

Advancing Breast Cancer Heterogeneity Analysis: Insights from Genomics, Transcriptomics and Proteomics at Bulk and Single-Cell Levels



Abstract
:Simple Summary
Abstract
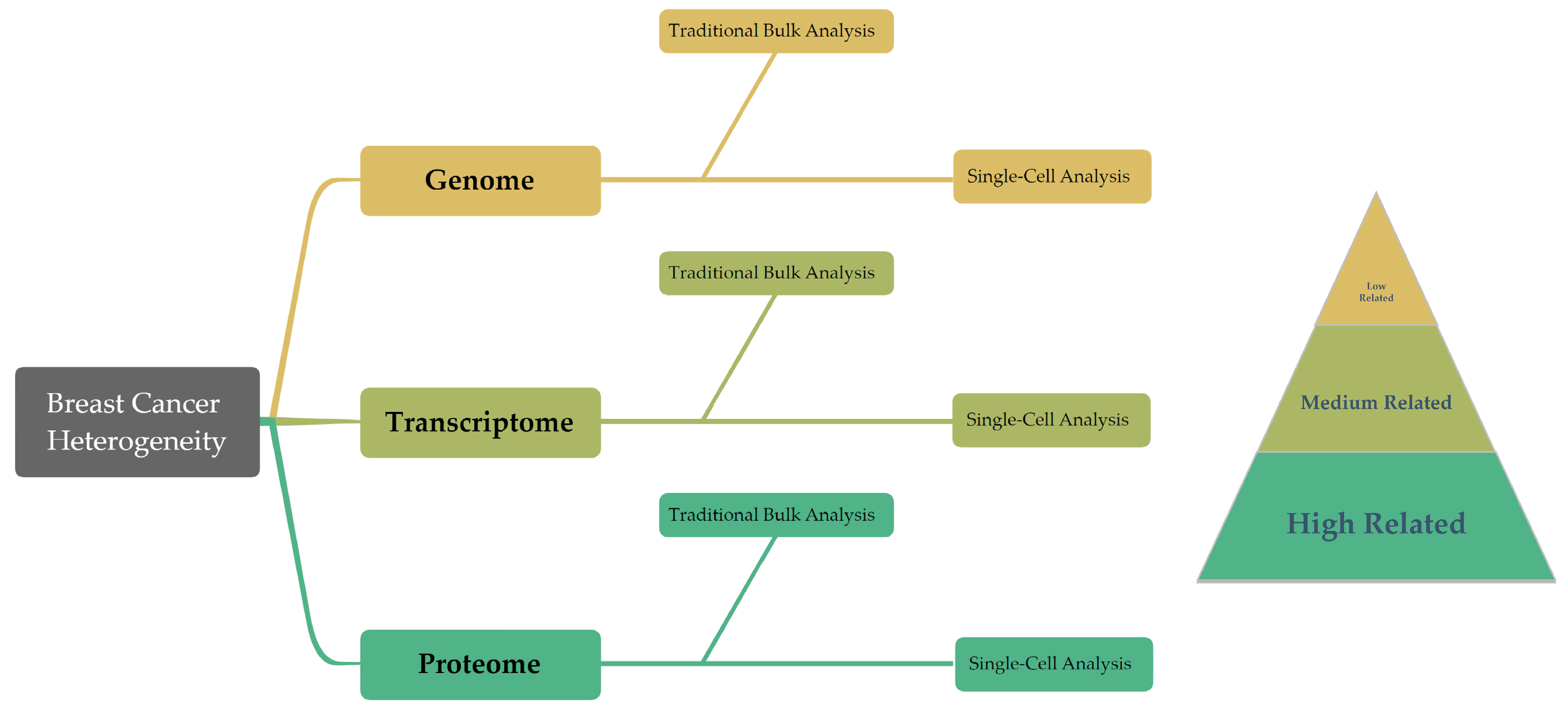
1. Introduction
2. Genomic Profiling
2.1. Traditional Genomic Profiling
2.2. Single-Cell Genomic Profiling
3. Transcriptional Profiling
3.1. Traditional Transcriptional Profiling
3.1.1. mRNA Expression Profiling in Breast Cancer Heterogeneity
3.1.2. mRNA Half-Life Detection in Breast Cancer Heterogeneity
3.1.3. Functional RNAs in Breast Cancer Heterogeneity
3.2. Single-Cell Transcriptome Profiling
4. Protein Profiling
4.1. Traditional Protein Profiling
4.2. Single-Cell Proteomic Profiling
- Flow cytometry, which is an approach that quantifies the fluorescence characteristics of individual cells or particles within a fluid stream when exposed to light sources [154]. Cells labeled with fluorescent antibodies are rapidly channeled through a detection region within a flow chamber. Subsequently, these stained cells are stimulated by lasers, and a detector captures the intensity of the emitted fluorescence. Over the decades, since its inception in the late 1960s, flow cytometry has evolved significantly. It has progressed from an initial capacity to measure 1–2 fluorescent substances within cells, to now being capable of analyzing 10–15 fluorescent substances within a single cell, enabling the assessment of entire cellular pathways.
- Single-cell mass spectrometry (MS), which is a method that offers the potential for a label-free quantitative analysis of the full proteome of a single cell, inclusive of proteins, peptides, and PTMs [155]. One key advantage of MS is that it does not necessitate molecular labeling, and it can attain sensitivity to the femtomolar level for pure proteins. Various mass spectrometry techniques, such as electrospray MS, laser/desorption/ionization MS, and secondary ion MS, are deployed in single-cell research. However, the utilization of MS for single-cell protein analysis faces challenges, primarily due to an inadequate sensitivity to detect the low-abundance proteins that are typically present in single cells.
- Reverse-phase protein array (RPA), which is a miniaturized protein imprinting technique that facilitates quantitative monitoring of protein expression in hundreds or even thousands of samples concurrently [156]. This method involves archiving whole-cell lysates in a microarray format for detecting proteins of interest via immunological detection. Notably, RPA obviates the need for protein sample separation via electrophoresis, thereby enabling the concurrent analysis of multiple samples. Additionally, RPA requires only a minimal sample volume for multiplex protein detection.
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.-W.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Desmedt, C.; Durbecq, V.; Dal Lago, L.; Lacroix, M.; Cardoso, F.; Piccart-Gebhart, M.; Ross, J.S.; Hortobagyi, G.N. Genomic and molecular classification of breast cancer. In Molecular Oncology of Breast Cancer; Jones and Barlett Publishers: Sudbury, MA, USA, 2005; pp. 81–95. [Google Scholar]
- Rivenbark, A.G.; O’Connor, S.M.; Coleman, W.B. Molecular and cellular heterogeneity in breast cancer: Challenges for personalized medicine. Am. J. Pathol. 2013, 183, 1113–1124. [Google Scholar] [CrossRef]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.-O.; Lee, K.-M.; Lee, H.-B.; Kim, K.-T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef]
- Kim, J.J.; Liang, W.; Kang, C.-C.; Pegram, M.D.; Herr, A.E. Single-cell immunoblotting resolves estrogen receptor-α isoforms in breast cancer. PLoS ONE 2021, 16, e0254783. [Google Scholar] [CrossRef]
- Gerlinger, M.; Swanton, C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br. J. Cancer 2010, 103, 1139–1143. [Google Scholar] [CrossRef]
- Shiovitz, S.; Korde, L.A. Genetics of breast cancer: A topic in evolution. Ann. Oncol. 2015, 26, 1291–1299. [Google Scholar] [CrossRef]
- Griseri, P.; Pagès, G. Regulation of the mRNA half-life in breast cancer. World J. Clin. Oncol. 2014, 5, 323. [Google Scholar] [CrossRef]
- Ellis, M.J.; Perou, C.M. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013, 3, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, L.; Lakhani, S.R. Pathology of hereditary breast cancer. Mod. Pathol. 2010, 23, S46–S51. [Google Scholar] [CrossRef]
- Wellings, S.; Jensen, H. On the origin and progression of ductal carcinoma in the human breast. J. Natl. Cancer Inst. 1973, 50, 1111–1118. [Google Scholar] [CrossRef]
- Sundah, N.R.; Ho, N.R.; Lim, G.S.; Natalia, A.; Ding, X.; Liu, Y.; Seet, J.E.; Chan, C.W.; Loh, T.P.; Shao, H. Barcoded DNA nanostructures for the multiplexed profiling of subcellular protein distribution. Nat. Biomed. Eng. 2019, 3, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.I.; King, M.-C. Inherited breast and ovarian cancer. Hum. Mol. Genet. 1995, 4, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef]
- Ford, D.; Easton, D.; Stratton, M.; Narod, S.; Goldgar, D.; Devilee, P.; Bishop, D.; Weber, B.; Lenoir, G.; Chang-Claude, J. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am. J. Hum. Genet. 1998, 62, 676–689. [Google Scholar] [CrossRef]
- Balmana, J.; Diez, O.; Rubio, I.; Cardoso, F. BRCA in breast cancer: ESMO Clinical Practice Guidelines. Ann. Oncol. 2011, 22, vi31–vi34. [Google Scholar] [CrossRef]
- Brooksby, B.; Jiang, S.; Dehghani, H.; Pogue, B.W.; Paulsen, K.D.; Weaver, J.; Kogel, C.; Poplack, S.P. Combining near-infrared tomography and magnetic resonance imaging to study in vivo breast tissue: Implementation of a Laplacian-type regularization to incorporate magnetic resonance structure. J. Biomed. Opt. 2005, 10, 051504. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J. Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Araki, M. Tumor suppressor PTEN: Modulator of cell signaling, growth, migration and apoptosis. J. Cell Sci. 2001, 114, 2375–2382. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Velculescu, V.E. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 2004, 3, 1221–1224. [Google Scholar] [CrossRef]
- Bhaskar, P.T.; Hay, N. The two TORCs and AKT. Dev. Cell 2007, 12, 487–502. [Google Scholar] [CrossRef]
- De Brakeleer, S.; De Grève, J.; Loris, R.; Janin, N.; Lissens, W.; Sermijn, E.; Teugels, E. Cancer predisposing missense and protein truncating BARD1 mutations in non-BRCA1 or BRCA2 breast cancer families. Hum. Mutat. 2010, 31, E1175–E1185. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Liu, M.L.; Shibata, M.A.; Von Lintig, F.C.; Wang, W.; Cassenaer, S.; Boss, G.R.; Green, J.E. Haploid loss of Ki-ras delays mammary tumor progression in C3 (1)/SV40 Tag transgenic mice. Oncogene 2001, 20, 2044–2049. [Google Scholar] [CrossRef]
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; Rasmussen, K.E.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007, 8, R76. [Google Scholar] [CrossRef] [PubMed]
- Silwal-Pandit, L.; Vollan, H.K.; Chin, S.F.; Rueda, O.M.; McKinney, S.; Osako, T.; Quigley, D.A.; Kristensen, V.N.; Aparicio, S.; Børresen-Dale, A.L.; et al. TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin. Cancer Res. 2014, 20, 3569–3580. [Google Scholar] [CrossRef] [PubMed]
- Brooks, M.D.; Burness, M.L.; Wicha, M.S. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell 2015, 17, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Kessenbrock, K.; Davis, R.T.; Pervolarakis, N.; Werb, Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 2018, 20, 1349–1360. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Carter, N.P.; Bebb, C.E.; Nordenskjo, M.; Ponder, B.A.; Tunnacliffe, A. Degenerate oligonucleotide-primed PCR: General amplification of target DNA by a single degenerate primer. Genomics 1992, 13, 718–725. [Google Scholar]
- Picher, A.J.; Budeus, B.; Wafzig, O.; Krüger, C.; García-Gómez, S.; Martínez-Jiménez, M.I.; Díaz-Talavera, A.; Weber, D.; Blanco, L.; Schneider, A. TruePrime is a novel method for whole-genome amplification from single cells based on Tth PrimPol. Nat. Commun. 2016, 7, 13296. [Google Scholar] [CrossRef]
- Beroukhim, R.; Getz, G.; Nghiemphu, L.; Barretina, J.; Hsueh, T.; Linhart, D.; Vivanco, I.; Lee, J.C.; Huang, J.H.; Alexander, S. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc. Natl. Acad. Sci. USA 2007, 104, 20007–20012. [Google Scholar] [CrossRef]
- Adey, A.; Morrison, H.G.; Xun, X.; Kitzman, J.O.; Turner, E.H.; Stackhouse, B.; MacKenzie, A.P.; Caruccio, N.C.; Zhang, X.; Shendure, J. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010, 11, R119. [Google Scholar] [CrossRef]
- Hou, Y.; Song, L.; Zhu, P.; Zhang, B.; Tao, Y.; Xu, X.; Li, F.; Wu, K.; Liang, J.; Shao, D. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 2012, 148, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef]
- Williams, C.H., III. Chemical Genetics of Vertebrate Development; Vanderbilt University: Nashville, TN, USA, 2017. [Google Scholar]
- Powell, A.A.; Talasaz, A.H.; Zhang, H.; Coram, M.A.; Reddy, A.; Deng, G.; Telli, M.L.; Advani, R.H.; Carlson, R.W.; Mollick, J.A. Single cell profiling of circulating tumor cells: Transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS ONE 2012, 7, e33788. [Google Scholar] [CrossRef] [PubMed]
- Cardiff, R.D.; Anver, M.R.; Gusterson, B.A.; Hennighausen, L.; Jensen, R.A.; Merino, M.J.; Rehm, S.; Russo, J.; Tavassoli, F.A.; Wakefield, L.M. The mammary pathology of genetically engineered mice: The consensus report and recommendations from the Annapolis meeting. Oncogene 2000, 19, 968–988. [Google Scholar] [CrossRef]
- Harrell, J.C.; Prat, A.; Parker, J.S.; Fan, C.; He, X.; Carey, L.; Anders, C.; Ewend, M.; Perou, C.M. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res. Treat. 2012, 132, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Casalini, P.; Tagliabue, E.; Ménard, S.; Croce, C.M. MicroRNA profiling as a tool to understand prognosis, therapy response and resistance in breast cancer. Eur. J. Cancer 2008, 44, 2753–2759. [Google Scholar] [CrossRef]
- Brosius, J.; Raabe, C.A. What is an RNA? A top layer for RNA classification. RNA Biol. 2016, 13, 140–144. [Google Scholar] [CrossRef]
- Gurdon, J.; Lane, C.; Woodland, H.; Marbaix, G. Use of frog eggs and oocytes for the study of messenger RNA and its translation in living cells. Nature 1971, 233, 177–182. [Google Scholar] [CrossRef]
- Dsouza, V.L.; Adiga, D.; Sriharikrishnaa, S.; Suresh, P.S.; Chatterjee, A.; Kabekkodu, S.P. Small nucleolar RNA and its potential role in breast cancer–A comprehensive review. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1875, 188501. [Google Scholar] [CrossRef]
- Verghese, E.; Hanby, A.; Speirs, V.; Hughes, T. Small is beautiful: microRNAs and breast cancer—Where are we now? J. Pathol. J. Pathol. Soc. Great Br. Irel. 2008, 215, 214–221. [Google Scholar] [CrossRef]
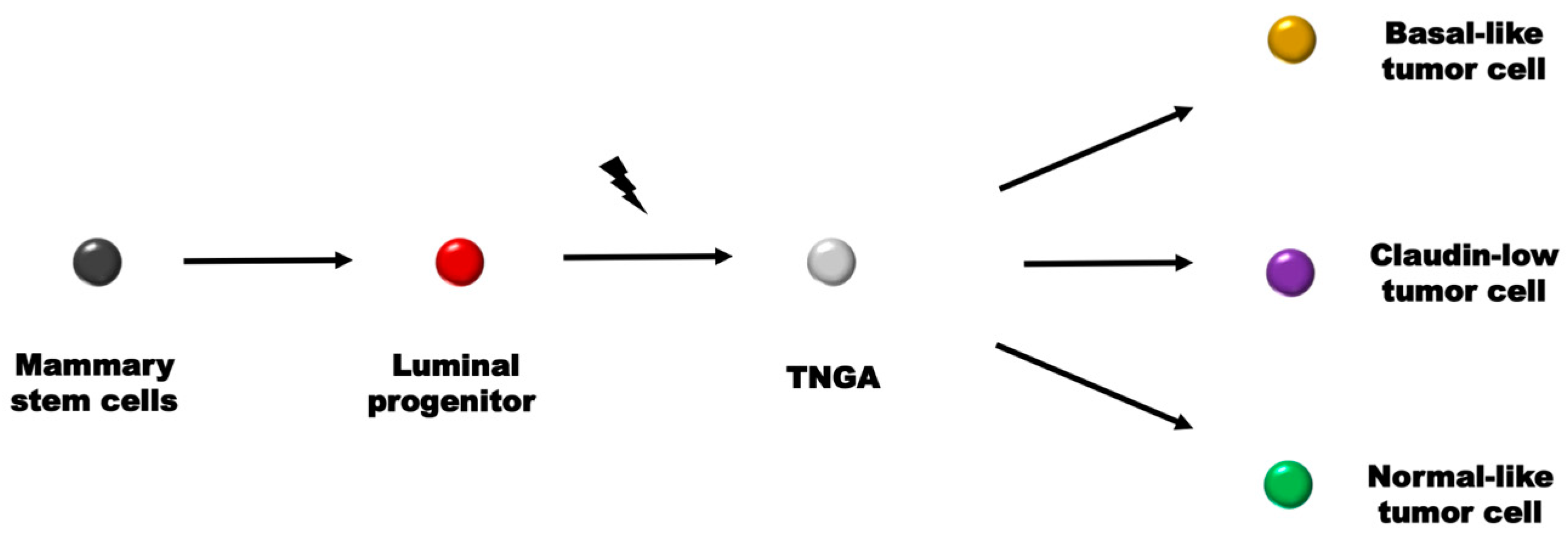
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.-Y. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160. [Google Scholar] [CrossRef]
- Myhre, L.; Alm, K.; Johansson, M.C.; Oredsson, S.M. Normal-like breast cells, but not breast cancer cells, recovered from treatment with N′, N′′-diethylnorspermine. Anti-Cancer Drugs 2009, 20, 230–237. [Google Scholar] [CrossRef]
- Casero, R.A., Jr.; Marton, L.J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007, 6, 373–390. [Google Scholar] [CrossRef]
- Hahm, H.A.; Ettinger, D.S.; Bowling, K.; Hoker, B.; Chen, T.L.; Zabelina, Y.; Casero Jr, R.A. Phase I study of N 1, N 11-diethylnorspermine in patients with non-small cell lung cancer. Clin. Cancer Res. 2002, 8, 684–690. [Google Scholar]
- Fan, C.; Oh, D.S.; Wessels, L.; Weigelt, B.; Nuyten, D.S.; Nobel, A.B.; Van’t Veer, L.J.; Perou, C.M. Concordance among gene-expression–based predictors for breast cancer. N. Engl. J. Med. 2006, 355, 560–569. [Google Scholar] [CrossRef]
- Rakha, E.; Ellis, I.; Reis-Filho, J. Are triple-negative and basal-like breast cancer synonymous? Clin. Cancer Res. 2008, 14, 618. [Google Scholar] [CrossRef]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K. Differences in breast carcinoma characteristics in newly diagnosed African–American and Caucasian patients: A single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and end results database. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2007, 110, 876–884. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Cervera, N.; Esterni, B.; Hermitte, F.; Viens, P.; Birnbaum, D. How basal are triple-negative breast cancers? Int. J. Cancer 2008, 123, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Elsheikh, S.E.; Aleskandarany, M.A.; Habashi, H.O.; Green, A.R.; Powe, D.G.; El-Sayed, M.E.; Benhasouna, A.; Brunet, J.-S.; Akslen, L.A. Triple-negative breast cancer: Distinguishing between basal and nonbasal subtypes. Clin. Cancer Res. 2009, 15, 2302–2310. [Google Scholar] [CrossRef]
- Kreike, B.; van Kouwenhove, M.; Horlings, H.; Weigelt, B.; Peterse, H.; Bartelink, H.; van de Vijver, M.J. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007, 9, R65. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.; Meszoely, I.; Sanders, M.; Shyr, Y.; Abramson, V.; Means-Powell, J.; Chakravarthy, A.; Arteaga, C.; Pietenpol, J. A phase II neoadjuvant study of cisplatin/paclitaxel with or without RAD001 in patients with triple-negative (TN) locally advanced breast cancer (BC). J. Clin. Oncol. 2010, 28, TPS119. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Gonzalez-Angulo, A.-M.; Stemke-Hale, K.; Gilcrease, M.Z.; Krishnamurthy, S.; Lee, J.-S.; Fridlyand, J.; Sahin, A.; Agarwal, R.; Joy, C. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009, 69, 4116–4124. [Google Scholar] [CrossRef]
- Andreassi, C.; Riccio, A. To localize or not to localize: mRNA fate is in 3′ UTR ends. Trends Cell Biol. 2009, 19, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J. From birth to death: The complex lives of eukaryotic mRNAs. Science 2005, 309, 1514–1518. [Google Scholar] [CrossRef]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef]
- Hamilton, T.; Li, X.; Novotny, M.; Pavicic, P.G., Jr.; Datta, S.; Zhao, C.; Hartupee, J.; Sun, D. Cell type-and stimulus-specific mechanisms for post-transcriptional control of neutrophil chemokine gene expression. J. Leukoc. Biol. 2012, 91, 377–383. [Google Scholar] [CrossRef]
- Shaw, G.; Kamen, R. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 1986, 46, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Khabar, K.S. Post-transcriptional control during chronic inflammation and cancer: A focus on AU-rich elements. Cell. Mol. Life Sci. 2010, 67, 2937–2955. [Google Scholar] [CrossRef] [PubMed]
- Audic, Y.; Hartley, R.S. Post-transcriptional regulation in cancer. Biol. Cell 2004, 96, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.E.; Kuwano, Y.; Alkharouf, N.; Blackshear, P.J.; Gorospe, M.; Wilson, G.M. The mRNA-destabilizing protein tristetraprolin is suppressed in many cancers, altering tumorigenic phenotypes and patient prognosis. Cancer Res. 2009, 69, 5168–5176. [Google Scholar] [CrossRef]
- Lembo, A.; Di Cunto, F.; Provero, P. Shortening of 3′ UTRs correlates with poor prognosis in breast and lung cancer. PLoS ONE 2012, 7, e31129. [Google Scholar] [CrossRef] [PubMed]
- Liaw, H.-H.; Lin, C.-C.; Juan, H.-F.; Huang, H.-C. Differential microRNA regulation correlates with alternative polyadenylation pattern between breast cancer and normal cells. PLoS ONE 2013, 8, e56958. [Google Scholar] [CrossRef]
- Akman, B.H.; Can, T.; Erson-Bensan, A.E. Estrogen-induced upregulation and 3′-UTR shortening of CDC6. Nucleic Acids Res. 2012, 40, 10679–10688. [Google Scholar] [CrossRef]
- Farazi, T.A.; Horlings, H.M.; Ten Hoeve, J.J.; Mihailovic, A.; Halfwerk, H.; Morozov, P.; Brown, M.; Hafner, M.; Reyal, F.; van Kouwenhove, M. MicroRNA Sequence and Expression Analysis in Breast Tumors by Deep SequencingmiRNA Sequence and Expression Analysis in Breast Tumors. Cancer Res. 2011, 71, 4443–4453. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, Y.; Hu, N.; Shan, C.; Zhang, S.; Zhang, W.; Zhang, X.; Ye, L. miRNA-520b and miR-520e sensitize breast cancer cells to complement attack via directly targeting 3′ UTR of CD46. Cancer Biol. Ther. 2010, 10, 232–241. [Google Scholar] [CrossRef]
- Iorio, M.V.; Ferracin, M.; Liu, C.-G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Yasuda, T.; Saigo, K.; Urashima, T.; Toyoda, H.; Okanoue, T.; Shimotohno, K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 2006, 25, 2537–2545. [Google Scholar] [CrossRef]
- Mittal, S.; Kaur, H.; Gautam, N.; Mantha, A.K. Biosensors for breast cancer diagnosis: A review of bioreceptors, biotransducers and signal amplification strategies. Biosens. Bioelectron. 2017, 88, 217–231. [Google Scholar] [CrossRef] [PubMed]
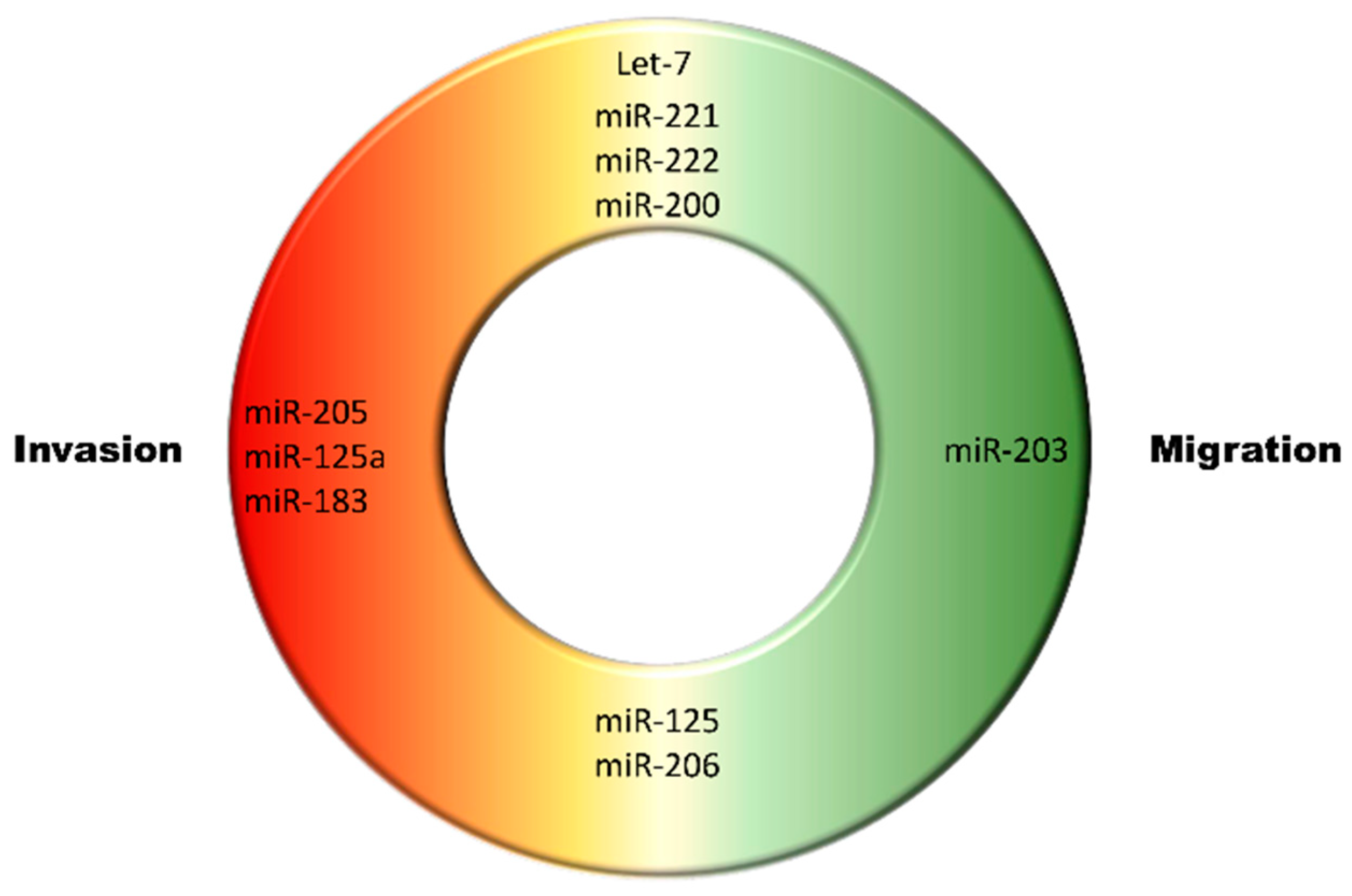
- Tang, J.; Ahmad, A.; Sarkar, F. The Role of MicroRNAs in Breast Cancer Migration, Invasion and Metastasis. Int. J. Mol. Sci. 2012, 13, 13414. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-T.; Li, S.-C.; Ho, M.-R.; Pan, H.-W.; Ger, L.-P.; Hu, L.-Y.; Yu, S.-Y.; Li, W.-H.; Tsai, K.-W. Comprehensive analysis of microRNAs in breast cancer. BMC Genom. 2012, 13, S18. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Adams, B.D.; Furneaux, H.; White, B.A. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-α (ERα) and represses ERα messenger RNA and protein expression in breast cancer cell lines. Mol. Endocrinol. 2007, 21, 1132–1147. [Google Scholar] [CrossRef]
- Stinson, S.; Lackner, M.R.; Adai, A.T.; Yu, N.; Kim, H.-J.; O’Brien, C.; Spoerke, J.; Jhunjhunwala, S.; Boyd, Z.; Januario, T. TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci. Signal. 2011, 4, ra41. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Paranjape, T.; Ullrich, R.; Nallur, S.; Gillespie, E.; Keane, K.; Esquela-Kerscher, A.; Weidhaas, J.; Slack, F. The mir-34 microRNA is required for the DNA damage response in vivo in C. elegans and in vitro in human breast cancer cells. Oncogene 2009, 28, 2419–2424. [Google Scholar] [CrossRef]
- Mattie, M.D.; Benz, C.C.; Bowers, J.; Sensinger, K.; Wong, L.; Scott, G.K.; Fedele, V.; Ginzinger, D.; Getts, R.; Haqq, C. Optimized high-throughput microRNA expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol. Cancer 2006, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef]
- Ahmad, A.; Ali, A.; Ali, S.; Wang, Z.; Kong, D.; Sarkar, F.; Mulligan, J. MicroRNAs: Targets of interest in breast cancer research. In MicroRNA: Expression, Detection and Therapeutic Strategies; Nova Publishers: New York, NY, USA, 2011; pp. 59–78. [Google Scholar]
- Shell, S.; Park, S.-M.; Radjabi, A.R.; Schickel, R.; Kistner, E.O.; Jewell, D.A.; Feig, C.; Lengyel, E.; Peter, M.E. Let-7 expression defines two differentiation stages of cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 11400–11405. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Deng, C.; Wang, J.; Xiao, J.; Gatalica, Z.; Recker, R.R.; Xiao, G.G. Let-7 family miRNAs regulate estrogen receptor alpha signaling in estrogen receptor positive breast cancer. Breast Cancer Res. Treat. 2011, 127, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.-P.; Tong, F. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Prat, A.; Cheang, M.; Lenburg, M.; Paik, S.; Perou, C. Breast cancer molecular subtypes predict response to anthracycline/taxane-based chemotherapy. Cancer Res 2009, 69, 2019. [Google Scholar] [CrossRef]
- Ginestier, C.; Liu, S.; Diebel, M.E.; Korkaya, H.; Luo, M.; Brown, M.; Wicinski, J.; Cabaud, O.; Charafe-Jauffret, E.; Birnbaum, D. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Investig. 2010, 120, 485–497. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Gross, A.; Schoendube, J.; Zimmermann, S.; Steeb, M.; Zengerle, R.; Koltay, P. Technologies for single-cell isolation. Int. J. Mol. Sci. 2015, 16, 16897–16919. [Google Scholar] [CrossRef]
- Kamme, F.; Salunga, R.; Yu, J.; Tran, D.-T.; Zhu, J.; Luo, L.; Bittner, A.; Guo, H.-Q.; Miller, N.; Wan, J. Single-cell microarray analysis in hippocampus CA1: Demonstration and validation of cellular heterogeneity. J. Neurosci. 2003, 23, 3607–3615. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef]
- Hu, P.; Zhang, W.; Xin, H.; Deng, G. Single cell isolation and analysis. Front. Cell Dev. Biol. 2016, 4, 116. [Google Scholar] [CrossRef]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef]
- Sasagawa, Y.; Nikaido, I.; Hayashi, T.; Danno, H.; Uno, K.D.; Imai, T.; Ueda, H.R. Quartz-Seq: A highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013, 14, 3097. [Google Scholar] [CrossRef]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Gao, R.; Kim, C.; Sei, E.; Foukakis, T.; Crosetto, N.; Chan, L.-K.; Srinivasan, M.; Zhang, H.; Meric-Bernstam, F.; Navin, N. Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer. Nat. Commun. 2017, 8, 228. [Google Scholar] [CrossRef]
- Svensson, V.; Natarajan, K.N.; Ly, L.-H.; Miragaia, R.J.; Labalette, C.; Macaulay, I.C.; Cvejic, A.; Teichmann, S.A. Power analysis of single-cell RNA-sequencing experiments. Nat. Methods 2017, 14, 381–387. [Google Scholar] [CrossRef]
- Picelli, S.; Björklund, Å.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Bhargava, V.; Ko, P.; Willems, E.; Mercola, M.; Subramaniam, S. Quantitative transcriptomics using designed primer-based amplification. Sci. Rep. 2013, 3, 1740. [Google Scholar] [CrossRef]
- Dalerba, P.; Kalisky, T.; Sahoo, D.; Rajendran, P.S.; Rothenberg, M.E.; Leyrat, A.A.; Sim, S.; Okamoto, J.; Johnston, D.M.; Qian, D. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat. Biotechnol. 2011, 29, 1120–1127. [Google Scholar] [CrossRef]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef]
- Malik, N.; Canfield, V.A.; Beckers, M.-C.; Gros, P.; Levenson, R. Identification of the mammalian Na, K-ATPase β3 subunit. J. Biol. Chem. 1996, 271, 22754–22758. [Google Scholar] [CrossRef]
- Horiuchi, D.; Kusdra, L.; Huskey, N.E.; Chandriani, S.; Lenburg, M.E.; Gonzalez-Angulo, A.M.; Creasman, K.J.; Bazarov, A.V.; Smyth, J.W.; Davis, S.E. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med. 2012, 209, 679–696. [Google Scholar] [CrossRef]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bäuerle, T.; Wallwiener, M. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef]
- Keller, L.; Pantel, K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat. Rev. Cancer 2019, 19, 553–567. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Aranda, V.; Bardelli, A.; Blanpain, C.; Bock, C.; Borowski, C.; Caldas, C.; Califano, A.; Doherty, M.; Elsner, M. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015, 21, 846–853. [Google Scholar] [CrossRef]
- Gao, Y.; Ni, X.; Guo, H.; Su, Z.; Ba, Y.; Tong, Z.; Guo, Z.; Yao, X.; Chen, X.; Yin, J. Single-cell sequencing deciphers a convergent evolution of copy number alterations from primary to circulating tumor cells. Genome Res. 2017, 27, 1312–1322. [Google Scholar] [CrossRef]
- Ameri, K.; Luong, R.; Zhang, H.; Powell, A.; Montgomery, K.; Espinosa, I.; Bouley, D.; Harris, A.; Jeffrey, S. Circulating tumour cells demonstrate an altered response to hypoxia and an aggressive phenotype. Br. J. Cancer 2010, 102, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Savage, P.; Blanchet-Cohen, A.; Revil, T.; Badescu, D.; Saleh, S.M.; Wang, Y.-C.; Zuo, D.; Liu, L.; Bertos, N.R.; Munoz-Ramos, V. A targetable EGFR-dependent tumor-initiating program in breast cancer. Cell Rep. 2017, 21, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef]
- Pfoertner, S.; Jeron, A.; Probst-Kepper, M.; Guzman, C.A.; Hansen, W.; Westendorf, A.M.; Toepfer, T.; Schrader, A.J.; Franzke, A.; Buer, J. Signatures of human regulatory T cells: An encounter with old friends and new players. Genome Biol. 2006, 7, R54. [Google Scholar] [CrossRef]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.-L.; Gyorki, D.E.; Ward, T.; Partanen, A. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Thompson, E.W.; Williams, E.D. Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs 2007, 185, 7–19. [Google Scholar] [CrossRef]
- Prat, A.; Ellis, M.J.; Perou, C.M. Practical implications of gene-expression-based assays for breast oncologists. Nat. Rev. Clin. Oncol. 2012, 9, 48–57. [Google Scholar] [CrossRef]
- Huo, D.; Ikpatt, F.; Khramtsov, A.; Dangou, J.-M.; Nanda, R.; Dignam, J.; Zhang, B.; Grushko, T.; Zhang, C.; Oluwasola, O. Population differences in breast cancer: Survey in indigenous African women reveals over-representation of triple-negative breast cancer. J. Clin. Oncol. 2009, 27, 4515. [Google Scholar] [CrossRef]
- Love, R.R. Defining a global research agenda for breast cancer. Cancer 2008, 113, 2366–2371. [Google Scholar] [CrossRef]
- Hammond, M.E.H.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch. Pathol. Lab. Med. 2010, 134, e48–e72. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.H.; Schwartz, J.N.; Hagerty, K.L.; Allred, D.C.; Cote, R.J.; Dowsett, M.; Fitzgibbons, P.L.; Hanna, W.M.; Langer, A. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch. Pathol. Lab. Med. 2007, 131, 18–43. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Singh, A.K. Single-cell protein analysis. Curr. Opin. Biotechnol. 2012, 23, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Patani, N.; Martin, L.A.; Dowsett, M. Biomarkers for the clinical management of breast cancer: International perspective. Int. J. Cancer 2013, 133, 1–13. [Google Scholar] [CrossRef]
- Rosen, P.P. The pathological classification of human mammary carcinoma: Past, present and future. Ann. Clin. Lab. Sci. 1979, 9, 144–156. [Google Scholar]
- Van Bogaert, L. Recent progress in the histological typing of human breast tumours. Diagn. Histopathol. 1981, 4, 349–353. [Google Scholar]
- Sobin, L.H.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumours; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Balslev, I.; Axelsson, C.K.; Zedeler, K.; Rasmussen, B.B.; Carstensen, B.; Mouridsen, H.T. The Nottingham prognostic index applied to 9,149 patients from the studies of the Danish Breast Cancer Cooperative Group (DBCG). Breast Cancer Res. Treat. 1994, 32, 281–290. [Google Scholar] [CrossRef]
- Charles, M.P.; Therese, S.; Michael, B.E.; Matt, v.d.R.; Stefanie, S.J.; Christian, A.R.; Jonathan, R.P.; Douglas, T.R.; Hilde, J.; Lars, A.A. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar]
- Anderson, W.F.; Chatterjee, N.; Ershler, W.B.; Brawley, O.W. Estrogen receptor breast cancer phenotypes in the Surveillance, Epidemiology, and End Results database. Breast Cancer Res. Treat. 2002, 76, 27–36. [Google Scholar] [CrossRef]
- Byar, D.P.; Sears, M.E.; McGuire, W.L. Relationship between estrogen receptor values and clinical data in predicting the response to endocrine therapy for patients with advanced breast cancer. Eur. J. Cancer 1979, 15, 299–310. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Combinatorial biomarker expression in breast cancer. Breast Cancer Res. Treat. 2010, 120, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, K.B.; Koseki, Y.; McGuire, W.L. Estrogen control of progesterone receptor in human breast cancer: Role of estradiol and antiestrogen. Endocrinology 1978, 103, 1742–1751. [Google Scholar] [CrossRef] [PubMed]
- Viale, G.; Regan, M.M.; Maiorano, E.; Mastropasqua, M.G.; Dell’Orto, P.; Rasmussen, B.B.; Raffoul, J.; Neven, P.; Orosz, Z.; Braye, S. Prognostic and predictive value of centrally reviewed expression of estrogen and progesterone receptors in a randomized trial comparing letrozole and tamoxifen adjuvant therapy for postmenopausal early breast cancer: BIG 1-98. J. Clin. Oncol. 2007, 25, 3846–3852. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group. Tamoxifen for early breast cancer: An overview of the randomised trials. Lancet 1998, 351, 1451–1467. [Google Scholar] [CrossRef]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Chia, S.; Norris, B.; Speers, C.; Cheang, M.; Gilks, B.; Gown, A.M.; Huntsman, D.; Olivotto, I.A.; Nielsen, T.O.; Gelmon, K. Human epidermal growth factor receptor 2 overexpression as a prognostic factor in a large tissue microarray series of node-negative breast cancers. J. Clin. Oncol. 2008, 26, 5697–5704. [Google Scholar] [CrossRef]
- Rakha, E.A.; Abd El-Rehim, D.; Paish, C.; Green, A.R.; Lee, A.H.; Robertson, J.F.; Blamey, R.W.; Macmillan, D.; Ellis, I.O. Basal phenotype identifies a poor prognostic subgroup of breast cancer of clinical importance. Eur. J. Cancer 2006, 42, 3149–3156. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Macey, M.G.; Macey, M.G. Flow Cytometry; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Rubakhin, S.S.; Sweedler, J.V. Quantitative measurements of cell–cell signaling peptides with single-cell MALDI MS. Anal. Chem. 2008, 80, 7128–7136. [Google Scholar] [CrossRef]
- Spurrier, B.; Ramalingam, S.; Nishizuka, S. Reverse-phase protein lysate microarrays for cell signaling analysis. Nat. Protoc. 2008, 3, 1796–1808. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.-L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef]
- O’Neill, R.A.; Bhamidipati, A.; Bi, X.; Deb-Basu, D.; Cahill, L.; Ferrante, J.; Gentalen, E.; Glazer, M.; Gossett, J.; Hacker, K. Isoelectric focusing technology quantifies protein signaling in 25 cells. Proc. Natl. Acad. Sci. USA 2006, 103, 16153–16158. [Google Scholar] [CrossRef]
- Chokkalingam, V.; Tel, J.; Wimmers, F.; Liu, X.; Semenov, S.; Thiele, J.; Figdor, C.G.; Huck, W.T. Probing cellular heterogeneity in cytokine-secreting immune cells using droplet-based microfluidics. Lab Chip 2013, 13, 4740–4744. [Google Scholar] [CrossRef]
- Wu, M.; Perroud, T.D.; Srivastava, N.; Branda, C.S.; Sale, K.L.; Carson, B.D.; Patel, K.D.; Branda, S.S.; Singh, A.K. Microfluidically-unified cell culture, sample preparation, imaging and flow cytometry for measurement of cell signaling pathways with single cell resolution. Lab Chip 2012, 12, 2823–2831. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Routenberg Love, K.; Gong, Y.; Gierahn, T.M.; Love, J.C. Immuno-hybridization chain reaction for enhancing detection of individual cytokine-secreting human peripheral mononuclear cells. Anal. Chem. 2011, 83, 6890–6895. [Google Scholar] [CrossRef]
- Ma, C.; Fan, R.; Ahmad, H.; Shi, Q.; Comin-Anduix, B.; Chodon, T.; Koya, R.C.; Liu, C.-C.; Kwong, G.A.; Radu, C.G. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat. Med. 2011, 17, 738–743. [Google Scholar] [CrossRef]
- Hughes, A.J.; Herr, A.E. Microfluidic western blotting. Proc. Natl. Acad. Sci. USA 2012, 109, 21450–21455. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.J.; Spelke, D.P.; Xu, Z.; Kang, C.-C.; Schaffer, D.V.; Herr, A.E. Single-cell western blotting. Nat. Methods 2014, 11, 749–755. [Google Scholar] [CrossRef]
- Duncombe, T.A.; Kang, C.C.; Maity, S.; Ward, T.M.; Pegram, M.D.; Murthy, N.; Herr, A.E. Hydrogel Pore-Size Modulation for Enhanced Single-Cell Western Blotting. Adv. Mater. 2016, 28, 327–334. [Google Scholar] [CrossRef]
- Zardavas, D.; Baselga, J.; Piccart, M. Emerging targeted agents in metastatic breast cancer. Nat. Rev. Clin. Oncol. 2013, 10, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Gumuscu, B.; Herr, A.E. Separation-encoded microparticles for single-cell western blotting. Lab Chip 2020, 20, 64–73. [Google Scholar] [CrossRef]
- Beech, J.P.; Holm, S.H.; Adolfsson, K.; Tegenfeldt, J.O. Sorting cells by size, shape and deformability. Lab Chip 2012, 12, 1048–1051. [Google Scholar] [CrossRef]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen–estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef]
- Manna, S.; Holz, M.K. Tamoxifen action in ER-negative breast cancer. Signal Transduct. Insights 2016, 5, STI-S29901. [Google Scholar] [CrossRef]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen signaling multiple pathways to impact gene transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef]
- Li, L.; Haynes, M.P.; Bender, J.R. Plasma membrane localization and function of the estrogen receptor α variant (ER46) in human endothelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4807–4812. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. A variant of estrogen receptor-α, hER-α36: Transduction of estrogen-and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 9063–9068. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, M.; Syed Khaja, A.S.; Aleskandarany, M.; Karlsson, R.; Althobiti, M.; Ødum, N.; Mongan, N.P.; Dizeyi, N.; Johnson, H.; Green, A.R. The role of PIP5K1α/pAKT and targeted inhibition of growth of subtypes of breast cancer using PIP5K1α inhibitor. Oncogene 2019, 38, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef]
- Washburn, M.P.; Koller, A.; Oshiro, G.; Ulaszek, R.R.; Plouffe, D.; Deciu, C.; Winzeler, E.; Yates III, J.R. Protein pathway and complex clustering of correlated mRNA and protein expression analyses in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2003, 100, 3107–3112. [Google Scholar] [CrossRef]
- Fehm, T.; Becker, S.; Duerr-Stoerzer, S.; Sotlar, K.; Mueller, V.; Wallwiener, D.; Lane, N.; Solomayer, E.; Uhr, J. Determination of HER2 status using both serum HER2 levels and circulating tumor cells in patients with recurrent breast cancer whose primary tumor was HER2 negative or of unknown HER2 status. Breast Cancer Res. 2007, 9, R74. [Google Scholar] [CrossRef] [PubMed]
- Sinkala, E.; Sollier-Christen, E.; Renier, C.; Rosas-Canyelles, E.; Che, J.; Heirich, K.; Duncombe, T.A.; Vlassakis, J.; Yamauchi, K.A.; Huang, H. Profiling protein expression in circulating tumour cells using microfluidic western blotting. Nat. Commun. 2017, 8, 14622. [Google Scholar] [CrossRef]
- Abdulla, A.; Zhang, T.; Ahmad, K.Z.; Li, S.; Lou, J.; Ding, X. Label-free Separation of Circulating Tumor Cells Using a Self-Amplified Inertial Focusing (SAIF) Microfluidic Chip. Anal. Chem. 2020, 92, 16170–16179. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, A.; Zhang, Z.; Ahmad, K.Z.; Warden, A.R.; Li, H.; Ding, X. Rapid and efficient capturing of circulating tumor cells from breast cancer Patient’s whole blood via the antibody functionalized microfluidic (AFM) chip. Biosens. Bioelectron. 2022, 201, 113965. [Google Scholar] [CrossRef]
- Abdulla, A.; Zhang, T.; Li, S.; Guo, W.; Warden, A.R.; Xin, Y.; Maboyi, N.; Lou, J.; Xie, H.; Ding, X. Integrated microfluidic single-cell immunoblotting chip enables high-throughput isolation, enrichment and direct protein analysis of circulating tumor cells. Microsyst. Nanoeng. 2022, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Abdulla, A.; Wang, A.; Warden, A.R.; Ahmad, K.Z.; Xin, Y.; Ding, X. Sickle-like Inertial Microfluidic System for Online Rare Cell Separation and Tandem Label-Free Quantitative Proteomics (Orcs-Proteomics). Anal. Chem. 2022, 94, 6026–6035. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D. A single-cell atlas of the tumor and immune ecosystem of human breast cancer. Cell 2019, 177, 1330–1345.e1318. [Google Scholar] [CrossRef]
- Shahi, P.; Kim, S.C.; Haliburton, J.R.; Gartner, Z.J.; Abate, A.R. Abseq: Ultrahigh-throughput single cell protein profiling with droplet microfluidic barcoding. Sci. Rep. 2017, 7, 44447. [Google Scholar] [CrossRef]
- Yu, Y.; Dang, J.; Liu, X.; Wang, L.; Li, S.; Zhang, T.; Ding, X. Metal-Labeled Aptamers as Novel Nanoprobes for Imaging Mass Cytometry Analysis. Anal. Chem. 2020, 92, 6312–6320. [Google Scholar] [CrossRef]
- Dang, J.; Li, H.; Zhang, L.; Li, S.; Zhang, T.; Huang, S.; Li, Y.; Huang, C.; Ke, Y.; Shen, G.; et al. New Structure Mass Tag based on Zr-NMOF for Multiparameter and Sensitive Single-Cell Interrogating in Mass Cytometry. Adv Mater 2021, 33, e2008297. [Google Scholar] [CrossRef]
- van den Brink, S.C.; Sage, F.; Vértesy, Á.; Spanjaard, B.; Peterson-Maduro, J.; Baron, C.S.; Robin, C.; Van Oudenaarden, A. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 2017, 14, 935–936. [Google Scholar] [CrossRef] [PubMed]
- Alexovič, M.; Sabo, J.; Longuespée, R. Automation of single-cell proteomic sample preparation. Proteomics 2021, 21, 2100198. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, T.; Tan, Z.; Warden, A.R.; Li, S.; Cheung, E.; Ding, X. A Hashing-Based Framework for Enhancing Cluster Delineation of High-Dimensional Single-Cell Profiles. Phenomics 2022, 2, 323–335. [Google Scholar] [CrossRef]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Irmisch, A.; Bonilla, X.; Chevrier, S.; Lehmann, K.-V.; Singer, F.; Toussaint, N.C.; Esposito, C.; Mena, J.; Milani, E.S.; Casanova, R. The Tumor Profiler Study: Integrated, multi-omic, functional tumor profiling for clinical decision support. Cancer Cell 2021, 39, 288–293. [Google Scholar] [CrossRef]
- Nevedomskaya, E.; Haendler, B. From Omics to Multi-Omics Approaches for In-Depth Analysis of the Molecular Mechanisms of Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 6281. [Google Scholar] [CrossRef]
- Yu, T.J.; Ma, D.; Liu, Y.Y.; Xiao, Y.; Gong, Y.; Jiang, Y.Z.; Shao, Z.M.; Hu, X.; Di, G.H. Bulk and single-cell transcriptome profiling reveal the metabolic heterogeneity in human breast cancers. Mol. Ther. 2021, 29, 2350–2365. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Bakker, O.B.; Aguirre-Gamboa, R.; Sanna, S.; Oosting, M.; Smeekens, S.P.; Jaeger, M.; Zorro, M.; Võsa, U.; Withoff, S.; Netea-Maier, R.T. Integration of multi-omics data and deep phenotyping enables prediction of cytokine responses. Nat. Immunol. 2018, 19, 776–786. [Google Scholar] [CrossRef]
- Rosàs-Canyelles, E.; Modzelewski, A.J.; Geldert, A.; He, L.; Herr, A.E. Multimodal detection of protein isoforms and nucleic acids from mouse pre-implantation embryos. Nat. Protoc. 2021, 16, 1062–1088. [Google Scholar] [CrossRef] [PubMed]
- Sammut, S.-J.; Crispin-Ortuzar, M.; Chin, S.-F.; Provenzano, E.; Bardwell, H.A.; Ma, W.; Cope, W.; Dariush, A.; Dawson, S.-J.; Abraham, J.E.; et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 2022, 601, 623–629. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Discovery | Involved Process | Mutation Risk | Reference |
|---|---|---|---|---|
| PTEN | 1997 | apoptosis, cell cycle, and signal transduction | activation of proliferation and survival signals | [23] |
| STK11 | 1997 | cell cycle, metabolism, and energy balance | activation of cell proliferation and metabolic pathways | [24] |
| CHEK2 | 1999 | DNA repair and cell apoptosis | impairments in DNA repair and cell apoptosis processes | [25] |
| PIK3CA | 2004 | regulation of signaling pathway | activation of survival signals | [26] |
| AKT1 | 2007 | regulation of signaling pathway | activation of cell proliferation and survival signals | [27] |
| BARD1 | 2010 | DNA repair and cell apoptosis | increased susceptibility to breast cancer | [28] |
| NF1 | 2015 | regulation of signaling pathway | increased rate of developing breast cancer | [29] |
| Omics Field | Analysis Type | Advantages | Limitations | Refs. |
|---|---|---|---|---|
| Genomics | Bulk | Lower cost; matured analytical methods; provides comprehensive sequence information. | Averages over cell populations; misses information about rare cell populations; limited prediction of the ultimate biological effect. | [19,22,23,24,25] |
| Single cell | Detects mutations and structural variations in individual cells; highlights cell-to-cell heterogeneity and rare cell populations; enables study of intra-tumoral heterogeneity in cancer. | Requires substantial sequencing depth for accurate results; higher costs; greater complexity of data analysis; limited information on the ultimate biological effect. | [7,43,44,45] | |
| Transcriptomics | Bulk | Lower cost; matured techniques and analytical methods; global expression analysis; detects all splice variants. | Averages over cell populations; misses cell-to-cell heterogeneity; only represents an intermediate step; correlation with protein levels is not always linear. | [32,57,58,59,82,83,84] |
| Single cell | Captures cell-to-cell variability in gene expression; detects all splice variants; sensitive, high dynamic range, and quantitative; parses cell-specific transcriptomes in single-cell experiments. | Data can be noisy; more complex data analysis; only represents an intermediate step; correlation with protein levels is not always linear. | [107,110,116,117,118] | |
| Proteomics | Bulk | Comprehensive coverage of the proteome; mature techniques; resolves the final regulatory level. | Averages over cell populations; less sensitivity to low-abundance proteins; certain proteins difficult to isolate; high dynamic range of proteome makes detection difficult. | [137,141,145,147] |
| Single cell | Potential to capture protein-level heterogeneity across individual cells; proteins are the main effectors of cellular function. | Technically challenging; limited coverage of the proteome; less mature techniques; certain proteins difficult to isolate; high dynamic range of proteome makes detection difficult; post-translational modifications may greatly influence activity but can be challenging to analyze. | [160,161,162,163,170,175,180] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Z.; Jiang, L.; Ding, X. Advancing Breast Cancer Heterogeneity Analysis: Insights from Genomics, Transcriptomics and Proteomics at Bulk and Single-Cell Levels. Cancers 2023, 15, 4164. https://doi.org/10.3390/cancers15164164
Zhu Z, Jiang L, Ding X. Advancing Breast Cancer Heterogeneity Analysis: Insights from Genomics, Transcriptomics and Proteomics at Bulk and Single-Cell Levels. Cancers. 2023; 15(16):4164. https://doi.org/10.3390/cancers15164164
Chicago/Turabian StyleZhu, Zijian, Lai Jiang, and Xianting Ding. 2023. "Advancing Breast Cancer Heterogeneity Analysis: Insights from Genomics, Transcriptomics and Proteomics at Bulk and Single-Cell Levels" Cancers 15, no. 16: 4164. https://doi.org/10.3390/cancers15164164
APA StyleZhu, Z., Jiang, L., & Ding, X. (2023). Advancing Breast Cancer Heterogeneity Analysis: Insights from Genomics, Transcriptomics and Proteomics at Bulk and Single-Cell Levels. Cancers, 15(16), 4164. https://doi.org/10.3390/cancers15164164