
Brown Tumour in Chronic Kidney Disease: Revisiting an Old Disease with a New Perspective

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Epidemiology and Risk Factors
3. The Mechanisms: Beyond Hyperparathyroidism
3.1. Secondary Hyperparathyroidism
3.2. RAAS Hyperactivity
3.3. Inflammatory Factors Induce Osteoclast Activation
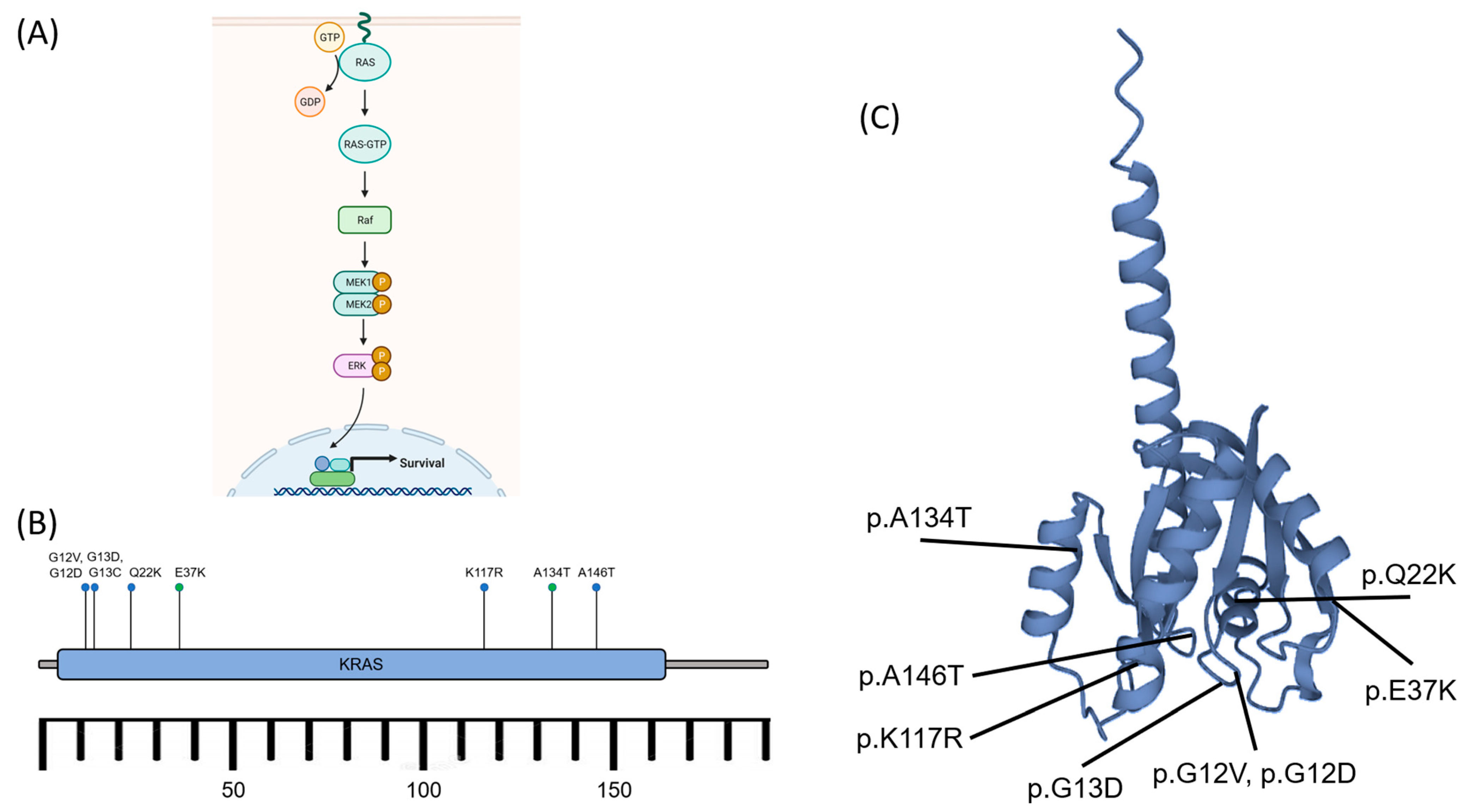
3.4. Molecular Drivers of Tumour Formation

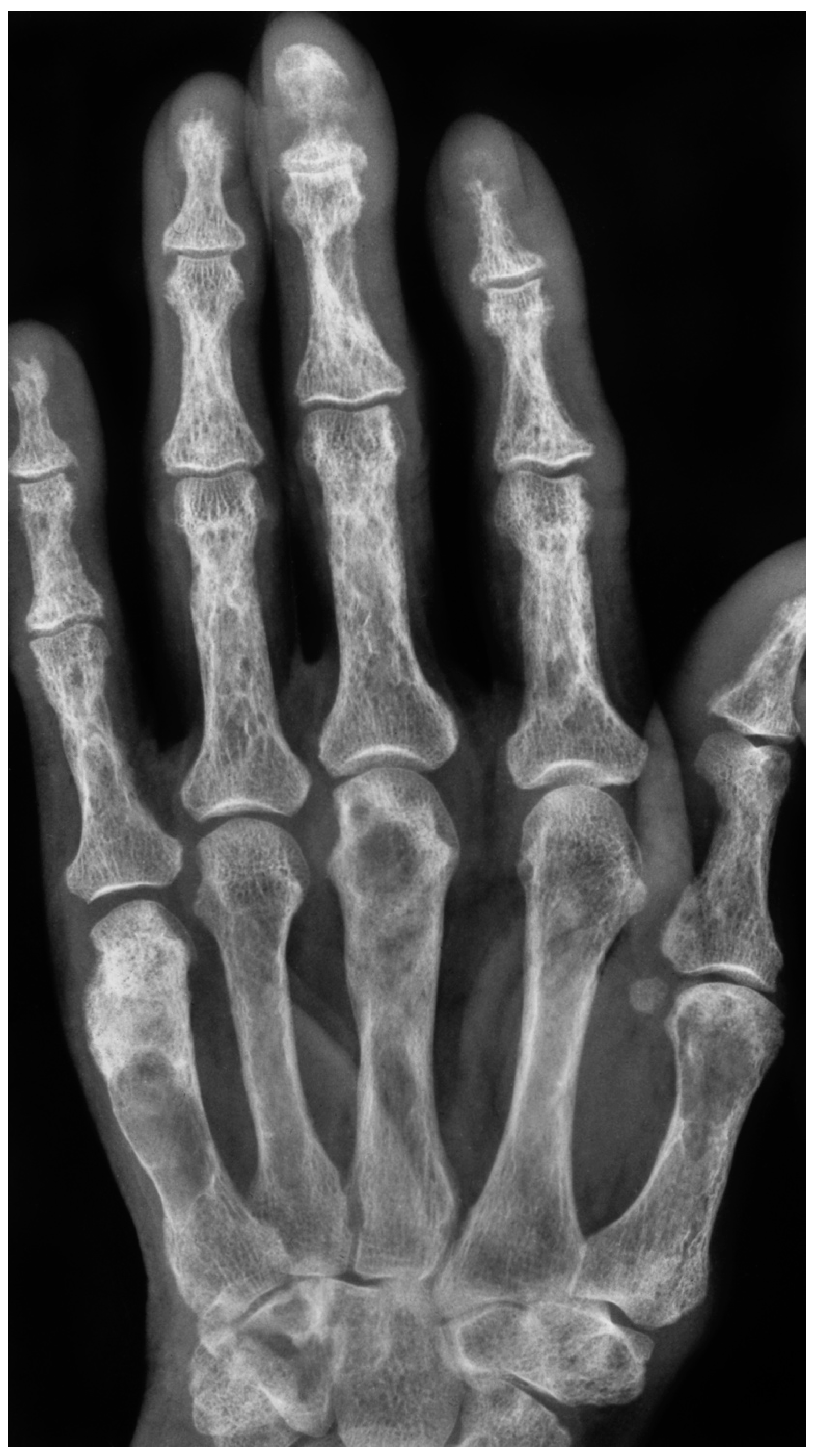
4. Diagnosis: The Role of Radiological Imaging and Lesion Biopsy
4.1. Radiological Imaging
4.2. Tissue Biopsy
5. Treatment: Beyond Removal of the Tumour
5.1. Pharmacological Prevention
5.2. Surgical Treatment
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seyedzadeh, A.; Tohidi, M.R.; Golmohamadi, S.; Omrani, H.R.; Seyedzadeh, M.S.; Amiri, S.; Hookari, S. Prevalence of Renal Osteodystrophy and its Related Factors among End-stage Renal Disease Patients Undergoing Hemodialysis: Report from Imam Reza Referral Hospital of Medical University of Kermanshah, Iran. Oman Med. J. 2022, 37, e335. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, G.; Ott, U.; Stein, G.; Steiner, T.; Wolf, G. Renal osteodystrophy after successful renal transplantation: A histomorphometric analysis in 57 patients. In Proceedings of the Transplantation Proceedings; Elsevier: Amsterdam, The Netherlands, 2007; Volume 39, pp. 3153–3158. [Google Scholar]
- Tarrass, F.; Benjelloun, M.; Bensaha, T. Severe jaw enlargement associated with uremic hyperparathyroidism. Hemodial. Int. 2008, 12, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Endel, G. Ueber Einen Fall von Cystoider Entartung des Ganzen Skelettes; Brühl: Giessen, Germany, 1864. [Google Scholar]
- Von Recklinghausen, F. Die Fibrose Oder Deformierende Ostitis, Die Osteomalacic und Die Osteoplastische Carcinose in Ihren Gegenseitigen Beziehungen; Rudolf Virchow Festschr: Berlin, Germany, 1891; pp. 1–89. Available online: https://www.abebooks.com/first-edition/fibr%C3%B6se-deformierende-Ostitis-Osteomalacie-osteoplastische-Carcinose/1239516015/bd (accessed on 11 August 2023).
- Fatma, L.B.; Barbouch, S.; Fethi, B.H.; Imen, B.A.; Karima, K.; Imed, H.; Fatma, B.M.; Rim, G.; Taieb, B.A.; Maiz, H. Ben Brown tumors in patients with chronic renal failure and secondary hyperparathyroidism: Report of 12 cases. Saudi J. Kidney Dis. Transplant. 2010, 21, 772. [Google Scholar]
- Morrone, L.F.; Ettorre, G.C.; Passavanti, G.; Tampoia, M.; Schiavone, P.; Coratelli, P. Maxillary brown tumor in secondary hyperparathyroidism requiring urgent parathyroidectomy. J. Nephrol. 2001, 14, 415–419. [Google Scholar] [PubMed]
- Jaffe, H.L.; Lichtenstein, L. Benign chondroblastoma of bone: A reinterpretation of the so-called calcifying or chondromatous giant cell tumor. Am. J. Pathol. 1942, 18, 969. [Google Scholar]
- Singh, S.; Padhy, S.K.; Mohapatra, S.S.; Panda, A.; Shahi, P. Chronic Kidney Disease Presenting With Brown Tumors in the Mandible. Cureus 2022, 14, 3–7. [Google Scholar] [CrossRef]
- Wiederkehr, M. Brown tumor complicating end-stage kidney disease. Clin. Nephrol.—Case Stud. 2020, 8, 72–79. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.L.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Chen, T.K.; Knicely, D.H.; Grams, M.E. Chronic Kidney Disease Diagnosis and Management. JAMA 2019, 322, 1294. [Google Scholar] [CrossRef]
- Antonelli, J.R.; Hottei, T.L. Oral manifestations of renal osteodystrophy: Case report and review of the literature. Spec. Care Dent. 2003, 23, 28–34. [Google Scholar] [CrossRef]
- Baracaldo, R.M.; Bao, D.; Iampornpipopchai, P.; Fogel, J.; Rubinstein, S. Facial disfigurement due to osteitis fibrosa cystica or brown tumor from secondary hyperparathyroidism in patients on dialysis: A systematic review and an illustrative case report. Hemodial. Int. 2015, 19, 583–592. [Google Scholar]
- Nunes, T.B.; Bologna, S.B.; Witzel, A.L.; Nico, M.M.S.; Lourenço, S.V. A rare case of concomitant maxilla and mandible Brown tumours, papillary thyroid carcinoma, parathyroid adenoma, and osteitis fibrosa cystica. Case Rep. Dent. 2016, 2016, 5320298. [Google Scholar] [PubMed]
- Lee, J.H.; Chung, S.M.; Kim, H.S. Osteitis fibrosa cystica mistaken for malignant disease. Clin. Exp. Otorhinolaryngol. 2013, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Thompson, J.R. The regulation of parathyroid hormone secretion and synthesis. J. Am. Soc. Nephrol. 2011, 22, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Quinn, S.J.; Thomsen, A.R.B.; Pang, J.L.; Kantham, L.; Bräuner-Osborne, H.; Pollak, M.; Goltzman, D.; Brown, E.M. Interactions between calcium and phosphorus in the regulation of the production of fibroblast growth factor 23 in vivo. Am. J. Physiol. Metab. 2013, 304, E310–E320. [Google Scholar]
- Jat, J.A.; Mal, P.; Kumar, D. Renal osteodystrophy in end stage renal failure patients on maintenance haemodialysis. J. Clin. Exp. Nephrol. 2016, 1, 25. [Google Scholar]
- Satpathy, A.S.; Dasgupta, A.; Dutta, C.; Mohan, N.V.K.; Satpathy, S. Osteitis fibrosa cystica of mandible in hyperparathyroidism-jaw tumor syndrome: A rare presentation and review of literature. Natl. J. Maxillofac. Surg. 2017, 8, 162. [Google Scholar] [CrossRef]
- Jervis, L.; James, M.; Howe, W.; Richards, S. Osteolytic lesions: Osteitis fibrosa cystica in the setting of severe primary hyperparathyroidism. Case Rep. 2017, 2017, bcr2017220603. [Google Scholar] [CrossRef]
- Mellouli, N.; Belkacem Chebil, R.; Darej, M.; Hasni, Y.; Oualha, L.; Douki, N. Mandibular osteitis fibrosa cystica as first sign of vitamin D deficiency. Case Rep. Dent. 2018, 2018, 6814803. [Google Scholar] [CrossRef]
- Kemp, A.M.C.; Bukvic, M.; Sturgis, C.D. Fine Needle Aspiration Diagnosis of Osteitis Fibrosa Cystica (Brown Tumor of Bone). Acta Cytol. 2008, 52, 471–474. [Google Scholar] [CrossRef]
- Crutchlow, W.P.; David, D.S.; Whitsell, J. Multiple skeletal complications in a case of chronic renal failure treated by kidney homotransplantation. Am. J. Med. 1971, 50, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, M.; Begerow, B.; Minne, H.W.; Abrams, C.; Nachtigall, D.; Hansen, C. Effects of a short-term vitamin D and calcium supplementation on body sway and secondary hyperparathyroidism in elderly women. J. Bone Miner. Res. 2000, 15, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Aravindhan, R.; Magesh, K.T.; Vivek, N.; Saravanan, C. Alteration of cellular metabolism in cancer cells and its therapeutic. J. Oral Maxillofac. Pathol. 2021, 25, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Livingston, S.; Sabir, Z.L.; Haussler, C.A.; Jurutka, P.W. Vitamin D Receptor Mediates a Myriad of Biological Actions Dependent on Its 1,25-Dihydroxyvitamin D Ligand: Distinct Regulatory Themes Revealed by Induction of Klotho and Fibroblast Growth Factor-23. JBMR Plus 2021, 5, e10432. [Google Scholar] [CrossRef] [PubMed]
- Bricker, N.S.; Slatopolsky, E.; Reiss, E.; Avioli, L.V. Calcium, Phosphorus, and Bone in Renal Disease and Transplantation. Arch. Intern. Med. 1969, 123, 543–553. [Google Scholar] [CrossRef]
- Reiss, E.; Canterbury, J.M.; Kanter, A. Circulating Parathyroid Hormone Concentration in Chronic Renal Insufficiency. Arch. Intern. Med. 1969, 124, 417–422. [Google Scholar] [CrossRef]
- Brown, A.J.; Dusso, A.; Lopez-Hilker, S.; Lewis-Finch, J.; Grooms, P.; Slatopolsky, E. 1,25-(OH)2D receptors are decreased in parathyroid glands from chronically uremic dogs. Kidney Int. 1989, 35, 19–23. [Google Scholar] [CrossRef]
- Cantley, L.K.; Russell, J.; Lettieri, D.; Sherwood, L.M. 1,25-Dihydroxyvitamin D3 Suppresses Parathyroid Hormone Secretion from Bovine Parathyroid Cells in Tissue Culture. Endocrinology 1985, 117, 2114–2119. [Google Scholar] [CrossRef]
- Naveh-Many, T.; Bell, O.; Silver, J.; Kilav, R. Cis and trans acting factors in the regulation of parathyroid hormone (PTH) mRNA stability by calcium and phosphate. FEBS Lett. 2002, 529, 60–64. [Google Scholar] [CrossRef]
- Ritter, C.S.; Martin, D.R.; Lu, Y.; Slatopolsky, E.; Brown, A.J. Reversal of secondary hyperparathyroidism by phosphate restriction restores parathyroid calcium-sensing receptor expression and function. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2002, 17, 2206–2213. [Google Scholar] [CrossRef]
- Shoben, A.B.; Rudser, K.D.; De Boer, I.H.; Young, B.; Kestenbaum, B. Association of oral calcitriol with improved survival in nondialyzed CKD. J. Am. Soc. Nephrol. 2008, 19, 1613–1619. [Google Scholar] [CrossRef]
- Block, G.A.; Martin, K.J.; De Francisco, A.L.M.; Turner, S.A.; Avram, M.M.; Suranyi, M.G.; Hercz, G.; Cunningham, J.; Abu-Alfa, A.K.; Messa, P. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N. Engl. J. Med. 2004, 350, 1516–1525. [Google Scholar] [CrossRef]
- Moe, S.M.; Drüeke, T.B. Management of secondary hyperparathyroidism: The importance and the challenge of controlling parathyroid hormone levels without elevating calcium, phosphorus, and calcium-phosphorus product. Am. J. Nephrol. 2003, 23, 369–379. [Google Scholar] [CrossRef]
- Locatelli, F.; Cannata-Andía, J.B.; Drüeke, T.B.; Hörl, W.H.; Fouque, D.; Heimburger, O.; Ritz, E. Management of disturbances of calcium and phosphate metabolism in chronic renal insufficiency, with emphasis on the control of hyperphosphataemia. Nephrol. Dial. Transplant. 2002, 17, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Takami, M.; Rho, J.; Josien, R.; Choi, Y. A novel member of the leukocyte receptor complex regulates osteoclast differentiation. J. Exp. Med. 2002, 195, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Zur, Y.; Rosenfeld, L.; Keshelman, C.A.; Dalal, N.; Guterman-Ram, G.; Orenbuch, A.; Einav, Y.; Levaot, N.; Papo, N. A dual-specific macrophage colony-stimulating factor antagonist of c-FMS and α v β 3 integrin for osteoporosis therapy. PLoS Biol. 2018, 16, e2002979. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.P. M-CSF, c-Fms, and signaling in osteoclasts and their precursors. Ann. N. Y. Acad. Sci. 2006, 1068, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Huang, Y.; Luo, J.; Ye, Y. Misdiagnosis of brown tumour caused by primary hyperparathyroidism: A case report with literature review. BMC Endocr. Disord. 2022, 22, 66. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, B.; Purohit, S.; Harsh, A.; Vangala, N. A complex case of brown tumors as initial manifestation of primary hyperparathyroidism in a young female. J. Oral. Maxillofac. Pathol. 2019, 23, 477. [Google Scholar] [CrossRef]
- Zou, H.; Song, L.; Jia, M.; Wang, L.; Sun, Y. Brown tumor of multiple facial bones associated with primary hyperparathyroidism: A clinical case report. Medicine 2018, 97, e11877. [Google Scholar] [CrossRef]
- Gaggar, P.; Raju, S.B.; Akkiraju, P.; Madipalli, R.T. POS-239 Removal of brown tumor associated with normalization of serum parathyroid levels in a 19 year old Indian female on maintenance hemodialysis– A serendipitous discovery. Kidney Int. Rep. 2022, 7, S104–S105. [Google Scholar] [CrossRef]
- Vimalraj, S. Alkaline phosphatase: Structure, expression and its function in bone mineralization. Gene 2020, 754, 144855. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, L.; Huang, L.; Sun, X.; Ji, H.; Lu, Y. Risk factors for elevated preoperative alkaline phosphatase in patients with refractory secondary hyperparathyroidism. Am. Surg. 2017, 83, 1368–1372. [Google Scholar] [CrossRef]
- Kang, B.H.; Hwang, S.Y.; Kim, J.Y.; Hong, Y.A.; Jung, M.Y.; Lee, E.A.; Lee, J.E.; Lee, J.B.; Ko, G.J.; Pyo, H.J.; et al. Predicting postoperative total calcium requirements after parathyroidectomy in secondary hyperparathyroidism. Korean J. Intern. Med. 2015, 30, 856–864. [Google Scholar] [CrossRef]
- Ge, P.; Liu, S.; Sheng, X.; Li, S.; Xu, M.; Jiang, J.; Chen, S. Serum parathyroid hormone and alkaline phosphatase as predictors of calcium requirements after total parathyroidectomy for hypocalcemia in secondary hyperparathyroidism. Head. Neck 2018, 40, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Fineman, I.; Johnson, J.P.; Di-Patre, P.-L.; Sandhu, H. Chronic renal failure causing brown tumors and myelopathy: Case report and review of pathophysiology and treatment. J. Neurosurg. Spine 1999, 90, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Leelahavanichkul, A.; Yan, Q.; Hu, X.; Eisner, C.; Huang, Y.; Chen, R.; Mizel, D.; Zhou, H.; Wright, E.C.; Kopp, J.B. Angiotensin II overcomes strain-dependent resistance of rapid CKD progression in a new remnant kidney mouse model. Kidney Int. 2010, 78, 1136–1153. [Google Scholar] [CrossRef] [PubMed]
- Ruggenenti, P.; Cravedi, P.; Remuzzi, G. Mechanisms and treatment of CKD. J. Am. Soc. Nephrol. 2012, 23, 1917–1928. [Google Scholar] [CrossRef]
- Shimizu, H.; Nakagami, H.; Osako, M.K.; Hanayama, R.; Kunugiza, Y.; Kizawa, T.; Tomita, T.; Yoshikawa, H.; Ogihara, T.; Morishita, R. Angiotensin II accelerates osteoporosis by activating osteoclasts. FASEB J. 2008, 22, 2465–2475. [Google Scholar] [CrossRef]
- Hatton, R.; Stimpel, M.; Chambers, T.J. Angiotensin II is generated from angiotensin I by bone cells and stimulates osteoclastic bone resorption in vitro. J. Endocrinol. 1997, 152, 5–10. [Google Scholar] [CrossRef]
- Vuoti, E.; Lehenkari, P.; Tuukkanen, J.; Glumoff, V.; Kylmäoja, E. Osteoclastogenesis of human peripheral blood, bone marrow, and cord blood monocytes. Sci. Rep. 2023, 13, 3763. [Google Scholar] [CrossRef] [PubMed]
- Root, S.H.; Aguila, H.L. Novel population of human monocyte and osteoclast progenitors from pluripotent stem cells and peripheral blood. Blood Adv. 2021, 5, 4435–4448. [Google Scholar] [CrossRef] [PubMed]
- Hiruma, Y.; Inoue, A.; Hirose, S.; Hagiwara, H. Angiotensin II stimulates the proliferation of osteoblast-rich populations of cells from rat calvariae. Biochem. Biophys. Res. Commun. 1997, 230, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Rejnmark, L.; Vestergaard, P.; Mosekilde, L. Treatment with beta-blockers, ACE inhibitors, and calcium-channel blockers is associated with a reduced fracture risk: A nationwide case–control study. J. Hypertens. 2006, 24, 581–589. [Google Scholar]
- Lynn, H.; Kwok, T.; Wong, S.Y.S.; Woo, J.; Leung, P. Angiotensin converting enzyme inhibitor use is associated with higher bone mineral density in elderly Chinese. Bone 2006, 38, 584–588. [Google Scholar] [CrossRef]
- Shiratori, T.; Kyumoto-Nakamura, Y.; Kukita, A.; Uehara, N.; Zhang, J.; Koda, K.; Kamiya, M.; Badawy, T.; Tomoda, E.; Xu, X. IL-1β induces pathologically activated osteoclasts bearing extremely high levels of resorbing activity: A possible pathological subpopulation of osteoclasts, accompanied by suppressed expression of Kindlin-3 and Talin-1. J. Immunol. 2018, 200, 218–228. [Google Scholar]
- Zewinger, S.; Schumann, T.; Fliser, D.; Speer, T. Innate immunity in CKD-associated vascular diseases. Nephrol. Dial. Transplant. 2016, 31, 1813–1821. [Google Scholar] [CrossRef]
- Boswell, J.M.; Yui, M.A.; Burt, D.W.; Kelley, V.E. Increased tumor necrosis factor and IL-1 beta gene expression in the kidneys of mice with lupus nephritis. J. Immunol. 1988, 141, 3050–3054. [Google Scholar] [CrossRef]
- Xu, L.X.; Kukita, T.; Nakano, Y.; Yu, H.; Hotokebuchi, T.; Kuratani, T.; Iijima, T.; Koga, T. Osteoclasts in normal and adjuvant arthritis bone tissues express the mRNA for both type I and II interleukin-1 receptors. Lab. Investig. 1996, 75, 677–687. [Google Scholar]
- Turek, D.; Haefliger, S.; Ameline, B.; Alborelli, I.; Calgua, B.; Hartmann, W.; Harder, D.; Flanagan, A.M.; Amary, F.; Baumhoer, D. Brown tumors belong to the spectrum of KRAS-driven neoplasms. Am. J. Surg. Pathol. 2022, 46, 1577–1582. [Google Scholar] [CrossRef]
- Bovée, J.V.M.G.; Hogendoorn, P.C.W. Non-ossifying fibroma: A RAS-MAPK driven benign bone neoplasm. J. Pathol. 2019, 248, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, G.D.; Shi, Z.; Qi, L.L.; Zhou, L.Y.; Fu, Z.X. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC (Review). Oncol. Lett. 2016, 12, 3045–3050. [Google Scholar] [CrossRef] [PubMed]
- Pennanen, P.; Kallionpää, R.A.; Peltonen, S.; Nissinen, L.; Kähäri, V.M.; Heervä, E.; Peltonen, J. Signaling pathways in human osteoclasts differentiation: ERK1/2 as a key player. Mol. Biol. Rep. 2021, 48, 1243–1254. [Google Scholar] [CrossRef]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta—Mol. Cell Res. 2011, 1813, 1619–1633. [Google Scholar] [CrossRef]
- Guimarães, L.M.; Gomes, I.P.; Pereira, T.d.S.F.; de Andrade, B.A.B.; Romañach, M.J.; de Lacerda, J.C.T.; Pontes, H.A.R.; Brennan, P.A.; Rahimi, S.; Carlos, R.; et al. KRAS mutations in brown tumor of the jaws in hyperparathyroidism. J. Oral. Pathol. Med. 2020, 49, 796–802. [Google Scholar] [CrossRef]
- Gomes, C.C.; Gayden, T.; Bajic, A.; Harraz, O.F.; Pratt, J.; Nikbakht, H.; Bareke, E.; Diniz, M.G.; Castro, W.H.; St-Onge, P.; et al. TRPV4 and KRAS and FGFR1 gain-of-function mutations drive giant cell lesions of the jaw. Nat. Commun. 2018, 9, 4572. [Google Scholar] [CrossRef]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2022, 51, D523–D531. [Google Scholar] [CrossRef]
- Baumhoer, D.; Kovac, M.; Sperveslage, J.; Ameline, B.; Strobl, A.C.; Krause, A.; Trautmann, M.; Wardelmann, E.; Nathrath, M.; Höller, S.; et al. Activating mutations in the MAP-kinase pathway define non-ossifying fibroma of bone. J. Pathol. 2019, 248, 116–122. [Google Scholar] [CrossRef]
- Risques, R.A.; Kennedy, S.R. Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet. 2018, 14, e1007108. [Google Scholar] [CrossRef] [PubMed]
- Rad, S.N.; Barnett, M.J.; Anastasopoulou, C.; Deluxe, L. Osteitis Fibrosa Cystica. Available online: https://www.ncbi.nlm.nih.gov/books/NBK559097/ (accessed on 31 July 2023).
- Antin, F.; Bakhos, D.; Jegoux, F.; Merkouza, M.; Laccourreye, L. Maxillofacial brown tumours: Series of 5 cases. Eur. Ann. Otorhinolaryngol. Head. Neck Dis. 2018, 135, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zheng, S.; Zhou, L.; Yong, X.; Zhang, Y.; QinXia; Zhang, W.; Wang, C. Pathologic evaluation of the solid variant of aneurysmal bone cysts with USP6 rearrangement with an emphasis on the frequent diagnostic pitfalls. Pathol. Int. 2020, 70, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Viswanathan, V.K. Lytic Bone Lesions. Available online: https://www.ncbi.nlm.nih.gov/books/NBK539837/ (accessed on 31 July 2023).
- Ugga, L.; Cuocolo, R.; Cocozza, S.; Ponsiglione, A.; Stanzione, A.; Chianca, V.; D’Amico, A.; Brunetti, A.; Imbriaco, M. Spectrum of lytic lesions of the skull: A pictorial essay. Insights Imaging 2018, 9, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Vilanilam, G.K.; Nikpanah, M.; Vo, C.D.; Kearns, C. Osteitis Fibrosa Cystica: Brown Tumors of Hyperparathyroidism and End-Stage Renal Disease. Radiographics 2023, 43, e220211. [Google Scholar] [CrossRef] [PubMed]
- Misiorowski, W.; Czajka-Oraniec, I.; Kochman, M.; Zgliczyński, W.; Bilezikian, J.P. Osteitis fibrosa cystica—A forgotten radiological feature of primary hyperparathyroidism. Endocrine 2017, 58, 380–385. [Google Scholar] [CrossRef]
- Hsieh, M.-C.; Ko, J.-Y.; Eng, H.-L. Pathologic fracture of the distal femur in osteitis fibrosa cystica simulating metastatic disease. Arch. Orthop. Trauma. Surg. 2004, 124, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, M.; Damle, N.A. Fluorocholine PET imaging of parathyroid disease. Indian J. Endocrinol. Metab. 2018, 22, 535–541. [Google Scholar] [CrossRef]
- Tsushima, Y.; Sun, S.; Via, M.A. Brown Tumors Secondary to Parathyroid Carcinoma Masquerading as Skeletal Metastases on 18F-FDG PET/CT: A Case Report. AACE Clin. Case Rep. 2019, 5, e230–e232. [Google Scholar] [CrossRef]
- Kluijfhout, W.P.; Pasternak, J.D.; Drake, F.T.; Beninato, T.; Gosnell, J.E.; Shen, W.T.; Duh, Q.Y.; Allen, I.E.; Vriens, M.R.; de Keizer, B.; et al. Use of PET tracers for parathyroid localization: A systematic review and meta-analysis. Langenbeck’s Arch. Surg. 2016, 401, 925–935. [Google Scholar] [CrossRef]
- Hindié, E.; Ugur, Ö.; Fuster, D.; O’Doherty, M.; Grassetto, G.; Ureña, P.; Kettle, A.; Gulec, S.A.; Pons, F.; Rubello, D. 2009 EANM parathyroid guidelines. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1201–1216. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Feng, J.; Zhou, Q.; Zheng, W.; Meng, X.; Wang, Y.; Wang, J. Intraoperative 99mTc-MIBI-Guided Parathyroidectomy Improves Curative Effect of Parathyroidectomy, Bone Metabolism, and Bone Mineral Density. Am. Surg. 2021, 87, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.R.; Reading, C.C. Thyroid and Parathyroid Imaging. In Seminars in Ultrasound, CT and MRI; Springer International Publishing: Cham, Switzerland, 1995; Volume 16, pp. 279–295. ISBN 9783030394578. [Google Scholar]
- Caldarella, C.; Treglia, G.; Isgrò, M.A.; Giordano, A. Diagnostic performance of positron emission tomography using 11C-methionine in patients with suspected parathyroid adenoma: A meta-analysis. Endocrine 2013, 43, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Taywade, S.K.; Damle, N.A.; Behera, A.; Devasenathipathy, K.; Bal, C.; Tripathi, M.; Agarwal, S.; Tandon, N.; Chumber, S.; Seenu, V. Comparison of 18F-Fluorocholine positron emission tomography/computed tomography and four-dimensional computed tomography in the preoperative localization of parathyroid adenomas-initial results. Indian J. Endocrinol. Metab. 2017, 21, 399–403. [Google Scholar] [CrossRef]
- Michaud, L.; Balogova, S.; Burgess, A.; Ohnona, J.; Huchet, V.; Kerrou, K.; Lefèvre, M.; Tassart, M.; Montravers, F.; Périé, S.; et al. A pilot comparison of 18F-fluorocholine PET/CT, ultrasonography and 123I/99mTc-sestaMIBI dual-phase dual-isotope scintigraphy in the preoperative localization of hyperfunctioning parathyroid glands in primary or secondary hyperparathyroidism: Influence of thyroid anomalies. Medicine 2015, 94, e1701. [Google Scholar] [CrossRef]
- Deng, Y.; Shen, X.; Lei, L.; Zhang, W. Osteitis Fibrosa Cystica Caused by Hyperparathyroidism Shown on 18F-NaF PET/CT. Clin. Nucl. Med. 2020, 45, 577–579. [Google Scholar] [CrossRef]
- Graf, C.; Huellner, M.; Tschopp, O.; Bode-Lesniewska, B.; Schmid, C. 18F-NaF-PET/CT in patients with primary hyperparathyroidism and brown tumors. J. Bone Miner. Metab. 2020, 38, 299–309. [Google Scholar] [CrossRef]
- Win, A.Z.; Aparici, C.M. NaF18-PET/CT Imaging of Secondary Hyperparathyroidism. Nucl. Med. Mol. Imaging 2015, 49, 331–332. [Google Scholar] [CrossRef][Green Version]
- Park, P.S.U.; Raynor, W.Y.; Sun, Y.; Werner, T.J.; Rajapakse, C.S.; Alavi, A. 18F-sodium fluoride pet as a diagnostic modality for metabolic, autoimmune, and osteogenic bone disorders: Cellular mechanisms and clinical applications. Int. J. Mol. Sci. 2021, 22, 6504. [Google Scholar] [CrossRef]
- Van der Bijl, A.E.; Taminiau, A.H.M.; Hermans, J.; Beerman, H.; Hogendoorn, P.C.W. Accuracy of the Jamshidi trocar biopsy in the diagnosis of bone tumors. Clin. Orthop. Relat. Res. 1997, 334, 233–243. [Google Scholar] [CrossRef]
- Parfitt, J.; Harris, M.; Wright, J.M.; Kalamchi, S. Tumor suppressor gene mutation in a patient with a history of hyperparathyroidism–jaw tumor syndrome and healed generalized osteitis fibrosa cystica: A case report and genetic pathophysiology review. J. Oral. Maxillofac. Surg. 2015, 73, 194.e1–194.e9. [Google Scholar] [CrossRef] [PubMed]
- Kashkari, S.; Kelly, T.R.; Bethem, D.; Pepe, R.G. Osteitis fibrosa cystica (brown tumor) of the spine with cord compression: Report of a case with needle aspiration biopsy findings. Diagn. Cytopathol. 1990, 6, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.W.; Unger, P.; Kaneko, M.; Gabrilove, J.L. Fine needle aspiration of osteitis fibrosa cystica. Diagn. Cytopathol. 1985, 1, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, H.; Zhang, X.; Tang, Y.; Wu, Z.; Wang, Y.; Huang, H.; Fu, X.; Liu, J.; Hogendoorn, P.C.W.; et al. Clinicopathologic and molecular features of denosumab-treated giant cell tumour of bone (GCTB): Analysis of 21 cases. Ann. Diagn. Pathol. 2022, 57, 151882. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, N.D.; Elder, G.J.; Kerr, P.G. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin. J. Am. Soc. Nephrol. 2009, 4, 221–233. [Google Scholar] [CrossRef]
- Cremers, S.; Drake, M.T.; Ebetino, F.H.; Bilezikian, J.P.; Russell, R.G.G. Pharmacology of bisphosphonates. Br. J. Clin. Pharmacol. 2019, 85, 1052–1062. [Google Scholar] [CrossRef]
- Evenepoel, P.; Van Den Bergh, B.; Naesens, M.; De Jonge, H.; Bammens, B.; Claes, K.; Kuypers, D.; Vanrenterghem, Y. Calcium metabolism in the early posttransplantation period. Clin. J. Am. Soc. Nephrol. 2009, 4, 665–672. [Google Scholar] [CrossRef]
- Evenepoel, P. Calcimimetics in chronic kidney disease: Evidence, opportunities and challenges. Kidney Int. 2008, 74, 265–275. [Google Scholar] [CrossRef][Green Version]
- Ballinger, A.E.; Palmer, S.C.; Nistor, I.; Craig, J.C.; Strippoli, G.F.M. Calcimimetics for secondary hyperparathyroidism in chronic kidney disease patients. Cochrane Database Syst. Rev. 2014, CD006254. [Google Scholar] [CrossRef]
- Guillaume, J.; Souberbielle, J.C.; Chazot, C. Vitamin D in Chronic Kidney Disease and Dialysis Patients. Nutrients 2017, 9, 328. [Google Scholar] [CrossRef]
- González, E.A.; Sachdeva, A.; Oliver, D.A.; Martin, K.J. Vitamin D Insufficiency and Deficiency in Chronic Kidney Disease. Am. J. Nephrol. 2004, 24, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Bosworth, C.; De Boer, I.H. Impaired Vitamin D Metabolism in CKD. Semin. Nephrol. 2013, 33, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, S.; LeBoff, M.; Lewiecki, E.M. Denosumab for the treatment of osteoporosis. Expert Opin. Drug Metab. Toxicol. 2015, 11, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Gopaul, A.; Kanagalingam, T.; Thain, J.; Khan, T.; Cowan, A.; Sultan, N.; Clemens, K.K. Denosumab in chronic kidney disease: A narrative review of treatment efficacy and safety. Arch. Osteoporos. 2021, 16, 116. [Google Scholar] [CrossRef]
- Lau, W.L.; Obi, Y.; Kalantar-Zadeh, K. Parathyroidectomy in the Management of Secondary Hyperparathyroidism. Clin. J. Am. Soc. Nephrol. 2018, 13, 952–961. [Google Scholar] [CrossRef]
- Apetrii, M.; Goldsmith, D.; Nistor, I.; Siriopol, D.; Voroneanu, L.; Scripcariu, D.; Vervloet, M.; Covic, A. Impact of surgical parathyroidectomy on chronic kidney disease-mineral and bone disorder (CKD-MBD)—A systematic review and meta-analysis. PLoS ONE 2017, 12, e0187025. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santoso, D.; Thaha, M.; Empitu, M.A.; Kadariswantiningsih, I.N.; Suryantoro, S.D.; Haryati, M.R.; Hertanto, D.M.; Pramudya, D.; Bintoro, S.U.Y.; Nasronudin, N.; et al. Brown Tumour in Chronic Kidney Disease: Revisiting an Old Disease with a New Perspective. Cancers 2023, 15, 4107. https://doi.org/10.3390/cancers15164107
Santoso D, Thaha M, Empitu MA, Kadariswantiningsih IN, Suryantoro SD, Haryati MR, Hertanto DM, Pramudya D, Bintoro SUY, Nasronudin N, et al. Brown Tumour in Chronic Kidney Disease: Revisiting an Old Disease with a New Perspective. Cancers. 2023; 15(16):4107. https://doi.org/10.3390/cancers15164107
Chicago/Turabian StyleSantoso, Djoko, Mochammad Thaha, Maulana A. Empitu, Ika Nindya Kadariswantiningsih, Satriyo Dwi Suryantoro, Mutiara Rizki Haryati, Decsa Medika Hertanto, Dana Pramudya, Siprianus Ugroseno Yudho Bintoro, Nasronudin Nasronudin, and et al. 2023. "Brown Tumour in Chronic Kidney Disease: Revisiting an Old Disease with a New Perspective" Cancers 15, no. 16: 4107. https://doi.org/10.3390/cancers15164107
APA StyleSantoso, D., Thaha, M., Empitu, M. A., Kadariswantiningsih, I. N., Suryantoro, S. D., Haryati, M. R., Hertanto, D. M., Pramudya, D., Bintoro, S. U. Y., Nasronudin, N., Alsagaff, M. Y., Susilo, H., Wungu, C. D. K., Budhiparama, N. C., & Hogendoorn, P. C. W. (2023). Brown Tumour in Chronic Kidney Disease: Revisiting an Old Disease with a New Perspective. Cancers, 15(16), 4107. https://doi.org/10.3390/cancers15164107