
The Efficacy of CB-103, a First-in-Class Transcriptional Notch Inhibitor, in Preclinical Models of Breast Cancer

 , , ,
, , ,  , ,
, , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
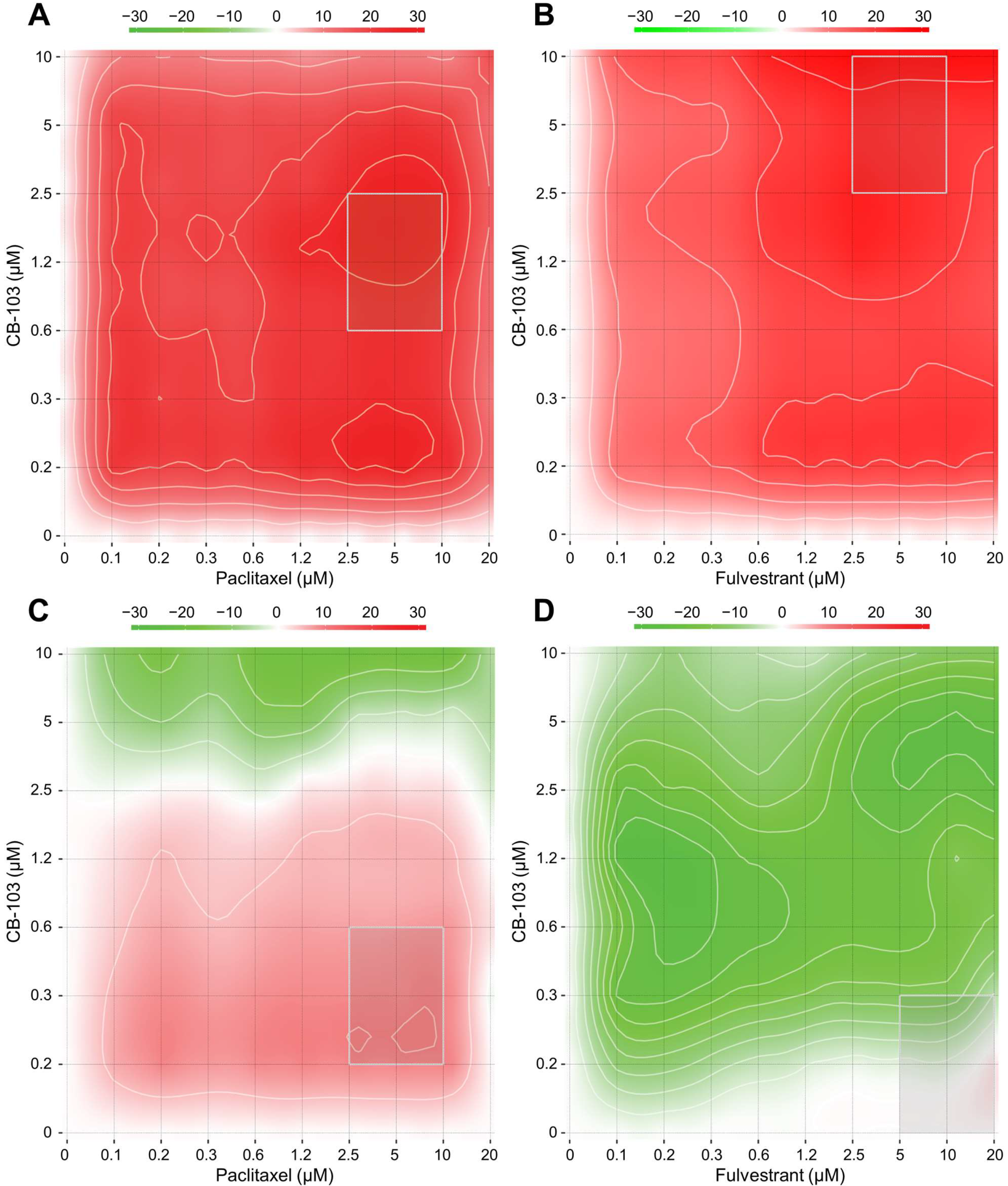
2.1. CB-103 Combination Screening
2.2. Cell Lines and Mammospheres
2.3. In Vivo Efficacy Study to Evaluate CB-103 vs. Fulvestrant and Combination in Therapy Resistant ER+ BC
2.4. ER+ BC PDX Combination with Fulvestrant
2.5. TNBC Xenograft Combination Study with Paclitaxel
2.6. Marginal Zone B Cell Assay
2.7. RNA Sequencing
2.8. Histology and Immunohistochemistry
2.9. Statistical Analysis
3. Results
3.1. CB-103 Shows Synergy with Several Anti-Neoplastic Drugs
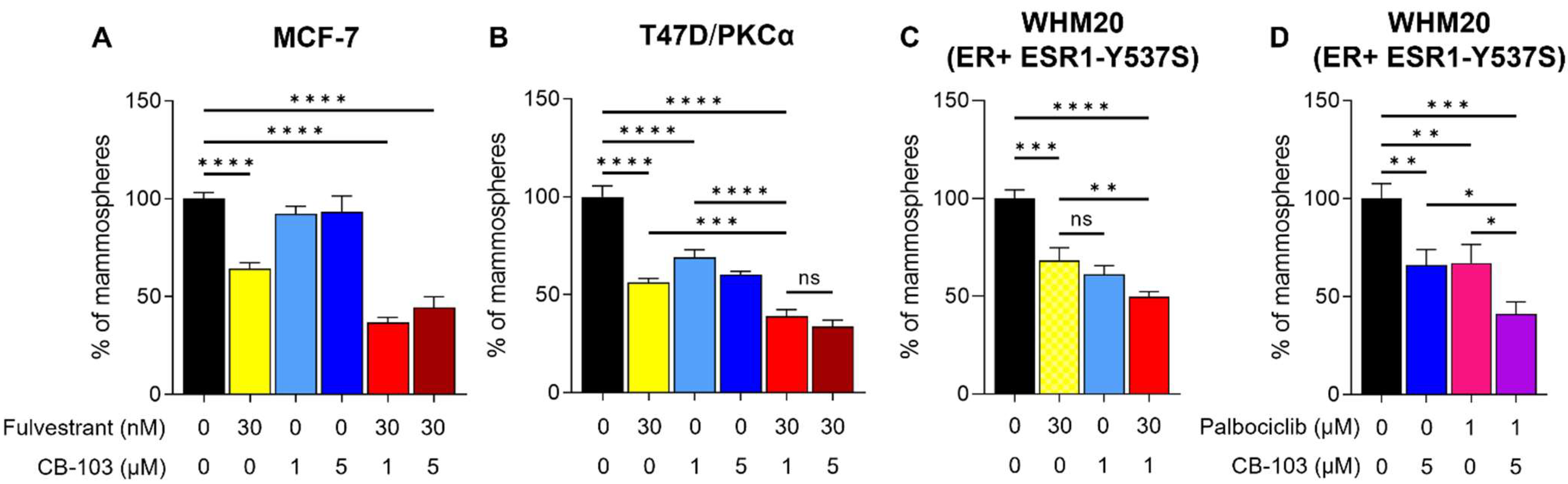
3.2. CB-103 Strongly Inhibits Mammosphere Formation when Combined with Fulvestrant
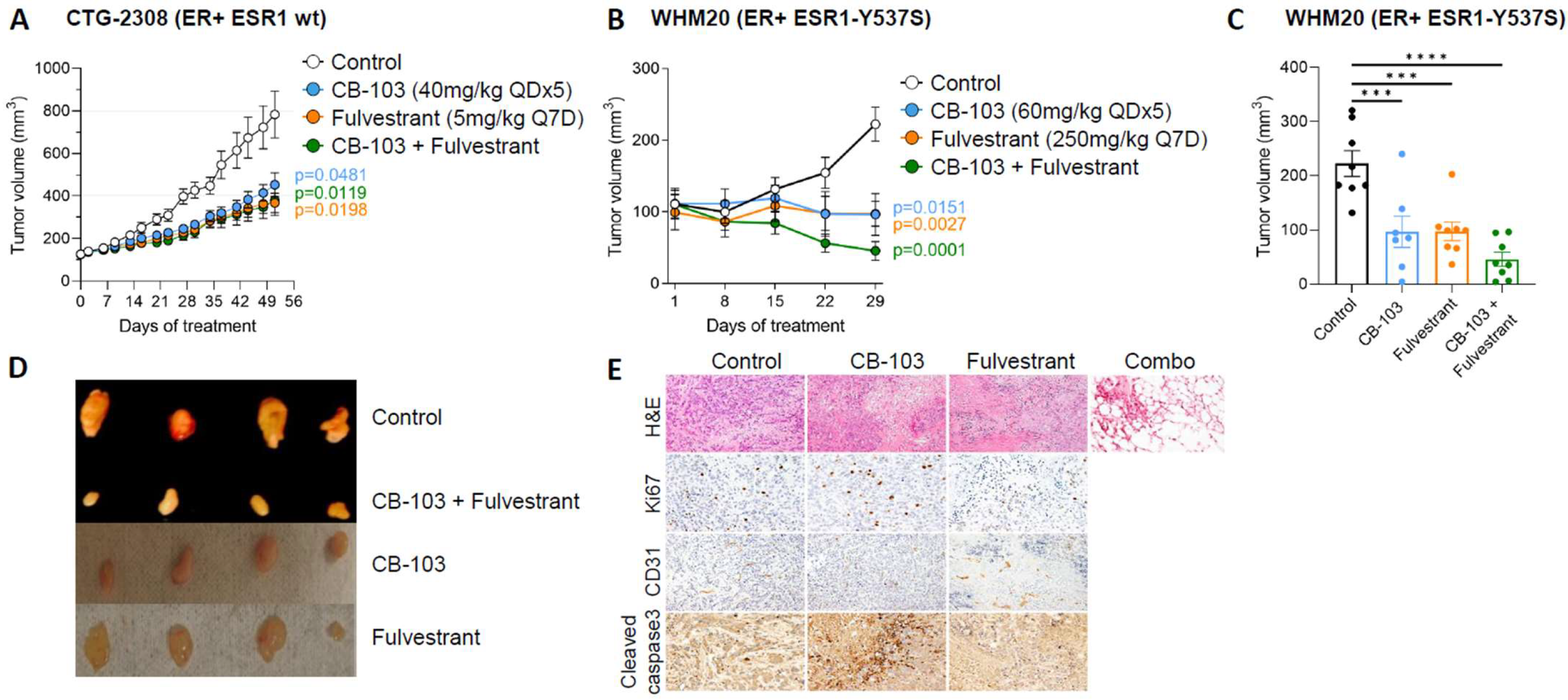
3.3. Combination Therapy with CB-103 Plus Fulvestrant Induces Tumor Regression in PDX Derived, ESR1-Mutant WHM20 Cell Line
3.4. RNAseq and Differential Gene Expression
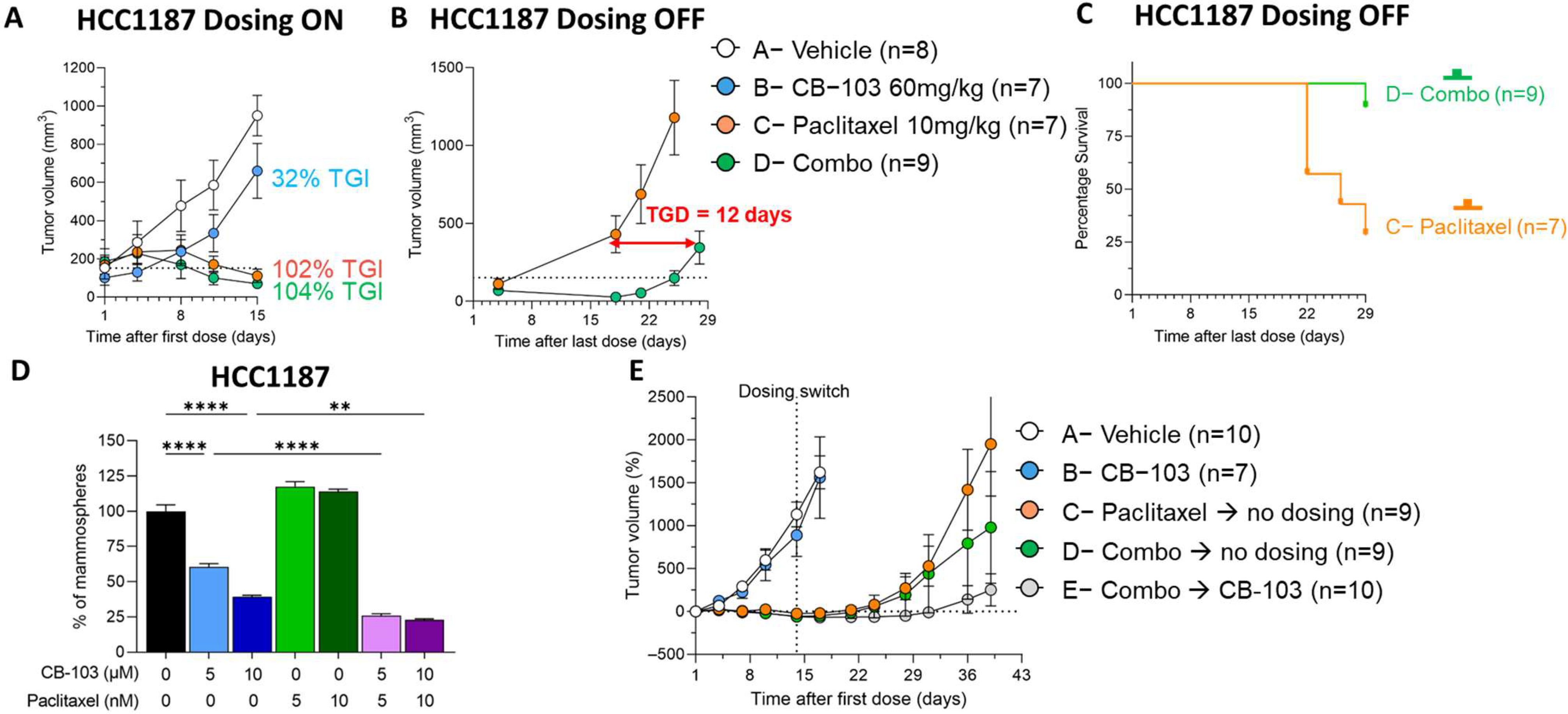
3.5. CB-103 Induces a Durable Response in Combination with Paclitaxel in TNBC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Hossain, F.; Majumder, S.; David, J.; Miele, L. Precision Medicine and Triple-Negative Breast Cancer: Current Landscape and Future Directions. Cancers 2021, 13, 3739. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.K.; Cheon, S.; Kim, H.; Lee, K.M.; Ryu, H.S.; Han, D. Discovery of Proteins Responsible for Resistance to Three Chemotherapy Drugs in Breast Cancer Cells Using Proteomics and Bioinformatics Analysis. Molecules 2022, 27, 1762. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Li, W.T.; Yang, Y.; Qi, Y.B.; Cheng, Y.; Wu, J.H. MiR-526b-3p Attenuates Breast Cancer Stem Cell Properties and Chemoresistance by Targeting HIF-2α/Notch Signaling. Front. Oncol. 2021, 11, 696269. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in oncology: The path forward. Nat. Rev. Drug Discov. 2021, 20, 125–144. [Google Scholar] [CrossRef]
- Rizzo, P.; Osipo, C.; Foreman, K.; Golde, T.; Osborne, B.; Miele, L. Rational targeting of Notch signaling in cancer. Oncogene 2008, 27, 5124–5131. [Google Scholar] [CrossRef] [PubMed]
- Mollen, E.W.J.; Ient, J.; Tjan-Heijnen, V.C.G.; Boersma, L.J.; Miele, L.; Smidt, M.L.; Vooijs, M. Moving Breast Cancer Therapy up a Notch. Front. Oncol. 2018, 8, 518. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, M.; Wu, H.; Xu, H.; Han, N.; Chu, Q.; Yu, S.; Chen, Y.; Wu, K. Expression of Notch1 Correlates with Breast Cancer Progression and Prognosis. PLoS ONE 2015, 10, e0131689. [Google Scholar] [CrossRef]
- Rizzo, P.; Miao, H.; D’Souza, G.; Osipo, C.; Song, L.L.; Yun, J.; Zhao, H.; Mascarenhas, J.; Wyatt, D.; Antico, G.; et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 2008, 68, 5226–5235. [Google Scholar] [CrossRef]
- Hao, L.; Rizzo, P.; Osipo, C.; Pannuti, A.; Wyatt, D.; Cheung, L.W.; Sonenshein, G.; Osborne, B.A.; Miele, L. Notch-1 activates estrogen receptor-α-dependent transcription via IKKα in breast cancer cells. Oncogene 2010, 29, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Pannuti, A.; Espinoza, I.; Zhu, H.; Hicks, C.; Zhu, X.; Caskey, M.; Rizzo, P.; D’Souza, G.; Backus, K.; et al. Crosstalk between PKCα and Notch-4 in endocrine-resistant breast cancer cells. Oncogenesis 2013, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Gelsomino, L.; Panza, S.; Giordano, C.; Barone, I.; Gu, G.; Spina, E.; Catalano, S.; Fuqua, S.; Ando, S. Mutations in the estrogen receptor alpha hormone binding domain promote stem cell phenotype through notch activation in breast cancer cell lines. Cancer Lett. 2018, 428, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Giuli, M.V.; Giuliani, E.; Screpanti, I.; Bellavia, D.; Checquolo, S. Notch Signaling Activation as a Hallmark for Triple-Negative Breast Cancer Subtype. J. Oncol. 2019, 2019, 8707053. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Peng, Q.; Jiang, I.; Carroll, C.; Han, G.; Rymer, I.; Lippincott, J.; Zachwieja, J.; Gajiwala, K.; Kraynov, E.; et al. Specific inhibition of Notch1 signaling enhances the antitumor efficacy of chemotherapy in triple negative breast cancer through reduction of cancer stem cells. Cancer Lett. 2013, 328, 261–270. [Google Scholar] [CrossRef]
- Bhola, N.E.; Jansen, V.M.; Koch, J.P.; Li, H.; Formisano, L.; Williams, J.A.; Grandis, J.R.; Arteaga, C.L. Treatment of Triple-Negative Breast Cancer with TORC1/2 Inhibitors Sustains a Drug-Resistant and Notch-Dependent Cancer Stem Cell Population. Cancer Res. 2016, 76, 440–452. [Google Scholar] [CrossRef]
- Guner, G.; Lichtenthaler, S.F. The substrate repertoire of γ-secretase/presenilin. Semin. Cell Dev. Biol. 2020, 105, 27–42. [Google Scholar] [CrossRef]
- Zhdanovskaya, N.; Firrincieli, M.; Lazzari, S.; Pace, E.; Scribani Rossi, P.; Felli, M.P.; Talora, C.; Screpanti, I.; Palermo, R. Targeting Notch to Maximize Chemotherapeutic Benefits: Rationale, Advanced Strategies, and Future Perspectives. Cancers 2021, 13, 5106. [Google Scholar] [CrossRef]
- Means-Powell, J.A.; Mayer, I.A.; Ismail-Khan, R.; Del Valle, L.; Tonetti, D.; Abramson, V.G.; Sanders, M.S.; Lush, R.M.; Sorrentino, C.; Majumder, S.; et al. A Phase Ib Dose Escalation Trial of RO4929097 (a γ-secretase inhibitor) in Combination with Exemestane in Patients with ER + Metastatic Breast Cancer (MBC). Clin. Breast Cancer 2022, 22, 103–114. [Google Scholar] [CrossRef]
- Fabbro, D.; Bauer, M.; Murone, M.; Lehal, R. Notch Inhibition in Cancer: Challenges and Opportunities. Chimia 2020, 74, 779–783. [Google Scholar] [CrossRef]
- Ianevski, A.; He, L.; Aittokallio, T.; Tang, J. SynergyFinder: A web application for analyzing drug combination dose-response matrix data. Bioinformatics 2017, 33, 2413–2415. [Google Scholar] [CrossRef]
- Reifel-Miller, A.E.; Conarty, D.M.; Valasek, K.M.; Iversen, P.W.; Burns, D.J.; Birch, K.A. Protein kinase C isozymes differentially regulate promoters containing PEA-3/12-O-tetradecanoylphorbol-13-acetate response element motifs. J. Biol. Chem. 1996, 271, 21666–21671. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Lehal, R.; Zaric, J.; Vigolo, M.; Urech, C.; Frismantas, V.; Zangger, N.; Cao, L.; Berger, A.; Chicote, I.; Loubery, S.; et al. Pharmacological disruption of the Notch transcription factor complex. Proc. Natl. Acad. Sci. USA 2020, 117, 16292–16301. [Google Scholar] [CrossRef]
- Paredes, J.; Zabaleta, J.; Garai, J.; Ji, P.; Imtiaz, S.; Spagnardi, M.; Alvarado, J.; Li, L.; Akadri, M.; Barrera, K.; et al. Immune-Related Gene Expression and Cytokine Secretion Is Reduced among African American Colon Cancer Patients. Front. Oncol. 2020, 10, 1498. [Google Scholar] [CrossRef]
- Harrison, H.; Farnie, G.; Howell, S.J.; Rock, R.E.; Stylianou, S.; Brennan, K.R.; Bundred, N.J.; Clarke, R.B. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010, 70, 709–718. [Google Scholar] [CrossRef]
- Crabtree, J.S.; Miele, L. Breast Cancer Stem Cells. Biomedicines 2018, 6, 77. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., III; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Ruiz-Borrego, M.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): One cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet Oncol. 2021, 22, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Belachew, E.B.; Sewasew, D.T. Molecular Mechanisms of Endocrine Resistance in Estrogen-Receptor-Positive Breast Cancer. Front. Endocrinol. 2021, 12, 599586, Correction in Front. Endocrinol. 2021, 12, 689705. [Google Scholar] [CrossRef]
- La Camera, G.; Gelsomino, L.; Caruso, A.; Panza, S.; Barone, I.; Bonofiglio, D.; Ando, S.; Giordano, C.; Catalano, S. The Emerging Role of Extracellular Vesicles in Endocrine Resistant Breast Cancer. Cancers 2021, 13, 1160. [Google Scholar] [CrossRef]
- Newman, S.; Howarth, K.D.; Greenman, C.D.; Bignell, G.R.; Tavare, S.; Edwards, P.A. The relative timing of mutations in a breast cancer genome. PLoS ONE 2013, 8, e64991. [Google Scholar] [CrossRef]
- Sardesai, S.; Badawi, M.; Mrozek, E.; Morgan, E.; Phelps, M.; Stephens, J.; Wei, L.; Kassem, M.; Ling, Y.; Lustberg, M.; et al. A phase I study of an oral selective gamma secretase (GS) inhibitor RO4929097 in combination with neoadjuvant paclitaxel and carboplatin in triple negative breast cancer. Investig. New Drugs 2020, 38, 1400–1410. [Google Scholar] [CrossRef]
- Tanei, T.; Choi, D.S.; Rodriguez, A.A.; Liang, D.H.; Dobrolecki, L.; Ghosh, M.; Landis, M.D.; Chang, J.C. Antitumor activity of Cetuximab in combination with Ixabepilone on triple negative breast cancer stem cells. Breast Cancer Res. 2016, 18, 6. [Google Scholar] [CrossRef]
- Aravilli, R.K.; Kohila, V.; Vikram, S.L. Heuristics in Role of Human Glutathione S-transferase Mu 1 as Nitric Oxide Carrier and its Engineered Variants for Enhanced Activity. Curr. Pharm. Biotechnol. 2021, 22, 2071–2084. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G mutation in estrogen receptor-α: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gomez, H.; et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef]
- Simoes, B.M.; O’Brien, C.S.; Eyre, R.; Silva, A.; Yu, L.; Sarmiento-Castro, A.; Alferez, D.G.; Spence, K.; Santiago-Gomez, A.; Chemi, F.; et al. Anti-estrogen Resistance in Human Breast Tumors Is Driven by JAG1-NOTCH4-Dependent Cancer Stem Cell Activity. Cell Rep. 2015, 12, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martin, M.; et al. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2020, 382, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, F.A.; Murray, P.J.; Ditsova, D.; Ferris, M.A.; Davies, P.; Dale, J.K. CDK1 and CDK2 regulate NICD1 turnover and the periodicity of the segmentation clock. EMBO Rep. 2019, 20, e46436. [Google Scholar] [CrossRef] [PubMed]
- Pallavi, S.K.; Ho, D.M.; Hicks, C.; Miele, L.; Artavanis-Tsakonas, S. Notch and Mef2 synergize to promote proliferation and metastasis through JNK signal activation in Drosophila. EMBO J. 2012, 31, 2895–2907. [Google Scholar] [CrossRef]
- Yao, L.; Tian, F. GRWD1 affects the proliferation, apoptosis, invasion and migration of triple negative breast cancer through the Notch signaling pathway. Exp. Ther. Med. 2022, 24, 473. [Google Scholar] [CrossRef]
- Liu, D.; Hofman, P. Expression of NOTCH1, NOTCH4, HLA-DMA and HLA-DRA is synergistically associated with T cell exclusion, immune checkpoint blockade efficacy and recurrence risk in ER-negative breast cancer. Cell Oncol. 2022, 45, 463–477. [Google Scholar] [CrossRef]
- De Santis, F.; Romero-Cordoba, S.L.; Castagnoli, L.; Volpari, T.; Faraci, S.; Fuca, G.; Tagliabue, E.; De Braud, F.; Pupa, S.M.; Di Nicola, M. BCL6 and the Notch pathway: A signaling axis leading to a novel druggable biotarget in triple negative breast cancer. Cell. Oncol. 2022, 45, 257–274. [Google Scholar] [CrossRef]
- Jaiswal, A.; Murakami, K.; Elia, A.; Shibahara, Y.; Done, S.J.; Wood, S.A.; Donato, N.J.; Ohashi, P.S.; Reedijk, M. Therapeutic inhibition of USP9x-mediated Notch signaling in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2101592118. [Google Scholar] [CrossRef]
- Pappas, K.; Martin, T.C.; Wolfe, A.L.; Nguyen, C.B.; Su, T.; Jin, J.; Hibshoosh, H.; Parsons, R. NOTCH and EZH2 collaborate to repress PTEN expression in breast cancer. Commun. Biol. 2021, 4, 312. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Lin, L.; Li, X.; Lu, A.; Hou, C.; Wu, Q.; Hu, X.; Zhou, Z.; Chen, Z.; Tang, F. ADAM10 is involved in the oncogenic process and chemo-resistance of triple-negative breast cancer via regulating Notch1 signaling pathway, CD44 and PrPc. Cancer Cell Int. 2021, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, J.; Gast, K.; Quiroga, D.; Lustberg, M.; Williams, N. Triple-negative breast cancer: Promising prognostic biomarkers currently in development. Expert Rev. Anticancer Ther. 2021, 21, 135–148. [Google Scholar] [CrossRef]
- Granit, R.Z.; Masury, H.; Condiotti, R.; Fixler, Y.; Gabai, Y.; Glikman, T.; Dalin, S.; Winter, E.; Nevo, Y.; Carmon, E.; et al. Regulation of Cellular Heterogeneity and Rates of Symmetric and Asymmetric Divisions in Triple-Negative Breast Cancer. Cell Rep. 2018, 24, 3237–3250. [Google Scholar] [CrossRef]
- Kang, L.; Mao, J.; Tao, Y.; Song, B.; Ma, W.; Lu, Y.; Zhao, L.; Li, J.; Yang, B.; Li, L. MicroRNA-34a suppresses the breast cancer stem cell-like characteristics by downregulating Notch1 pathway. Cancer Sci. 2015, 106, 700–708. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Most Synergistic Area Score | ||
|---|---|---|
| Drug Combination | ER+ BC (MCF-7) | TNB (HCC1187) |
| PD 0332991 (Palbociclib) + CB-103 | 15.85 | −0.01 |
| Paclitaxel + CB-103 | 24.87 | 8.73 |
| Fulvestrant + CB-103 | 24.64 | −2.49 |
| Relevant Pathway Enrichment | Enrichment Score | p Value |
|---|---|---|
| Control vs. Fulvestrant | ||
| cAMP signaling pathway | 10.64280 | 2.39 × 10−5 |
| TGF-β signaling pathway | 8.06829 | 0.000313 |
| Complement and coagulation cascades | 6.78715 | 0.001128 |
| Calcium signaling pathway | 6.60461 | 0.001354 |
| cGMP-PKG signaling pathway | 6.35896 | 0.001731 |
| Aldosterone synthesis and secretion | 5.47526 | 0.004189 |
| Control vs. CB-103 | ||
| TGF-β signaling pathway | 4.36199 | 0.012753 |
| Bladder cancer | 3.91465 | 0.019948 |
| Arginine and proline metabolism | 3.69323 | 0.024891 |
| Drug metabolism—other enzymes | 3.22466 | 0.039770 |
| Transcriptional misregulation in cancer | 3.02111 | 0.048747 |
| Control vs. Combo | ||
| Estrogen signaling pathway | 8.35942 | 0.000234 |
| Complement and coagulation cascades | 7.10170 | 0.000824 |
| Platelet activation | 5.41271 | 0.004460 |
| Cytokine-cytokine receptor interaction | 4.75188 | 0.008635 |
| cAMP signaling pathway | 4.51626 | 0.010930 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vigolo, M.; Urech, C.; Lamy, S.; Monticone, G.; Zabaleta, J.; Hossain, F.; Wyczechowska, D.; Del Valle, L.; O’Regan, R.M.; Miele, L.; et al. The Efficacy of CB-103, a First-in-Class Transcriptional Notch Inhibitor, in Preclinical Models of Breast Cancer. Cancers 2023, 15, 3957. https://doi.org/10.3390/cancers15153957
Vigolo M, Urech C, Lamy S, Monticone G, Zabaleta J, Hossain F, Wyczechowska D, Del Valle L, O’Regan RM, Miele L, et al. The Efficacy of CB-103, a First-in-Class Transcriptional Notch Inhibitor, in Preclinical Models of Breast Cancer. Cancers. 2023; 15(15):3957. https://doi.org/10.3390/cancers15153957
Chicago/Turabian StyleVigolo, Michele, Charlotte Urech, Sebastien Lamy, Giulia Monticone, Jovanny Zabaleta, Fokhrul Hossain, Dorota Wyczechowska, Luis Del Valle, Ruth M. O’Regan, Lucio Miele, and et al. 2023. "The Efficacy of CB-103, a First-in-Class Transcriptional Notch Inhibitor, in Preclinical Models of Breast Cancer" Cancers 15, no. 15: 3957. https://doi.org/10.3390/cancers15153957
APA StyleVigolo, M., Urech, C., Lamy, S., Monticone, G., Zabaleta, J., Hossain, F., Wyczechowska, D., Del Valle, L., O’Regan, R. M., Miele, L., Lehal, R., & Majumder, S. (2023). The Efficacy of CB-103, a First-in-Class Transcriptional Notch Inhibitor, in Preclinical Models of Breast Cancer. Cancers, 15(15), 3957. https://doi.org/10.3390/cancers15153957