
Proteomic Mapping of the Interactome of KRAS Mutants Identifies New Features of RAS Signalling Networks and the Mechanism of Action of Sotorasib


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Immunoblotting, Antibodies and Reagents
2.3. Constructs and siRNA
2.4. Immunoprecipitation (IP) and in Beads Tryptic Digestion
2.5. Mass Spectrometry Analysis of Affinity Purification-MS (AP-MS)
2.6. Analysis of AP-MS Data
2.7. Transformation Assay
2.8. Migration Assay
2.9. Survival Curves
2.10. Statistics
3. Results
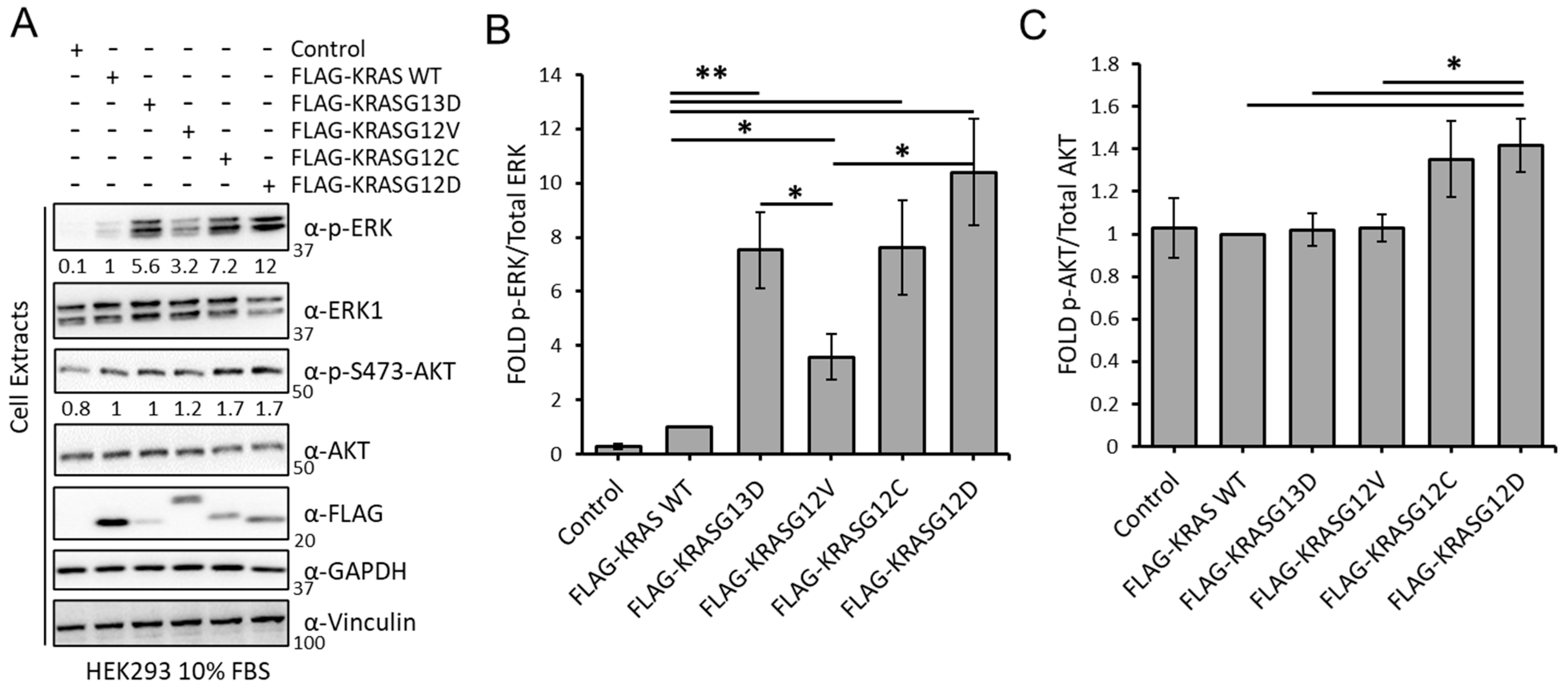
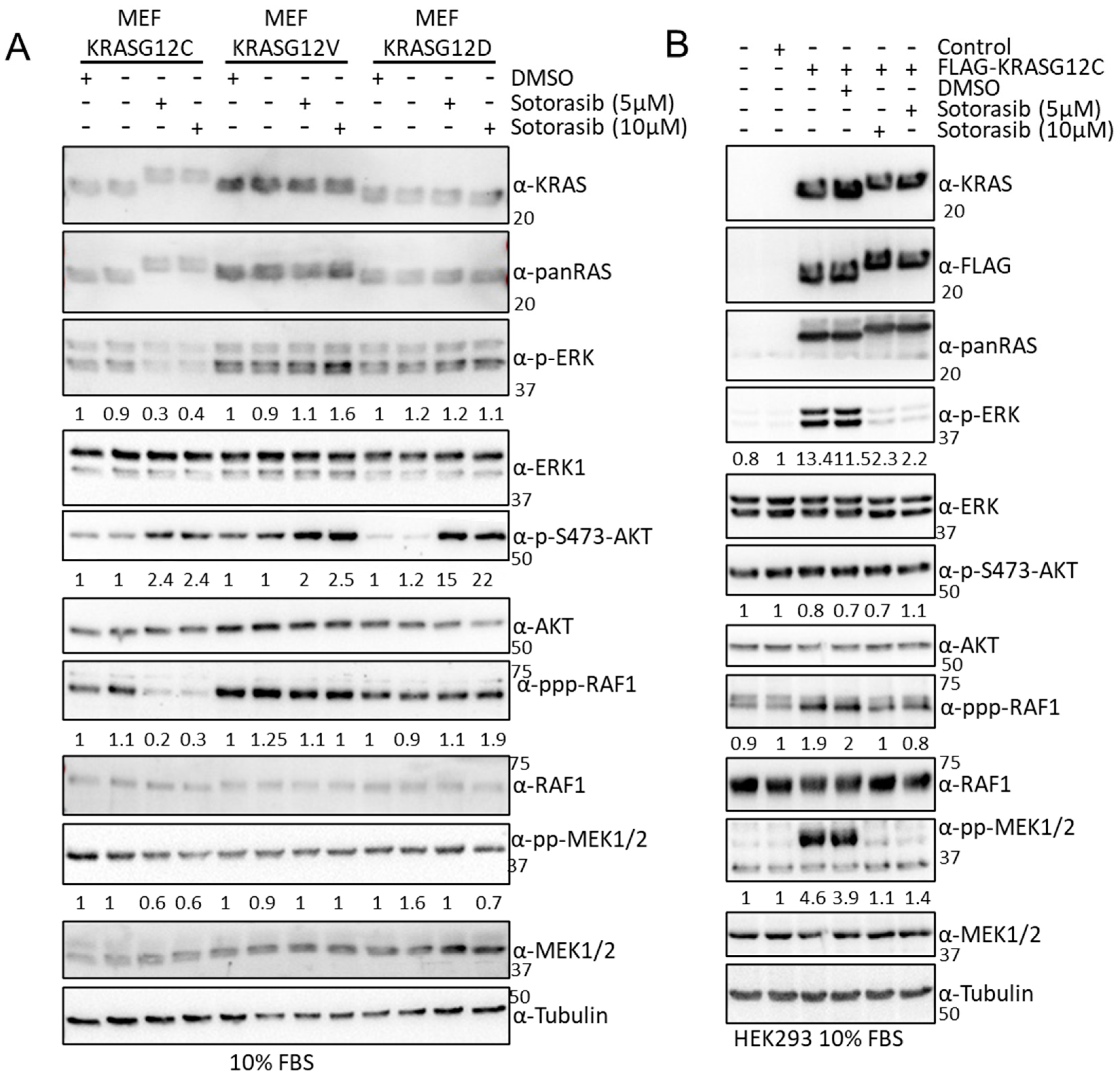
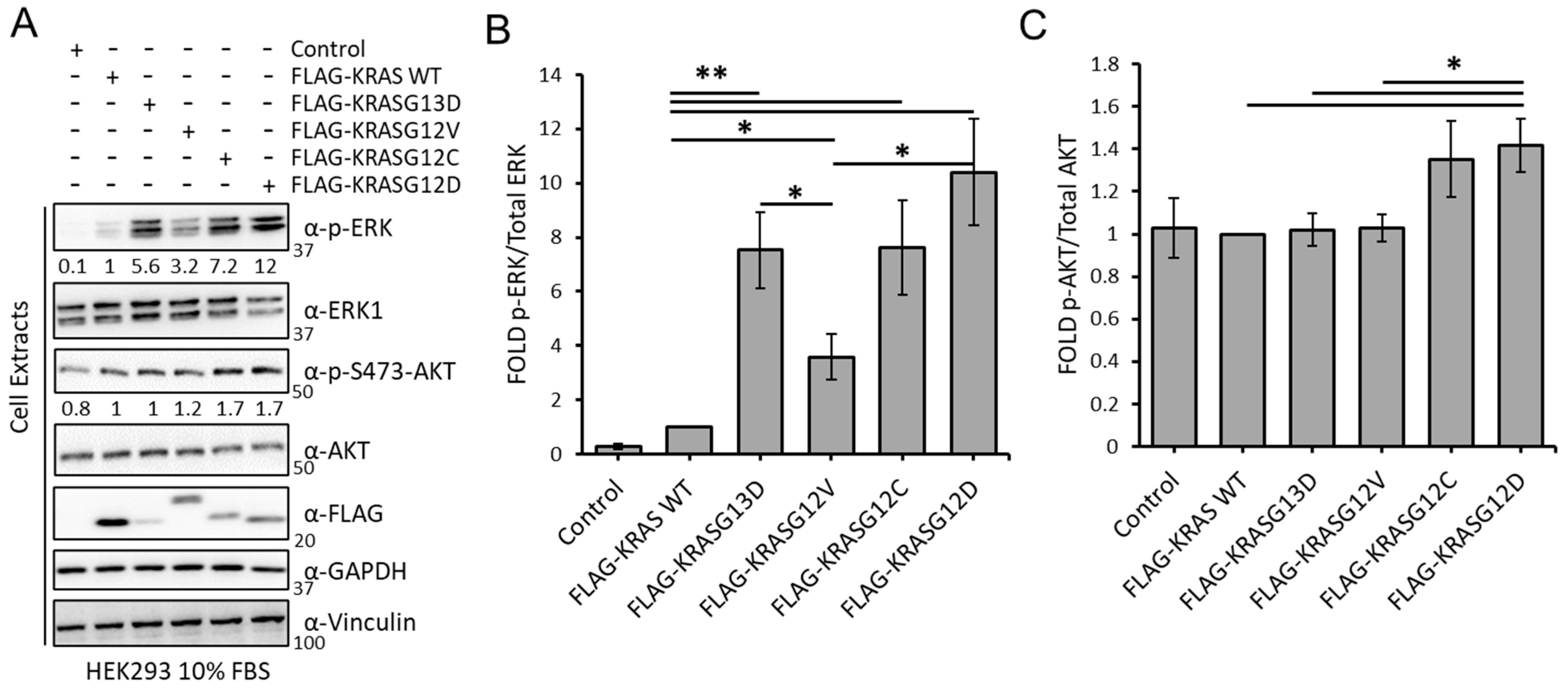
3.1. KRAS Mutants Differentially Regulate the RAF and AKT Effector Pathways in HEK293 Cells
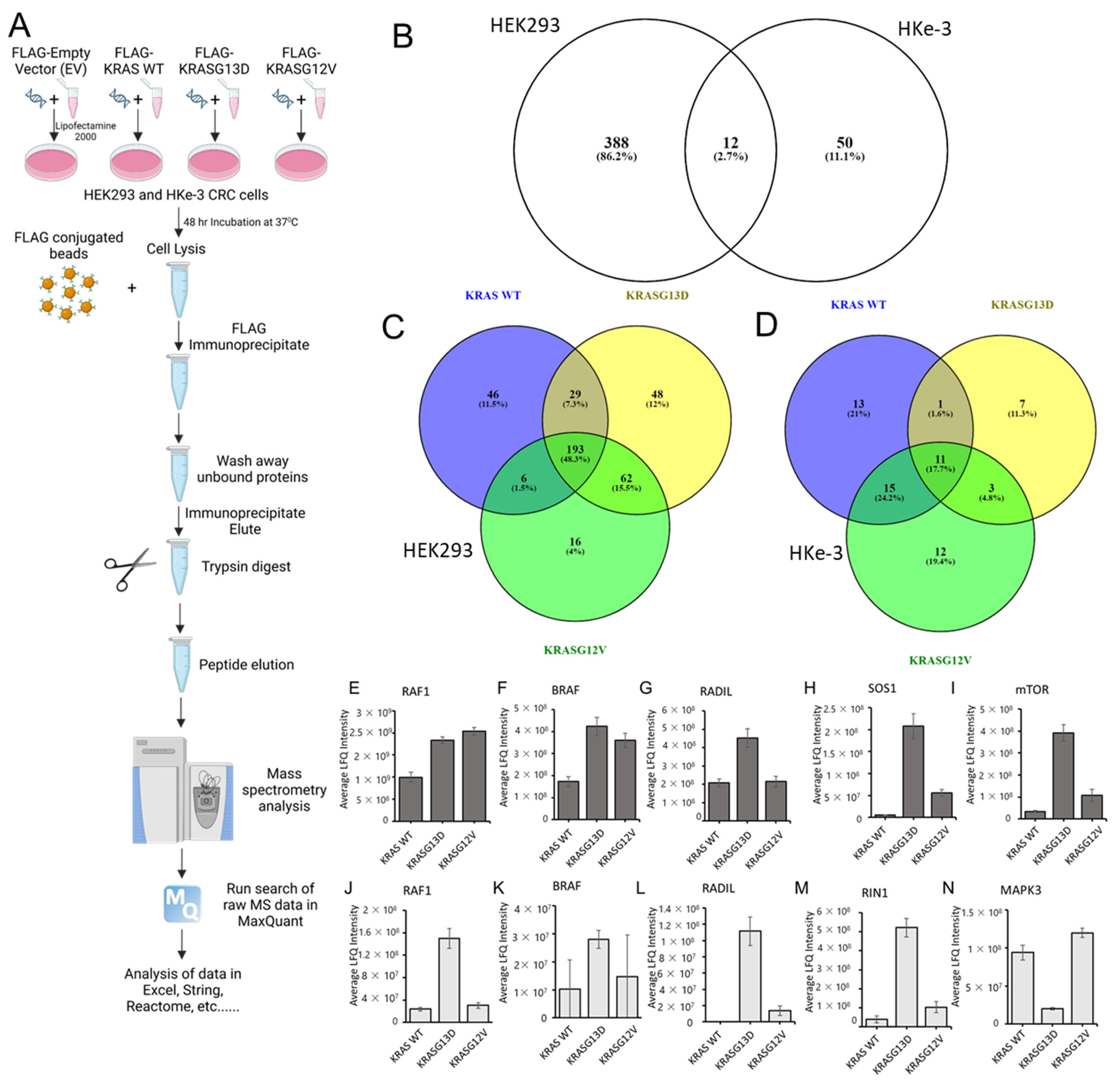
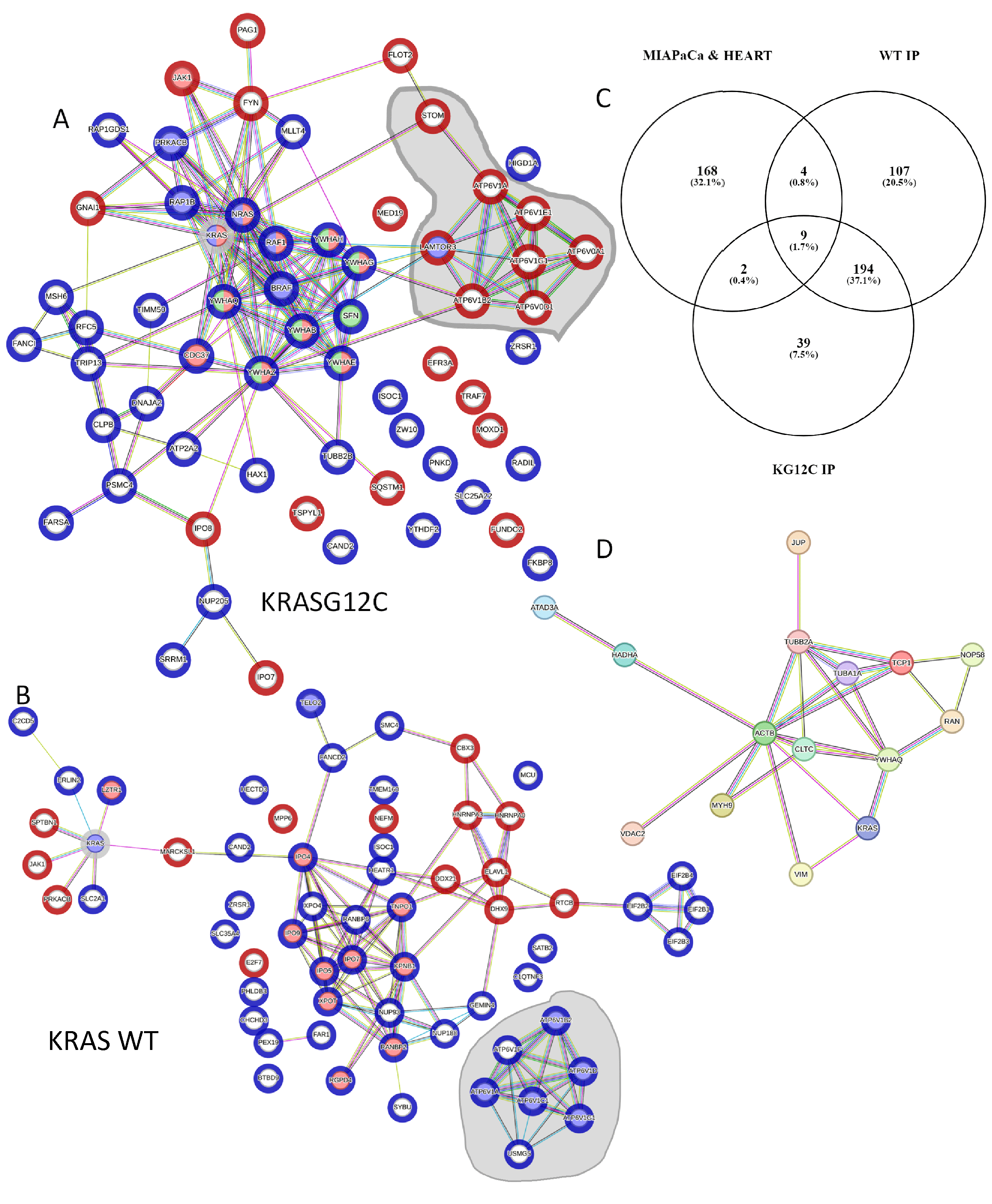
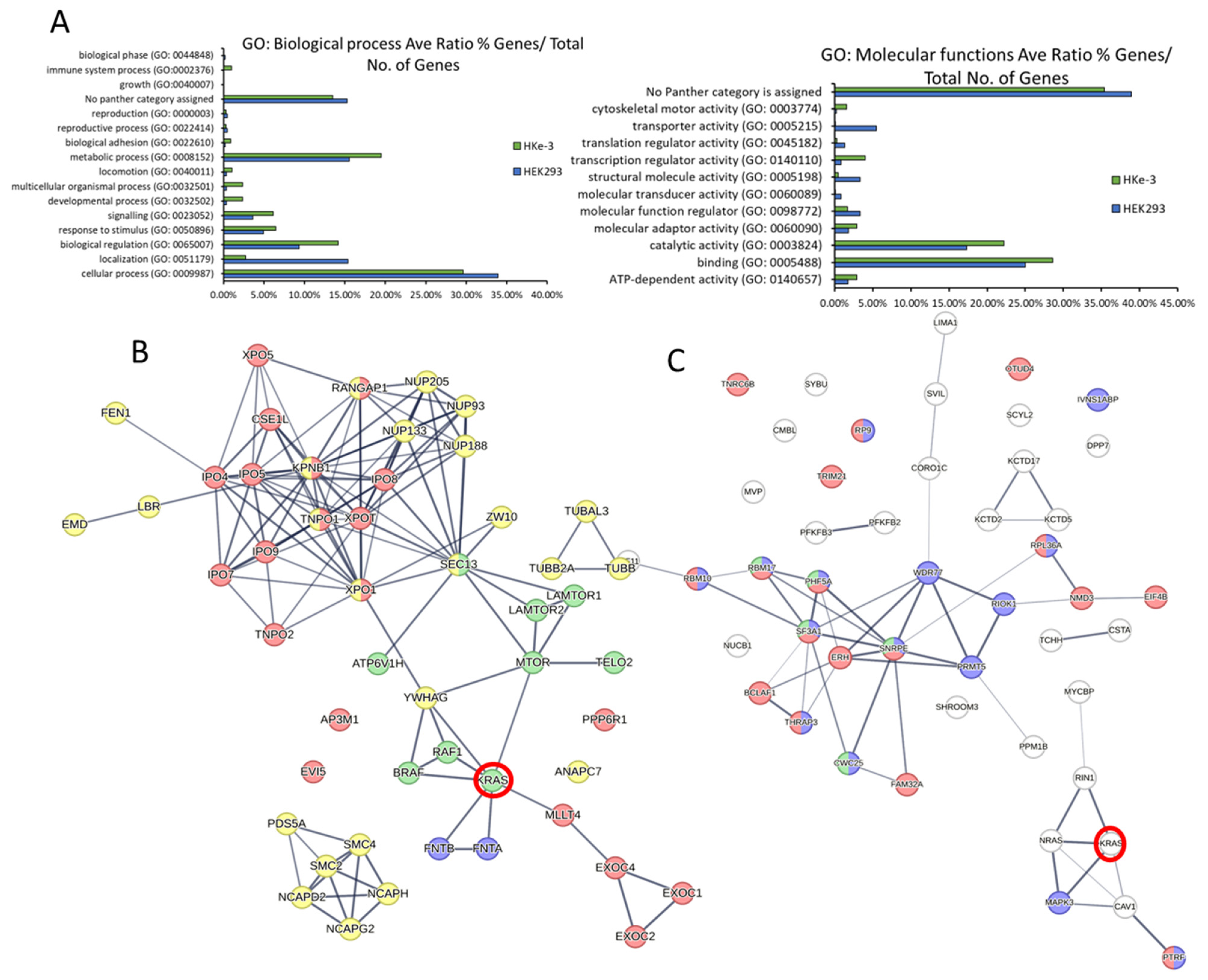
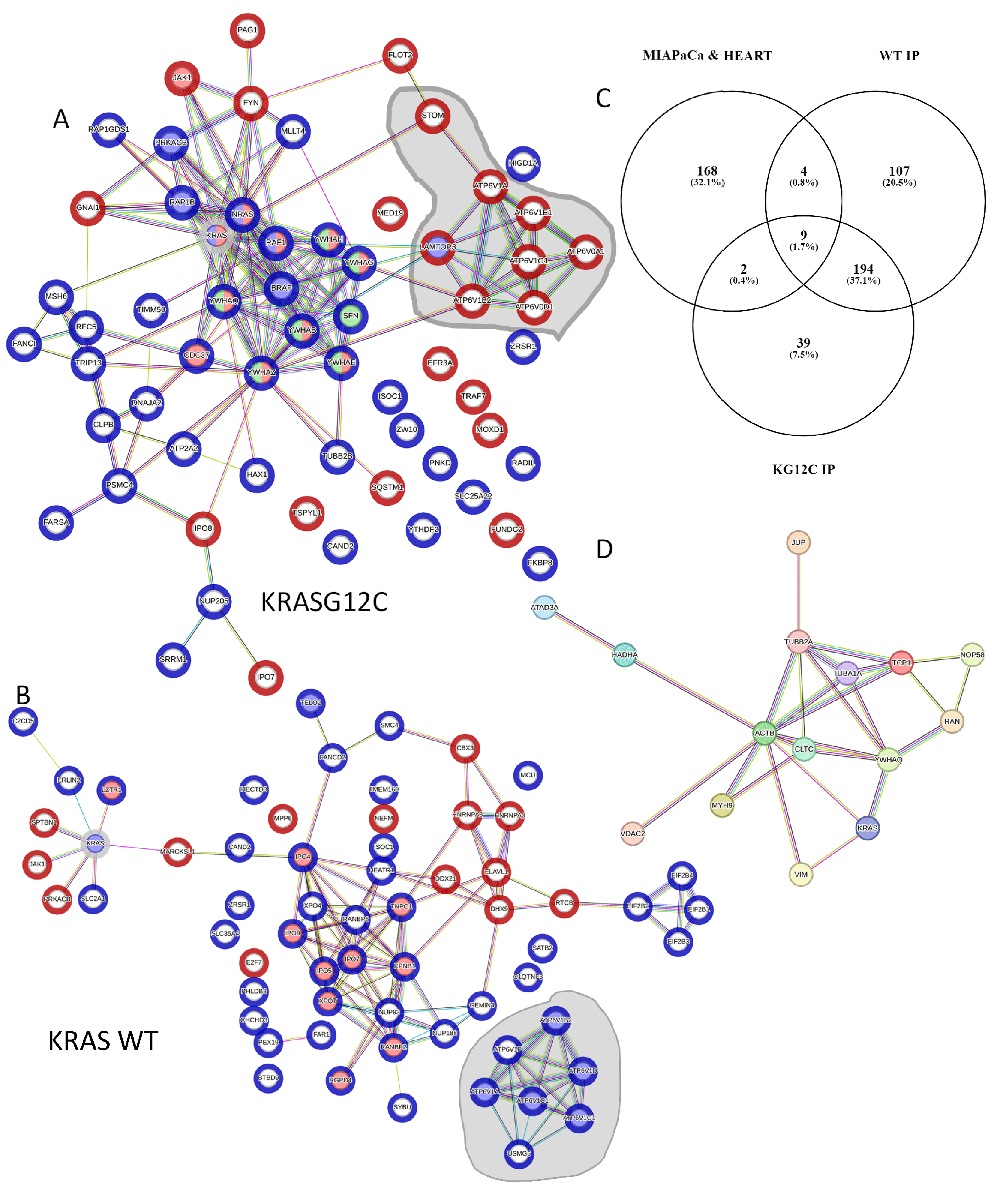
3.2. The KRAS Proteins Interactome Differs between Cell Types
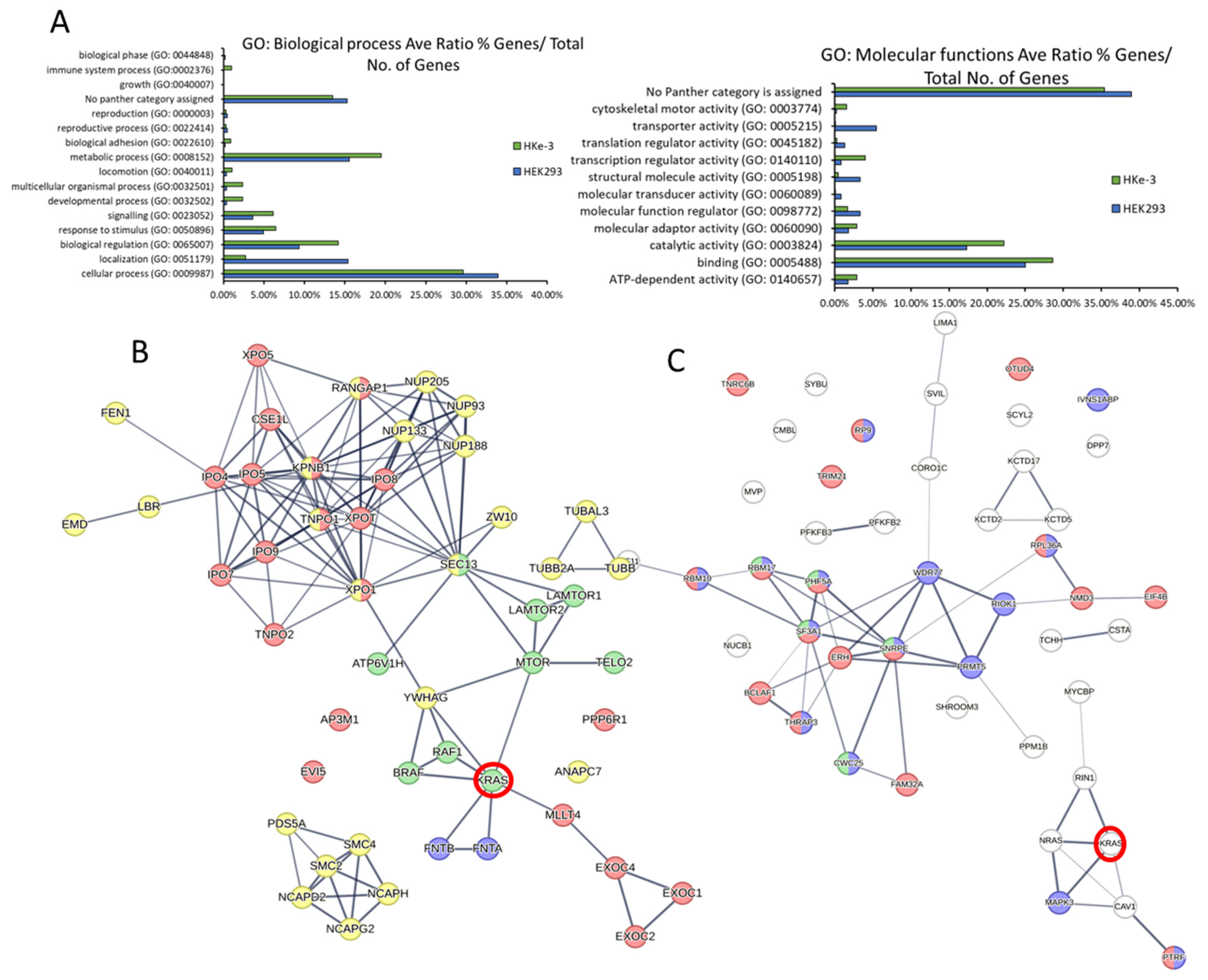
3.3. Bioinformatic Characterisation of KRAS Protein Signalling Networks Show Correlation between KRAS Protein Interactomes and Function
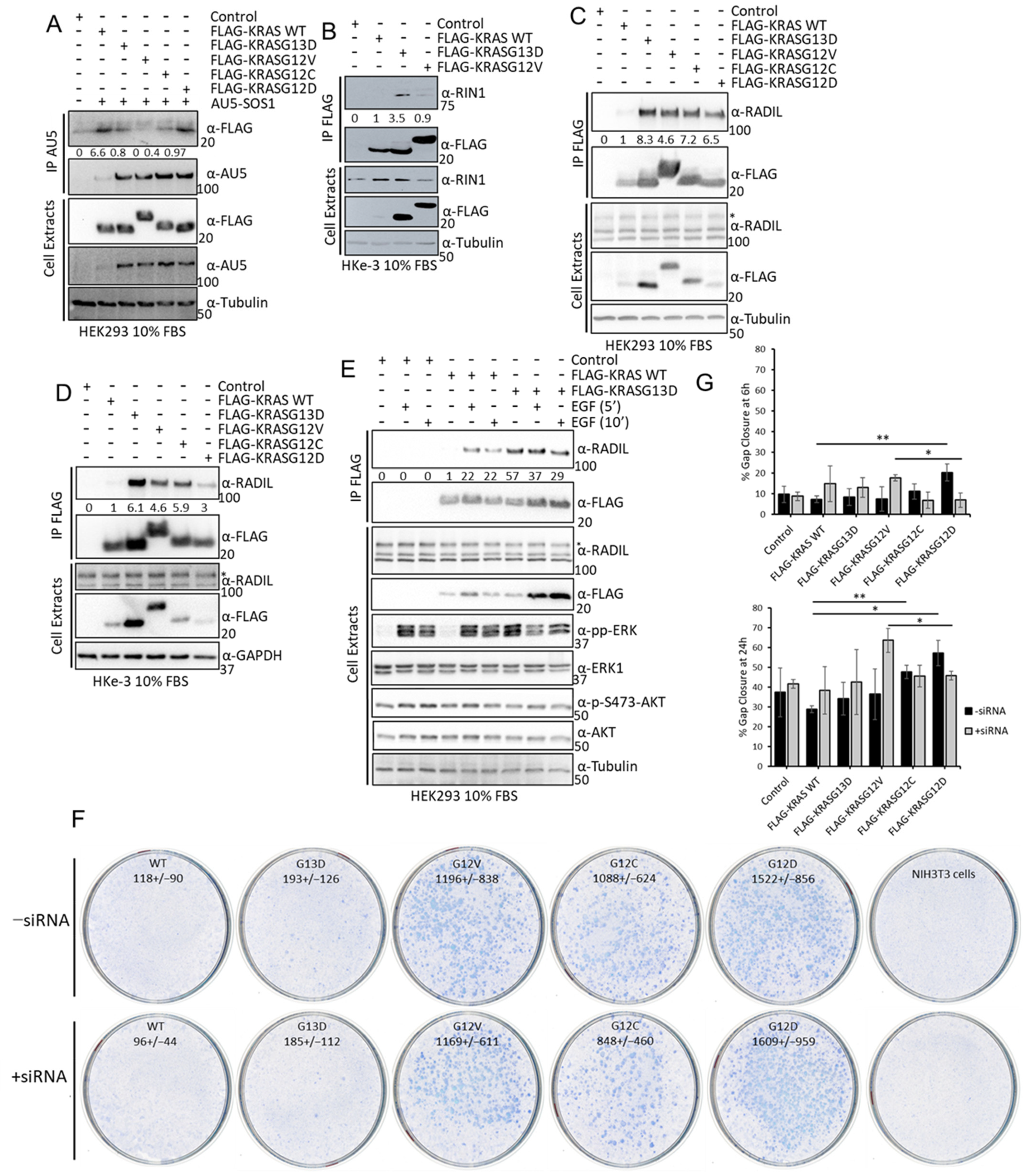
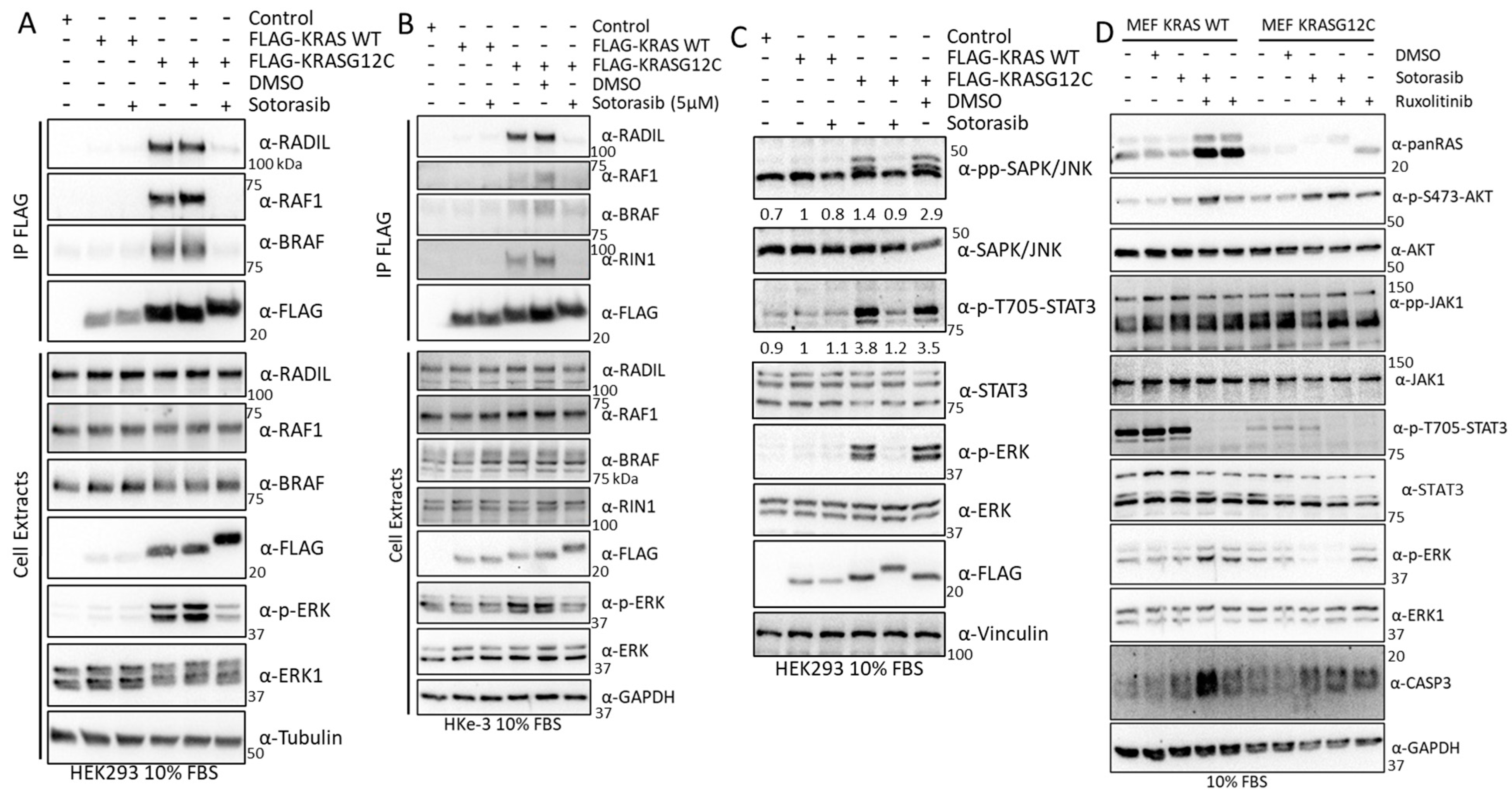
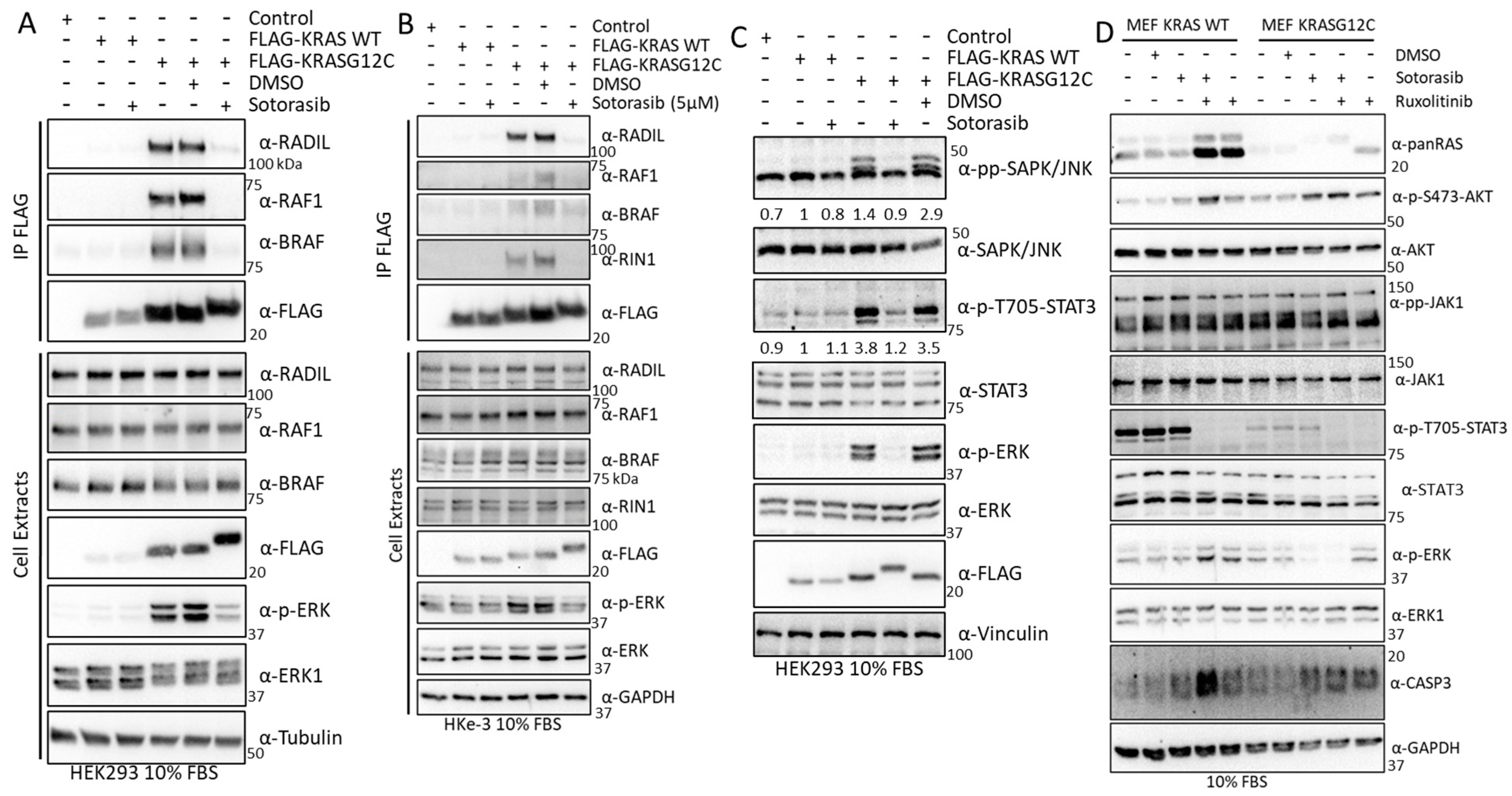
3.4. Experimental Validation Confirms That KRAS Proteins Interactomes Are Differentially Regulated by KRAS Point Mutation and Shows That RADIL Mediates KRAS-Dependent Migration in a Mutant Specific Manner
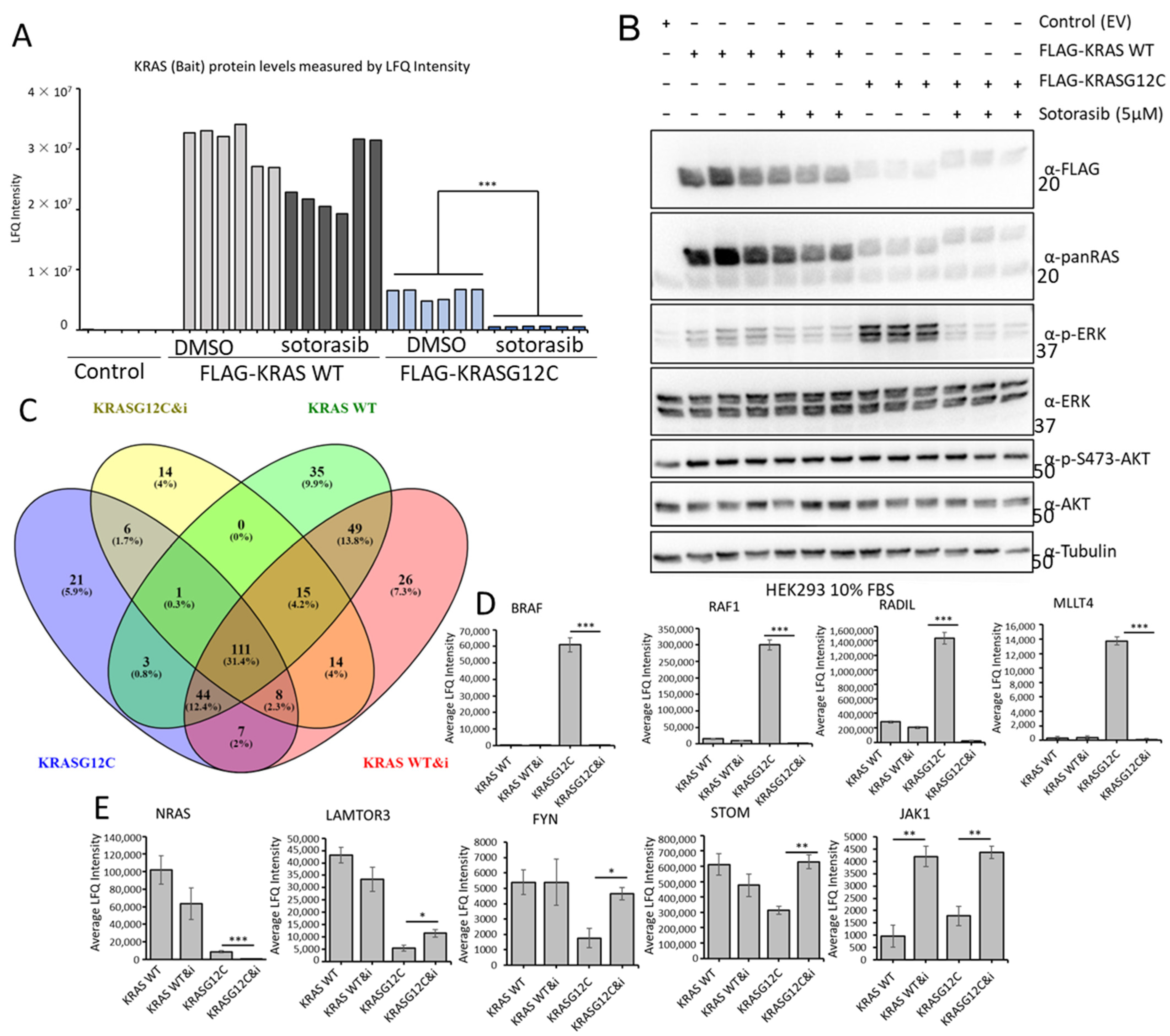
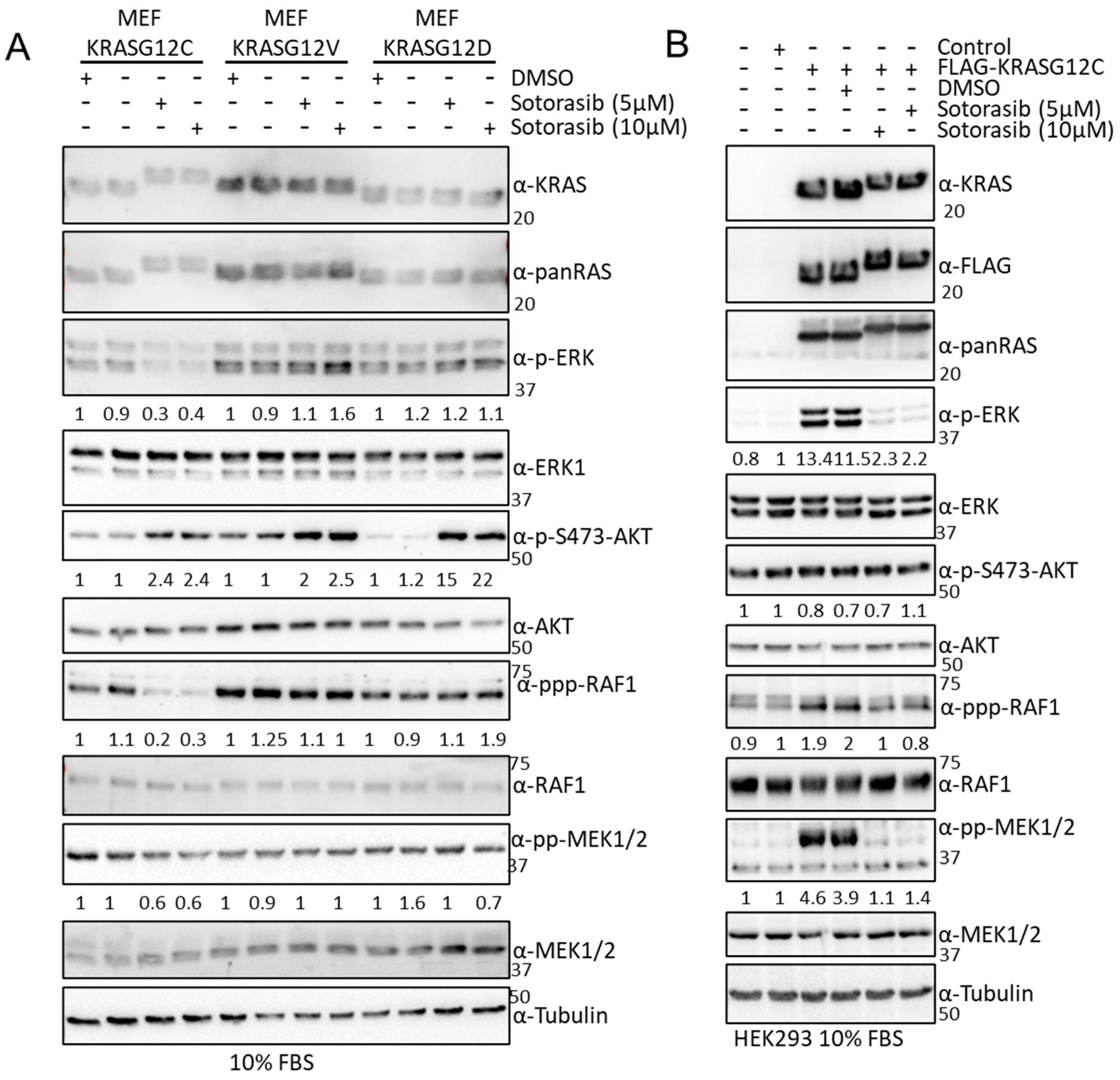
3.5. Sotorasib Shows Off-Target Effects in Cells That Do Not Express KRASG12C
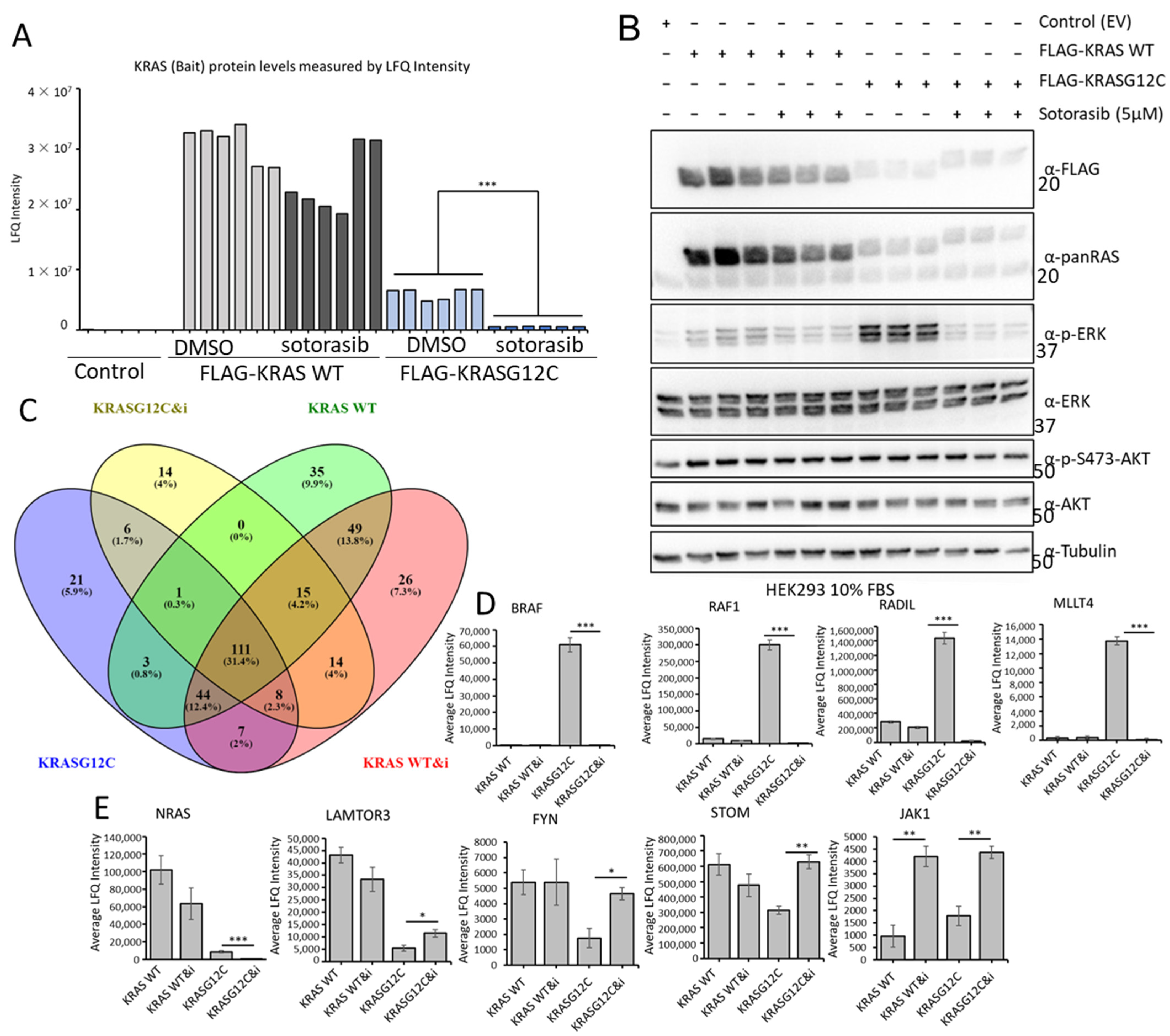
3.6. Sotorasib Regulates the KRASG12C Interactome and Has Unspecific Effects on the KRAS WT Interactome
3.7. Sotorasib Regulates KRASG12C and WT Signalling Networks
3.8. Validation Experiments Show a New Sotorasib-Regulated Crosstalk of RAS Signalling with JAK1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barbacid, M. Ras genes. Annu. Rev. Biochem. 1987, 56, 779–827. [Google Scholar] [CrossRef] [PubMed]
- Fey, D.; Matallanas, D.; Rauch, J.; Rukhlenko, O.S.; Kholodenko, B.N. The complexities and versatility of the RAS-to-ERK signalling system in normal and cancer cells. Semin. Cell Dev. Biol. 2016, 58, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Buday, L.; Downward, J. Many faces of Ras activation. Biochim. Biophys. Acta 2008, 1786, 178–187. [Google Scholar] [CrossRef]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Menzies, G.E.; Prior, I.A.; Brancale, A.; Reed, S.H.; Lewis, P.D. Carcinogen-induced DNA structural distortion differences in the RAS gene isoforms; the importance of local sequence. BMC Chem. 2021, 15, 51. [Google Scholar] [CrossRef]
- Smith, M.J.; Neel, B.G.; Ikura, M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc. Natl. Acad. Sci. USA 2013, 110, 4574–4579. [Google Scholar] [CrossRef]
- Stolze, B.; Reinhart, S.; Bulllinger, L.; Frohling, S.; Scholl, C. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci. Rep. 2015, 5, 8535. [Google Scholar] [CrossRef]
- Huang, H.; Daniluk, J.; Liu, Y.; Chu, J.; Li, Z.; Ji, B.; Logsdon, C.D. Oncogenic K-Ras requires activation for enhanced activity. Oncogene 2014, 33, 532–535. [Google Scholar] [CrossRef]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef]
- Kiel, C.; Matallanas, D.; Kolch, W. The Ins and Outs of RAS Effector Complexes. Biomolecules 2021, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Medarde, A.; De Las Rivas, J.; Santos, E. 40 Years of RAS-A Historic Overview. Genes 2021, 12, 681. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Rosen, J.C.; Sacher, A.; Tsao, M.S. Direct GDP-KRAS(G12C) inhibitors and mechanisms of resistance: The tip of the iceberg. Ther. Adv. Med. Oncol. 2023, 15, 17588359231160141. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, B.; Xu, C.; Zhan, D.; Zhao, S.; Wu, H.; Liu, M.; Lan, X.; Cai, D.; Ding, Q.; et al. Global profiling of AMG510 modified proteins identified tumor suppressor KEAP1 as an off-target. iScience 2023, 26, 106080. [Google Scholar] [CrossRef]
- Smith, G.; Bounds, R.; Wolf, H.; Steele, R.J.; Carey, F.A.; Wolf, C.R. Activating K-Ras mutations outwith ’hotspot’ codons in sporadic colorectal tumours—Implications for personalised cancer medicine. Br. J. Cancer 2010, 102, 693–703. [Google Scholar] [CrossRef]
- De Roock, W.; Jonker, D.J.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Tu, D.; Siena, S.; Lamba, S.; Arena, S.; Frattini, M.; Piessevaux, H.; et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010, 304, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Dhawahir, A.; Sum, E.Y.; Urosevic, J.; Lechuga, C.G.; Esteban, L.M.; Castellano, E.; Guerra, C.; Santos, E.; Barbacid, M. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010, 29, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef]
- Romano, D.; Maccario, H.; Doherty, C.; Quinn, N.P.; Kolch, W.; Matallanas, D. The differential effects of wild-type and mutated K-Ras on MST2 signaling are determined by K-Ras activation kinetics. Mol. Cell. Biol. 2013, 33, 1859–1868. [Google Scholar] [CrossRef]
- Matallanas, D.; Arozarena, I.; Berciano, M.T.; Aaronson, D.S.; Pellicer, A.; Lafarga, M.; Crespo, P. Differences on the inhibitory specificities of H-Ras, K-Ras, and N-Ras (N17) dominant negative mutants are related to their membrane microlocalization. J. Biol. Chem. 2003, 278, 4572–4581. [Google Scholar] [CrossRef]
- Romano, D.; Nguyen, L.K.; Matallanas, D.; Halasz, M.; Doherty, C.; Kholodenko, B.N.; Kolch, W. Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nat. Cell. Biol. 2014, 16, 673–684. [Google Scholar] [CrossRef]
- Turriziani, B.; Garcia-Munoz, A.; Pilkington, R.; Raso, C.; Kolch, W.; von Kriegsheim, A. On-beads digestion in conjunction with data-dependent mass spectrometry: A shortcut to quantitative and dynamic interaction proteomics. Biology 2014, 3, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Bache, N.; Geyer, P.E.; Bekker-Jensen, D.B.; Hoerning, O.; Falkenby, L.; Treit, P.V.; Doll, S.; Paron, I.; Muller, J.B.; Meier, F.; et al. A Novel LC System Embeds Analytes in Pre-formed Gradients for Rapid, Ultra-robust Proteomics. Mol. Cell. Proteom. 2018, 17, 2284–2296. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.; Wynne, K.; Moldenhauer, E.; Clarke, P.; Maguire, C.; Bollard, S.; Yin, X.; Brennan, L.; Mooney, L.; Fitzsimons, S.; et al. A comparative analysis of extracellular vesicles (EVs) from human and feline plasma. Sci. Rep. 2022, 12, 10851. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Rodriguez, J.; Pilkington, R.; Garcia Munoz, A.; Nguyen, L.K.; Rauch, N.; Kennedy, S.; Monsefi, N.; Herrero, A.; Taylor, C.T.; von Kriegsheim, A. Substrate-Trapped Interactors of PHD3 and FIH Cluster in Distinct Signaling Pathways. Cell. Rep. 2016, 14, 2745–2760. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Aaronson, S.A.; Todaro, G.J.; Freeman, A.E. Human sarcoma cells in culture. Identification by colony-forming ability on monolayers of normal cells. Exp. Cell. Res. 1970, 61, 1–5. [Google Scholar] [CrossRef]
- Herrero, A.; Reis-Cardoso, M.; Jimenez-Gomez, I.; Doherty, C.; Agudo-Ibanez, L.; Pinto, A.; Calvo, F.; Kolch, W.; Crespo, P.; Matallanas, D. Characterisation of HRas local signal transduction networks using engineered site-specific exchange factors. Small GTPases 2017, 11, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Cappiello, F.; Casciaro, B.; Mangoni, M.L. A Novel In Vitro Wound Healing Assay to Evaluate Cell Migration. J. Vis. Exp. 2018, 133, e56825. [Google Scholar] [CrossRef]
- Kelly, M.R.; Kostyrko, K.; Han, K.; Mooney, N.A.; Jeng, E.E.; Spees, K.; Dinh, P.T.; Abbott, K.L.; Gwinn, D.M.; Sweet-Cordero, E.A.; et al. Combined Proteomic and Genetic Interaction Mapping Reveals New RAS Effector Pathways and Susceptibilities. Cancer Discov. 2020, 10, 1950–1967. [Google Scholar] [CrossRef]
- Zhang, X.; Cao, J.; Miller, S.P.; Jing, H.; Lin, H. Cyomparative Nucleotide-Dependent Interactome Analysis Reveals Shared and Differential Properties of KRas4a and KRas4b. ACS Cent. Sci. 2018, 4, 71–80. [Google Scholar] [CrossRef]
- Kovalski, J.R.; Bhaduri, A.; Zehnder, A.M.; Neela, P.H.; Che, Y.; Wozniak, G.G.; Khavari, P.A. The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol. Cell. 2019, 73, 830–844.e812. [Google Scholar] [CrossRef]
- Ritchie, C.; Mack, A.; Harper, L.; Alfadhli, A.; Stork, P.J.S.; Nan, X.; Barklis, E. Analysis of K-Ras Interactions by Biotin Ligase Tagging. Cancer Genom. Proteom. 2017, 14, 225–239. [Google Scholar] [CrossRef]
- Adhikari, H.; Counter, C.M. Interrogating the protein interactomes of RAS isoforms identifies PIP5K1A as a KRAS-specific vulnerability. Nat. Commun. 2018, 9, 3646. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Navarrete-Perea, J.; Cannon, J.R.; Baltier, K.; Gebreab, F.; Gygi, M.P.; Thornock, A.; Zarraga, G.; Tam, S.; et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 2021, 184, 3022–3040.e3028. [Google Scholar] [CrossRef]
- Choi, B.H.; Kou, Z.; Colon, T.M.; Chen, C.H.; Chen, Y.; Dai, W. Identification of Radil as a Ras binding partner and putative activator. J. Biol. Chem. 2021, 296, 100314. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, S.; Furuse, M.; Yokoyama, N.; Sasazuki, T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 1993, 260, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.A.; Jarboui, M.A.; Srihari, S.; Raso, C.; Bryan, K.; Dernayka, L.; Charitou, T.; Bernal-Llinares, M.; Herrera-Montavez, C.; Krstic, A.; et al. Extensive rewiring of the EGFR network in colorectal cancer cells expressing transforming levels of KRAS(G13D). Nat. Commun. 2020, 11, 499. [Google Scholar] [CrossRef] [PubMed]
- Beganton, B.; Coyaud, E.; Laurent, E.M.N.; Mange, A.; Jacquemetton, J.; Le Romancer, M.; Raught, B.; Solassol, J. Proximal Protein Interaction Landscape of RAS Paralogs. Cancers 2020, 12, 3326. [Google Scholar] [CrossRef]
- Ternet, C.; Junk, P.; Sevrin, T.; Catozzi, S.; Wahlen, E.; Heldin, J.; Oliviero, G.; Wynne, K.; Kiel, C. Analysis of context-specific KRAS-effector (sub)complexes in Caco-2 cells. Life Sci. Alliance 2023, 6, e202201670. [Google Scholar] [CrossRef]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef]
- Mi, H.; Lazareva-Ulitsky, B.; Loo, R.; Kejariwal, A.; Vandergriff, J.; Rabkin, S.; Guo, N.; Muruganujan, A.; Doremieux, O.; Campbell, M.J.; et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005, 33, D284–D288. [Google Scholar] [CrossRef]
- Jeng, H.H.; Taylor, L.J.; Bar-Sagi, D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat. Commun. 2012, 3, 1168. [Google Scholar] [CrossRef]
- Zafra, M.P.; Parsons, M.J.; Kim, J.; Alonso-Curbelo, D.; Goswami, S.; Schatoff, E.M.; Han, T.; Katti, A.; Fernandez, M.T.C.; Wilkinson, J.E.; et al. An In Vivo Kras Allelic Series Reveals Distinct Phenotypes of Common Oncogenic Variants. Cancer Discov. 2020, 10, 1654–1671. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Chiou, L.W.; Chan, C.H.; Jhuang, Y.L.; Yang, C.Y.; Jeng, Y.M. DNA replication stress and mitotic catastrophe mediate sotorasib addiction in KRAS(G12C)-mutant cancer. J. Biomed. Sci. 2023, 30, 50. [Google Scholar] [CrossRef] [PubMed]
- Salmon, M.; Alvarez-Diaz, R.; Fustero-Torre, C.; Brehey, O.; Lechuga, C.G.; Sanclemente, M.; Fernandez-Garcia, F.; Lopez-Garcia, A.; Martin-Guijarro, M.C.; Rodriguez-Perales, S.; et al. Kras oncogene ablation prevents resistance in advanced lung adenocarcinomas. J. Clin. Investig. 2023, 133, e164413. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Suda, K.; Fujino, T.; Ohara, S.; Hamada, A.; Nishino, M.; Chiba, M.; Shimoji, M.; Takemoto, T.; Arita, T.; et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From In Vitro Experiments. J. Thorac. Oncol. 2021, 16, 1321–1332. [Google Scholar] [CrossRef]
- Brown, W.S.; McDonald, P.C.; Nemirovsky, O.; Awrey, S.; Chafe, S.C.; Schaeffer, D.F.; Li, J.; Renouf, D.J.; Stanger, B.Z.; Dedhar, S. Overcoming Adaptive Resistance to KRAS and MEK Inhibitors by Co-targeting mTORC1/2 Complexes in Pancreatic Cancer. Cell. Rep. Med. 2020, 1, 100131. [Google Scholar] [CrossRef]
- Chan, C.H.; Chiou, L.W.; Lee, T.Y.; Liu, Y.R.; Hsieh, T.H.; Yang, C.Y.; Jeng, Y.M. PAK and PI3K pathway activation confers resistance to KRAS(G12C) inhibitor sotorasib. Br. J. Cancer 2023, 128, 148–159. [Google Scholar] [CrossRef]
- McDonald, W.H.; Ohi, R.; Miyamoto, D.T.; Mitchison, T.J.; Yates III, J.R. Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: Single-dimension LC-MS/MS, 2-phase MudPIT, and 3-phase MudPIT. Int. J. Mass Spectrom. 2002, 219, 245–251. [Google Scholar] [CrossRef]
- Zhou, Y.; Hancock, J.F. Lipid Profiles of RAS Nanoclusters Regulate RAS Function. Biomolecules 2021, 11, 1439. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e517. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.C.; Levine, R.L. Molecular pathways: Molecular basis for sensitivity and resistance to JAK kinase inhibitors. Clin. Cancer Res. 2014, 20, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Mohrherr, J.; Haber, M.; Breitenecker, K.; Aigner, P.; Moritsch, S.; Voronin, V.; Eferl, R.; Moriggl, R.; Stoiber, D.; Gyorffy, B.; et al. JAK-STAT inhibition impairs K-RAS-driven lung adenocarcinoma progression. Int. J. Cancer 2019, 145, 3376–3388. [Google Scholar] [CrossRef]
- Bigenzahn, J.W.; Collu, G.M.; Kartnig, F.; Pieraks, M.; Vladimer, G.I.; Heinz, L.X.; Sedlyarov, V.; Schischlik, F.; Fauster, A.; Rebsamen, M.; et al. LZTR1 is a regulator of RAS ubiquitination and signaling. Science 2018, 362, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell. Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Sanz-Moreno, V.; Arozarena, I.; Calvo, F.; Agudo-Ibanez, L.; Santos, E.; Berciano, M.T.; Crespo, P. Distinct utilization of effectors and biological outcomes resulting from site-specific Ras activation: Ras functions in lipid rafts and Golgi complex are dispensable for proliferation and transformation. Mol. Cell. Biol. 2006, 26, 100–116. [Google Scholar] [CrossRef]
- Matallanas, D.; Romano, D.; Al-Mulla, F.; O’Neill, E.; Al-Ali, W.; Crespo, P.; Doyle, B.; Nixon, C.; Sansom, O.; Drosten, M.; et al. Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol. Cell. 2011, 44, 893–906. [Google Scholar] [CrossRef]
- Lampson, B.L.; Pershing, N.L.; Prinz, J.A.; Lacsina, J.R.; Marzluff, W.F.; Nicchitta, C.V.; MacAlpine, D.M.; Counter, C.M. Rare codons regulate KRas oncogenesis. Curr. Biol. 2013, 23, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Hood, F.E.; Sahraoui, Y.M.; Jenkins, R.E.; Prior, I.A. Ras protein abundance correlates with Ras isoform mutation patterns in cancer. Oncogene 2023, 42, 1224–1232. [Google Scholar] [CrossRef]
- Barrios-Bernal, P.; Lucio-Lozada, J.; Ramos-Ramirez, M.; Hernandez-Pedro, N.; Arrieta, O. A Novel Combination of Sotorasib and Metformin Enhances Cytotoxicity and Apoptosis in KRAS-Mutated Non-Small Cell Lung Cancer Cell Lines through MAPK and P70S6K Inhibition. Int. J. Mol. Sci. 2023, 24, 4331. [Google Scholar] [CrossRef]
- Ma, Y.; Schulz, B.; Trakooljul, N.; Al Ammar, M.; Sekora, A.; Sender, S.; Hadlich, F.; Zechner, D.; Weiss, F.U.; Lerch, M.M.; et al. Inhibition of KRAS, MEK and PI3K Demonstrate Synergistic Anti-Tumor Effects in Pancreatic Ductal Adenocarcinoma Cell Lines. Cancers 2022, 14, 4467. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Mezzadra, R.; Sinopoli, J.; Bian, Y.; Marasco, M.; Kaplun, E.; Gao, Y.; Zhao, H.; Paula, A.D.C.; Zhu, Y.; et al. Molecular Characterization of Acquired Resistance to KRASG12C-EGFR Inhibition in Colorectal Cancer. Cancer Discov. 2023, 13, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. The dark side of Ras: Regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [Google Scholar] [CrossRef]
- Dorard, C.; Madry, C.; Buhard, O.; Toifl, S.; Didusch, S.; Ratovomanana, T.; Letourneur, Q.; Dolznig, H.; Garnett, M.J.; Duval, A.; et al. RAF1 contributes to cell proliferation and STAT3 activation in colorectal cancer independently of microsatellite and KRAS status. Oncogene 2023, 42, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nolan, A.; Raso, C.; Kolch, W.; Kriegsheim, A.v.; Wynne, K.; Matallanas, D. Proteomic Mapping of the Interactome of KRAS Mutants Identifies New Features of RAS Signalling Networks and the Mechanism of Action of Sotorasib. Cancers 2023, 15, 4141. https://doi.org/10.3390/cancers15164141
Nolan A, Raso C, Kolch W, Kriegsheim Av, Wynne K, Matallanas D. Proteomic Mapping of the Interactome of KRAS Mutants Identifies New Features of RAS Signalling Networks and the Mechanism of Action of Sotorasib. Cancers. 2023; 15(16):4141. https://doi.org/10.3390/cancers15164141
Chicago/Turabian StyleNolan, Aoife, Cinzia Raso, Walter Kolch, Alex von Kriegsheim, Kieran Wynne, and David Matallanas. 2023. "Proteomic Mapping of the Interactome of KRAS Mutants Identifies New Features of RAS Signalling Networks and the Mechanism of Action of Sotorasib" Cancers 15, no. 16: 4141. https://doi.org/10.3390/cancers15164141
APA StyleNolan, A., Raso, C., Kolch, W., Kriegsheim, A. v., Wynne, K., & Matallanas, D. (2023). Proteomic Mapping of the Interactome of KRAS Mutants Identifies New Features of RAS Signalling Networks and the Mechanism of Action of Sotorasib. Cancers, 15(16), 4141. https://doi.org/10.3390/cancers15164141