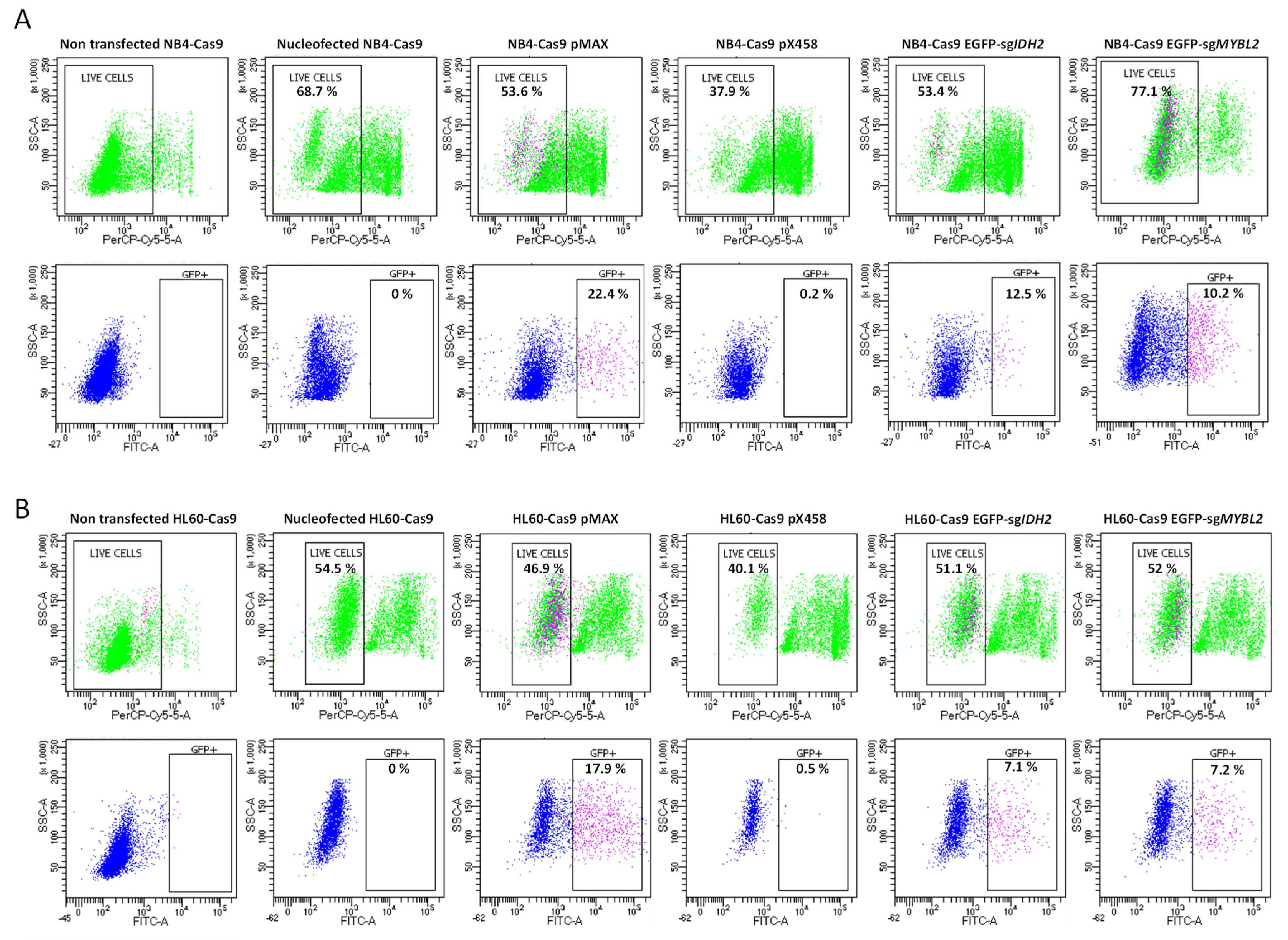
Cell Sorting to Analyze Transfection Efficiency and Cell Survival
3. Results
3.1. Target-Specific sgRNA Expression Constructs Created by Fusion PCR
3.2. Gene Editing in HEK293 Cells
3.3. Compatibility and Gene Editing in Leukemia Cell Lines
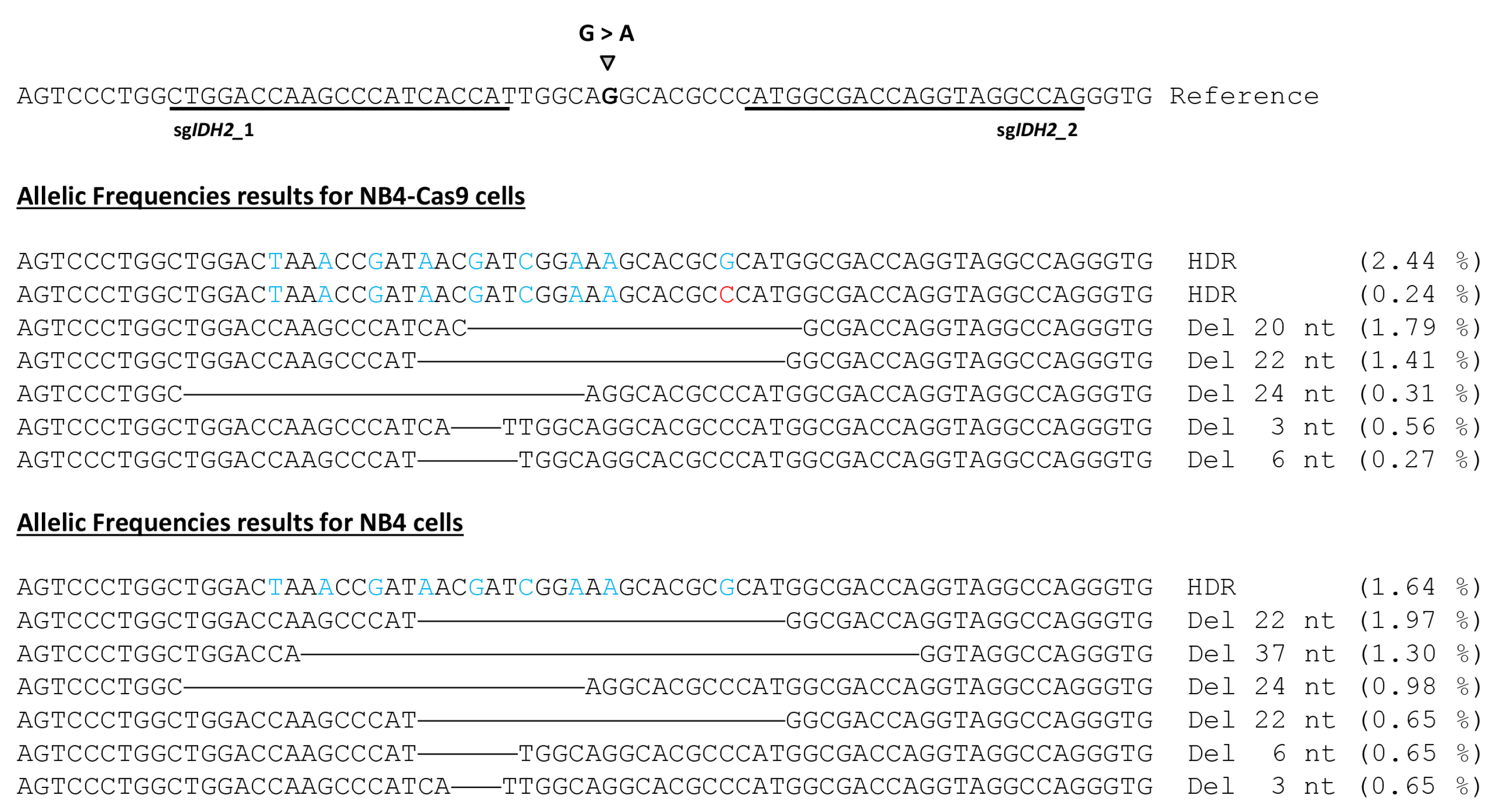
3.4. Deep Sequencing of CRISPR-Treated Cells Validates Efficacy and Specificity of Our Fusion PCR-Generated Constructs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mojica, F.J.M.; Juez, G.; Rodriguez-Valera, F. Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites. Mol. Microbiol. 1993, 9, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Ferrer, C.; Juez, G.; Rodriguez-Valera, F. Long stretches of short tandem repeats are present in the largest replicons of the Archaea Haloferax mediterranei and Haloferax volcanii and could be involved in replicon partitioning. Mol. Microbiol. 1995, 17, 85–93. [Google Scholar] [CrossRef]
- Mojica, F.J.M.; Diez-Villasenor, C.; Soria, E.; Juez, G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 2000, 36, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening Sequences of Regularly Spaced Prokaryotic Repeats Derive from Foreign Genetic Elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Bhattacharjee, O.; Mandal, D.; Sen, M.K.; Dey, D.; Dasgupta, A.; Kazi, T.A.; Gupta, R.; Sinharoy, S.; Acharya, K.; et al. CRISPR-Cas9 system: A new-fangled dawn in gene editing. Life Sci. 2019, 232, 116636. [Google Scholar] [CrossRef]
- Weber, J.; Rad, R. Engineering CRISPR mouse models of cancer. Curr. Opin. Genet. Dev. 2019, 54, 88–96. [Google Scholar] [CrossRef]
- Gantz, V.M.; Akbari, O.S. Gene editing technologies and applications for insects. Curr. Opin. Insect Sci. 2018, 28, 66–72. [Google Scholar] [CrossRef]
- Naert, T.; Vleminckx, K. CRISPR/Cas9 disease models in zebrafish and Xenopus: The genetic renaissance of fish and frogs. Drug Discov. Today Technol. 2018, 28, 41–52. [Google Scholar] [CrossRef]
- Farboud, B. Targeted genome editing in Caenorhabditis elegans using CRISPR/Cas9. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e287. [Google Scholar] [CrossRef]
- González-Romero, E.; Martínez-Valiente, C.; García-Ruiz, C.; Vázquez-Manrique, R.P.; Cervera, J.; Sanjuan-Pla, A. CRISPR to fix bad blood: A new tool in basic and clinical hematology. Haematologica 2019, 104, 881–893. [Google Scholar] [CrossRef]
- Reimer, J.; Knoess, S.; Labuhn, M.; Charpentier, E.M.; Göhring, G.; Schlegelberger, B.; Klusmann, J.-H.; Heckl, D. CRISPR-Cas9-induced t(11;19)/MLL-ENL translocations initiate leukemia in human hematopoietic progenitor cells in vivo. Haematologica 2017, 102, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Krill-Burger, J.M.; Popova, K.D.; Landers, C.C.; Sievers, Q.L.; Yudovich, D.; Belizaire, R.; Aster, J.C.; Morgan, E.A.; Tsherniak, A.; et al. Multiplex CRISPR-Cas9 Based Genome Editing in Human Hematopoietic Stem Cells Models Clonal Hematopoiesis and Myeloid Neoplasia. J. Autism. Dev. Disord. 2017, 21, 547–555.e8. [Google Scholar] [CrossRef] [PubMed]
- Sürün, D.; Schwäble, J.; Tomasovic, A.; Ehling, R.; Stein, S.; Kurrle, N.; von Melchner, H.; Schnütgen, F. High Efficiency Gene Correction in Hematopoietic Cells by Donor-Template-Free CRISPR/Cas9 Genome Editing. Mol. Ther. Nucleic Acids 2018, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef]
- Li, C.; Lieber, A. Adenovirus vectors in hematopoietic stem cell genome editing. FEBS Lett. 2019, 593, 3623–3648. [Google Scholar] [CrossRef]
- Dong, W.; Kantor, B. Lentiviral Vectors for Delivery of Gene-Editing Systems Based on CRISPR/Cas: Current State and Perspectives. Viruses 2021, 13, 1288. [Google Scholar] [CrossRef]
- Xu, C.L.; Ruan, M.Z.C.; Mahajan, V.B.; Tsang, S.H. Viral Delivery Systems for CRISPR. Viruses 2019, 11, 28. [Google Scholar] [CrossRef]
- Pelcovits, A.; Niroula, R. Acute Myeloid Leukemia: A Review. Rhode Isl. Med. J. 2013 2020, 103, 38–40. [Google Scholar]
- Glass, J.L.; Hassane, D.; Wouters, B.J.; Kunimoto, H.; Avellino, R.; Garrett-Bakelman, F.E.; Guryanova, O.A.; Bowman, R.; Redlich, S.; Intlekofer, A.M.; et al. Epigenetic Identity in AML Depends on Disruption of Nonpromoter Regulatory Elements and Is Affected by Antagonistic Effects of Mutations in Epigenetic Modifiers. Cancer Discov. 2017, 7, 868–883. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Musa, J.; Aynaud, M.-M.; Mirabeau, O.; Delattre, O.; Grünewald, T.G. MYBL2 (B-Myb): A central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017, 8, e2895. [Google Scholar] [CrossRef] [PubMed]
- Fuster, Ó.; Llop, M.; Dolz, S.; García, P.; Such, E.; Ibáñez, M.; Luna, I.; Gómez, I.; López, M.; Cervera, J.; et al. Adverse prognostic value of MYBL2 overexpression and association with microRNA-30 family in acute myeloid leukemia patients. Leuk. Res. 2013, 37, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Fuster-García, C.; García-García, G.; González-Romero, E.; Jaijo, T.; Sequedo, M.D.; Ayuso, C.; Vázquez-Manrique, R.P.; Millán, J.M.; Aller, E. USH2A Gene Editing Using the CRISPR System. Mol. Ther. Nucleic Acids 2017, 8, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef]
- Clement, K.; Rees, H.; Canver, M.C.; Gehrke, J.M.; Farouni, R.; Hsu, J.Y.; Cole, M.A.; Liu, D.R.; Joung, J.K.; Bauer, D.E.; et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019, 37, 224–226. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Castanotto, D.; Rossi, J.J. Construction and Transfection of PCR Products Expressing siRNAs or shRNAs in Mammalian Cells. Methods Mol. Biol. 2004, 252, 509–514. [Google Scholar] [CrossRef]
- Di Stazio, M.; Foschi, N.; Athanasakis, E.; Gasparini, P.; D’adamo, A.P. Systematic analysis of factors that improve homologous direct repair (HDR) efficiency in CRISPR/Cas9 technique. PLoS ONE 2021, 16, e0247603. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, E.; Milazzo, J.P.; Wang, Z.; Kinney, J.B.; Vakoc, C.R. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 2015, 33, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Heckl, D.; Kowalczyk, M.S.; Yudovich, D.; Belizaire, R.; Puram, R.V.; E McConkey, M.; Thielke, A.; Aster, J.C.; Regev, A.; Ebert, B.L. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 2014, 32, 941–946. [Google Scholar] [CrossRef]
- Rathe, S.K.; Moriarity, B.S.; Stoltenberg, C.B.; Kurata, M.; Aumann, N.K.; Rahrmann, E.P.; Bailey, N.J.; Melrose, E.G.; Beckmann, D.A.; Liska, C.R.; et al. Using RNA-seq and targeted nucleases to identify mechanisms of drug resistance in acute myeloid leukemia. Sci. Rep. 2014, 4, 6048. [Google Scholar] [CrossRef]
- Acosta, S.; Fiore, L.; Carota, I.A.; Oliver, G. Use of two gRNAs for CRISPR/Cas9 improves bi-allelic homologous recombination efficiency in mouse embryonic stem cells. Genesis 2018, 56, e23212. [Google Scholar] [CrossRef]
- Guo, Q.; Mintier, G.; Ma-Edmonds, M.; Storton, D.; Wang, X.; Xiao, X.; Kienzle, B.; Zhao, D.; Feder, J.N. ‘Cold shock’ increases the frequency of homology directed repair gene editing in induced pluripotent stem cells. Sci. Rep. 2018, 8, 2080. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, Y.; Berman, J.R.; Cooper, S.B.; Mayerl, S.J.; Chan, A.H.; Zhang, B.; Karlin-Neumann, G.A.; Conklin, B.R. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016, 6, 23549. [Google Scholar] [CrossRef]
- Castaño, J.; Herrero, A.B.; Bursen, A.; González, F.; Marschalek, R.; Gutiérrez, N.C.; Menendez, P. Expression of MLL-AF4 or AF4-MLL fusions does not impact the efficiency of DNA damage repair. Oncotarget 2016, 7, 30440–30452. [Google Scholar] [CrossRef]
- Brabetz, O.; Alla, V.; Angenendt, L.; Schliemann, C.; Berdel, W.E.; Arteaga, M.-F.; Mikesch, J.-H. RNA-Guided CRISPR-Cas9 System-Mediated Engineering of Acute Myeloid Leukemia Mutations. Mol. Ther. Nucleic Acids 2017, 6, 243–248. [Google Scholar] [CrossRef]
- Paix, A.; Folkmann, A.; Goldman, D.H.; Kulaga, H.; Grzelak, M.J.; Rasoloson, D.; Paidemarry, S.; Green, R.; Reed, R.R.; Seydoux, G. Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl. Acad. Sci. USA 2017, 114, E10745–E10754. [Google Scholar] [CrossRef]
- Raveux, A.; Vandormael-Pournin, S.; Cohen-Tannoudji, M. Optimization of the production of knock-in alleles by CRISPR/Cas9 microinjection into the mouse zygote. Sci. Rep. 2017, 7, srep42661. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2019, 9, 691. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Zhao, X.-J.; Wong, K.-W.; Fan, X.-Y. Comparison of plasmid DNA versus PCR amplified gene of insert DNA for nucleofection in Kasumi-1 cells. Cytotechnology 2015, 67, 275–283. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosomal Position | Gene Name | Sequence | Region | |
|---|---|---|---|---|
| sgIDH2_1 | ||||
| 1_off1 | chr3: 76082901 | ROBO2 | cGGACCAAGgCgATCACCATGGG | Intron |
| 1_off2 | chr4: 130711158 (-) | TGtACCAAGCtCATCAaCATTGG | Intergenic | |
| 1_off3 | chr1: 2405874 | PEX10 | TGGgCCAtGCCCATCcCCATCGG | Intron |
| 1_off4 | chr1: 54465152 | TGGACCAAGCCCcTCACCtTGGG | Intergenic | |
| 1_off5 | chr15: 70078666 | TLE3 | TGGcCCAAGCCCtTCACCAaCGG | Intron |
| 1_off6 | chr17: 83115842 (-) | TGGACCAAGtCCAcCtCCATGGG | Intergenic | |
| 1_off7 | chr10: 127909863 (-) | PTPRE | TGGACCAtGCCCATCcaCATCGG | Intron |
| 1_off8 | chr6: 31629230 | PRRC2A | gGGACCAAtCCCATCACCcTTGG | Exon |
| 1_off9 1 | chr14: 99534805 | CCNK/CCDC85C | TGGcCaAAGCCCtTCACCATAGG | Exon/Intron |
| 1_off10 | chr9: 38523669 | RP11-103F21.4 | gGGACCAgGCCCtTCACCATTGG | Pseudogene |
| 1_off11 | chr9: 97112588 | gGGACCAgGCCCtTCACCATTGG | Intergenic | |
| 1_off12 | chr18: 58014733 (-) | TGaACCAAGCCCATaACCcTTGG | Intergenic | |
| sgIDH2_2 | ||||
| 2_off1 | chr5: 173428070 | CTB-32H22 | CTGaCCTgCCTGGTCcCCATTGG | Intron |
| 2_off2 | chr1: 205830643 (-) | PM20D1 | tTGGCCTcCCTGGTCGtCATGGG | Intron |
| 2_off3 | chr2: 241745044 | D2HGD | CTGGCCTtCCTGGTgGtCATGGG | Intron |
| 2_off4 | chr17: 2879971(-) | RAP1GAP2 | CaGGCCTACCTGGTCcCCATTGG | Intron |
| Conditions | 150 ng hCas9 7 ng sgMYBL2 | 250 ng hCas9 12 ng sgMYBL2 | 250 ng hCas9 35 ng sgMYBL2 | 250 ng hCas9 60 ng sgMYBL2 | 500 ng hCas9 23.3 ng sgMYBL2 | 250 ng hCas9 35 ng sgMYBL2-P | 250 ng PCR Cas9 35 ng sgMYBL2 | PX458-MYBL2 |
|---|---|---|---|---|---|---|---|---|
| Exp.1 | 4.5 | 3.4 | 5.2 | 6.0 | 5.2 | 14.0 | 3.0 | 4.3 |
| Exp. 2 | 3.0 | 5.8 | 8.0 | 6.0 | 3.0 | 8.0 | 7.0 | 11.0 |
| Exp. 3 | 3.7 | 4.0 | 13.0 | 14.7 | 14 | 6.0 | 2.4 | 3.0 |
| Exp. 4 | 2.5 | 4.0 | 14.8 | 5.3 | 8.4 | 13.9 | - | 13.0 |
| Average NHEJ (% ± SEM) | 3.4 ± 0.4 | 4.3 ± 0.5 | 10.3 ± 2.2 | 8.0 ± 2.2 | 7.6 ± 2.3 | 10.5 ± 2.0 | 4.1 ± 1.4 | 7.8 ± 2.5 |
| Conditions 1 | sgIDH2_1 | sgIDH2_2 | sgIDH2_1 + sgIDH2_2 | PCR Cas9 sgIDH2_1 + sgIDH2_2 | sgIDH2_1 + sgIDH2_2 ssODN 2 |
|---|---|---|---|---|---|
| Exp.1 | 19.0 | 7.1 | 24.2 | 4.4 | 0.5 |
| Exp. 2 | 11.0 | 15.0 | 28.8 | 2.8 | 0.8 |
| Exp. 3 | 9.0 | 5.4 | 11.8 | 2.8 | 1.1 |
| Exp. 4 | 4.0 | 5.5 | 19.7 | - | - |
| Average NHEJ (% ± SEM) | 10.8 ± 3.10 | 8.3 ± 2.20 | 21.1 ± 3.60 | 3.36 ± 0.55 | |
| Average HDR (% ± SEM) | 1.01 ± 0.19 |
| Conditions 1 | sgIDH2_1 + sgIDH2_2 2 | RNPs 3 | sgIDH2_1 + sgIDH2_2 2 + 278 ng ssODN | RNPs 3 + 278 ng ssODN | sgMYBL2 2 |
|---|---|---|---|---|---|
| Exp. 1 | 21.5 | 28.7 | 2.8 | 2.5 | 4.0 |
| Exp. 2 | 7.0 | 34.16 | 2.3 | 1.6 | 2.0 |
| Exp. 3 | 15.0 | 26.7 | 1.5 | 2.0 | 0 |
| Average NHEJ (% ± SEM) | 14.5 ± 7.7 | 29.8 ± 3.8 | 2 ± 1.1 | ||
| HDR (% ± SEM) | 2.2 ± 0.4 | 2.0 ± 0.3 |
| Conditions 1 | sgIDH2_1 + sgIDH2_2 2 | sgMYBL2 2 |
|---|---|---|
| Exp. 1 | 2 | 8 |
| Exp. 2 | 5 | 11 |
| Exp. 3 | 4 | 0 |
| Average NHEJ (% ± SEM) | 3.7 ± 0.9 | 6.3 ± 3.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Romero, E.; Martínez-Valiente, C.; García-García, G.; Rosal-Vela, A.; Millán, J.M.; Sanz, M.Á.; Sanz, G.; Liquori, A.; Cervera, J.V.; Vázquez-Manrique, R.P. PCR-Based Strategy for Introducing CRISPR/Cas9 Machinery into Hematopoietic Cell Lines. Cancers 2023, 15, 4263. https://doi.org/10.3390/cancers15174263
González-Romero E, Martínez-Valiente C, García-García G, Rosal-Vela A, Millán JM, Sanz MÁ, Sanz G, Liquori A, Cervera JV, Vázquez-Manrique RP. PCR-Based Strategy for Introducing CRISPR/Cas9 Machinery into Hematopoietic Cell Lines. Cancers. 2023; 15(17):4263. https://doi.org/10.3390/cancers15174263
Chicago/Turabian StyleGonzález-Romero, Elisa, Cristina Martínez-Valiente, Gema García-García, Antonio Rosal-Vela, José María Millán, Miguel Ángel Sanz, Guillermo Sanz, Alessandro Liquori, José Vicente Cervera, and Rafael P. Vázquez-Manrique. 2023. "PCR-Based Strategy for Introducing CRISPR/Cas9 Machinery into Hematopoietic Cell Lines" Cancers 15, no. 17: 4263. https://doi.org/10.3390/cancers15174263
APA StyleGonzález-Romero, E., Martínez-Valiente, C., García-García, G., Rosal-Vela, A., Millán, J. M., Sanz, M. Á., Sanz, G., Liquori, A., Cervera, J. V., & Vázquez-Manrique, R. P. (2023). PCR-Based Strategy for Introducing CRISPR/Cas9 Machinery into Hematopoietic Cell Lines. Cancers, 15(17), 4263. https://doi.org/10.3390/cancers15174263