
Cellular Adaptation Takes Advantage of Atavistic Regression Programs during Carcinogenesis


{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Survival under Harsh Environmental Conditions: The Central Role of Mitochondria in Adaptive Mechanisms
3. Hypoxia and Adaptive Mechanisms in Cancer Metabolism
4. Cancer Cell Adaptation to Glucose Restriction
5. The Acidification of the TME
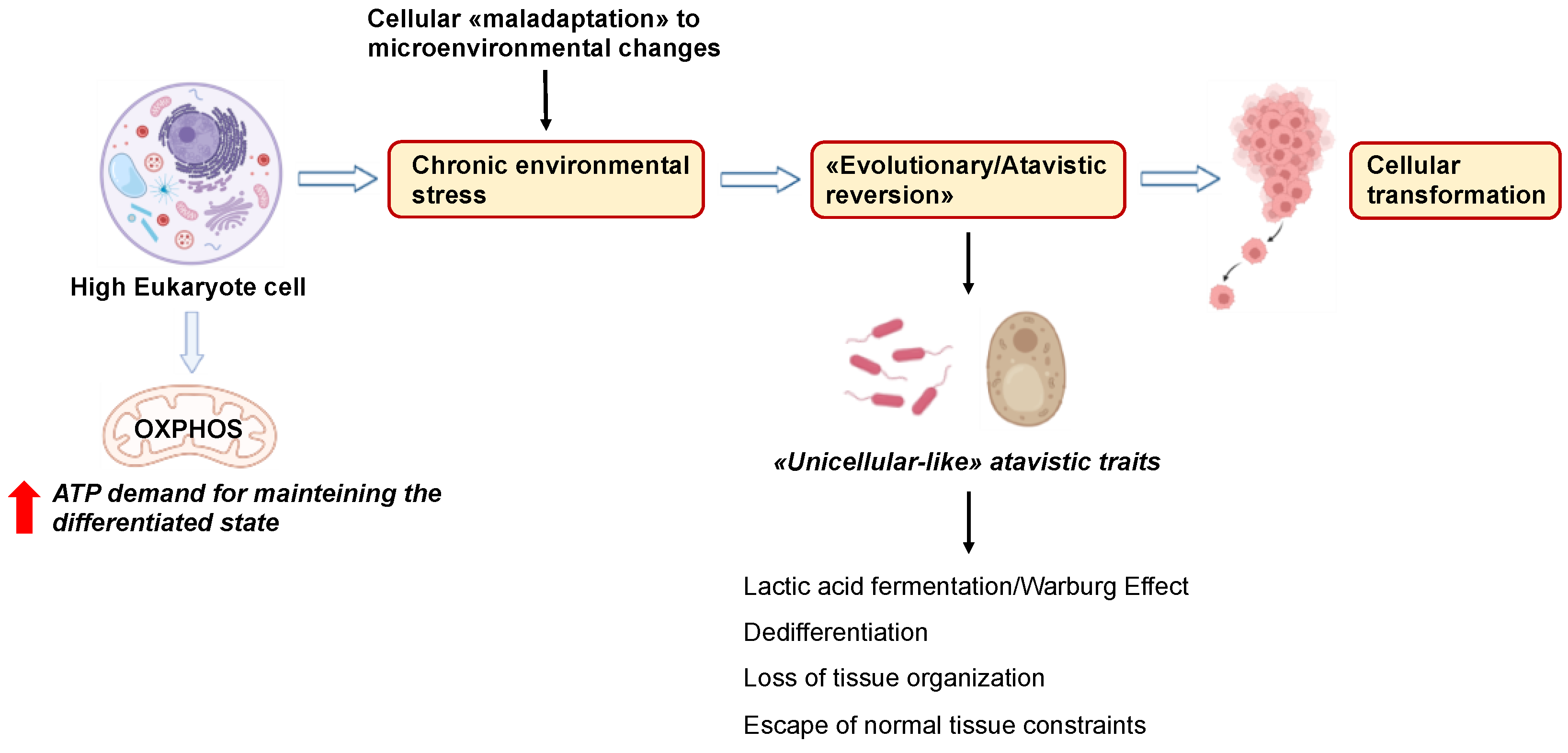
6. Cancer Cells Exploit “Atavistic Programs” for Their Adaptation and Plasticity to the Changed Microenvironment
6.1. “Atavistic Programs“ as a New Interpretive Framework in Cancer Biology
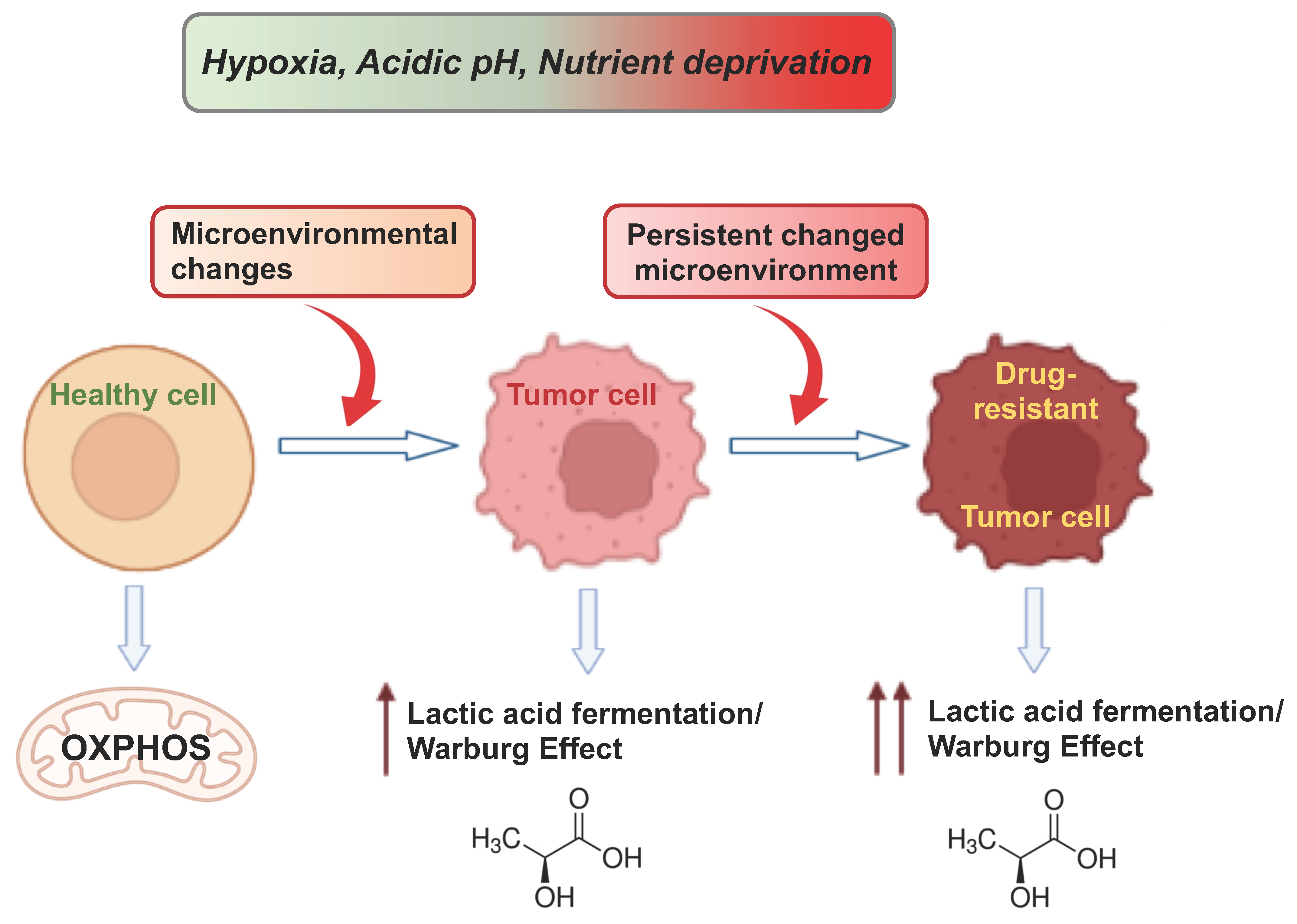
6.2. Atavistic Regression as an Adaptive Response to the Changed Microenvironment
6.3. Microenvironmental Changes Deregulate Nucleo-Cytoplasmic Crosstalk during Tumorigenesis
6.4. Atavism and Adaptation in Cancer Drug Resistance and Metastasis
7. Targeting Energy Metabolism and the Microenvironment in Cancer with Different Strategies
8. Discussion and Future Directions
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Eisenberg, L.; Eisenberg-Bord, M.; Eisenberg-Lerner, A.; Sagi-Eisenberg, R. Metabolic alterations in the tumor microenvironment and their role in oncogenesis. Cancer Lett. 2020, 484, 65–71. [Google Scholar] [CrossRef]
- Anastasiou, D. Tumour microenvironment factors shaping the cancer metabolism landscape. Br. J. Cancer 2017, 116, 277–286. [Google Scholar] [CrossRef]
- Marine, J.-C.; Dawson, S.-J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef]
- Bell, C.C.; Gilan, O. Principles and mechanisms of non-genetic resistance in cancer. Br. J. Cancer 2020, 122, 465–472. [Google Scholar] [CrossRef]
- Monieb, A.M.A.; Anika, N. Current developments in modelling the tumour microenvironment in vitro: Incorporation of biochemical and physical gradients. Organs-on-a-Chip 2021, 3, 100012. [Google Scholar] [CrossRef]
- Yee, P.P.; Li, W. Tumor necrosis: A synergistic consequence of metabolic stress and inflammation. Bioessays 2021, 43, e2100029. [Google Scholar] [CrossRef]
- Sistigu, A.; Musella, M.; Galassi, C.; Vitale, I.; De Maria, R. Tuning Cancer Fate: Tumor Microenvironment’s Role in Cancer Stem Cell Quiescence and Reawakening. Front. Immunol. 2020, 11, 2166. [Google Scholar] [CrossRef]
- Marescal, O.; Cheeseman, I.M. Cellular Mechanisms and Regulation of Quiescence. Dev. Cell 2020, 55, 259–271. [Google Scholar] [CrossRef]
- Boussadia, Z.; Gambardella, A.R.; Mattei, F.; Parolini, I. Acidic and Hypoxic Microenvironment in Melanoma: Impact of Tumour Exosomes on Disease Progression. Cells 2021, 10, 3311. [Google Scholar] [CrossRef]
- Palm, W. Metabolic plasticity allows cancer cells to thrive under nutrient starvation. Proc. Natl. Acad. Sci. USA 2021, 118, e2102057118. [Google Scholar] [CrossRef]
- Tsai, P.Y.; Lee, M.S.; Jadhav, U.; Naqvi, I.; Madha, S.; Adler, A.; Mistry, M.; Naumenko, S.; Lewis, C.A.; Hitchcock, D.S.; et al. Adaptation of pancreatic cancer cells to nutrient deprivation is reversible and requires glutamine synthetase stabilization by mTORC1. Proc. Natl. Acad. Sci. USA 2021, 118, e2003014118. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Vander Heiden, M.G.; Muir, A. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 2019, 8, e44235. [Google Scholar] [CrossRef]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef]
- Simoes, R.V.; Serganova, I.S.; Kruchevsky, N.; Leftin, A.; Shestov, A.A.; Thaler, H.T.; Sukenick, G.; Locasale, J.W.; Blasberg, R.G.; Koutcher, J.A.; et al. Metabolic plasticity of metastatic breast cancer cells: Adaptation to changes in the microenvironment. Neoplasia 2015, 17, 671–684. [Google Scholar] [CrossRef]
- Lineweaver, C.H.; Bussey, K.J.; Blackburn, A.C.; Davies, P.C.W. Cancer progression as a sequence of atavistic reversions. Bioessays 2021, 43, e2000305. [Google Scholar] [CrossRef]
- Mazzocca, A. The Systemic-Evolutionary Theory of the Origin of Cancer (SETOC): A New Interpretative Model of Cancer as a Complex Biological System. Int. J. Mol. Sci. 2019, 20, 4885. [Google Scholar] [CrossRef] [PubMed]
- Mazzocca, A. The SETOC: Visualizing Carcinogenesis through a Different Lens. Atlas of Science Another View on Science. Available online: https://atlasofscience.org (accessed on 25 June 2023).
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Oliveira, G.L.; Coelho, A.R.; Marques, R.; Oliveira, P.J. Cancer cell metabolism: Rewiring the mitochondrial hub. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166016. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Boukalova, S.; Hubackova, S.; Milosevic, M.; Ezrova, Z.; Neuzil, J.; Rohlena, J. Dihydroorotate dehydrogenase in oxidative phosphorylation and cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165759. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e359. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, K.M.; Scarbrough, P.M.; Ribeiro, A.; Richardson, R.; Yuan, H.; Sonveaux, P.; Landon, C.D.; Chi, J.T.; Pizzo, S.; Schroeder, T.; et al. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS ONE 2013, 8, e75154. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Guo, Y.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Zdralevic, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.M.; et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the "Warburg effect" restricting tumor growth to oxidative metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef]
- Haas, R.; Cucchi, D.; Smith, J.; Pucino, V.; Macdougall, C.E.; Mauro, C. Intermediates of Metabolism: From Bystanders to Signalling Molecules. Trends Biochem. Sci. 2016, 41, 460–471. [Google Scholar] [CrossRef]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Biophys. Acta 2016, 1857, 1086–1101. [Google Scholar] [CrossRef]
- Xu, H.; Chen, X.; Xu, X.; Shi, R.; Suo, S.; Cheng, K.; Zheng, Z.; Wang, M.; Wang, L.; Zhao, Y.; et al. Lysine Acetylation and Succinylation in HeLa Cells and their Essential Roles in Response to UV-induced Stress. Sci. Rep. 2016, 6, 30212. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, I.A.; Machado, N.G.; Skildum, A.J.; Scott, P.M.; Oliveira, P.J. Mitochondrial remodeling in cancer metabolism and survival: Potential for new therapies. Biochim. Biophys. Acta 2012, 1826, 238–254. [Google Scholar] [CrossRef]
- Oliva, C.R.; Markert, T.; Ross, L.J.; White, E.L.; Rasmussen, L.; Zhang, W.; Everts, M.; Moellering, D.R.; Bailey, S.M.; Suto, M.J.; et al. Identification of Small Molecule Inhibitors of Human Cytochrome c Oxidase That Target Chemoresistant Glioma Cells. J. Biol. Chem. 2016, 291, 24188–24199. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.W.; Srinivasan, S.; Guha, M.; Avadhani, N.G. Defects in cytochrome c oxidase expression induce a metabolic shift to glycolysis and carcinogenesis. Genom. Data 2015, 6, 99–107. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Dong, D.W.; Whelan, K.A.; Ruthel, G.; Uchikado, Y.; Natsugoe, S.; Nakagawa, H.; Avadhani, N.G. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 2016, 35, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Chang, J.; Lee, H.S.; Kwon, H.J. Mitochondrial UQCRB as a new molecular prognostic biomarker of human colorectal cancer. Exp. Mol. Med. 2017, 49, e391. [Google Scholar] [CrossRef]
- Herrmann, P.C.; Gillespie, J.W.; Charboneau, L.; Bichsel, V.E.; Paweletz, C.P.; Calvert, V.S.; Kohn, E.C.; Emmert-Buck, M.R.; Liotta, L.A.; Petricoin, E.F., 3rd. Mitochondrial proteome: Altered cytochrome c oxidase subunit levels in prostate cancer. Proteomics 2003, 3, 1801–1810. [Google Scholar] [CrossRef]
- Gnocchi, D.; Sabba, C.; Mazzocca, A. Lactic acid fermentation: A maladaptive mechanism and an evolutionary throwback boosting cancer drug resistance. Biochimie 2023, 208, 180–185. [Google Scholar] [CrossRef]
- Sotgia, F.; Lisanti, M.P. Mitochondrial markers predict survival and progression in non-small cell lung cancer (NSCLC) patients: Use as companion diagnostics. Oncotarget 2017, 8, 68095–68107. [Google Scholar] [CrossRef]
- Sotgia, F.; Lisanti, M.P. Mitochondrial mRNA transcripts predict overall survival, tumor recurrence and progression in serous ovarian cancer: Companion diagnostics for cancer therapy. Oncotarget 2017, 8, 66925–66939. [Google Scholar] [CrossRef]
- Sotgia, F.; Fiorillo, M.; Lisanti, M.P. Mitochondrial markers predict recurrence, metastasis and tamoxifen-resistance in breast cancer patients: Early detection of treatment failure with companion diagnostics. Oncotarget 2017, 8, 68730–68745. [Google Scholar] [CrossRef]
- De Heer, E.C.; Jalving, M.; Harris, A.L. HIFs, angiogenesis, and metabolism: Elusive enemies in breast cancer. J. Clin. Investig. 2020, 130, 5074–5087. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef]
- Paredes, F.; Williams, H.C.; San Martin, A. Metabolic adaptation in hypoxia and cancer. Cancer Lett. 2021, 502, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Sebestyen, A.; Kopper, L.; Danko, T.; Timar, J. Hypoxia Signaling in Cancer: From Basics to Clinical Practice. Pathol. Oncol. Res. 2021, 27, 1609802. [Google Scholar] [CrossRef]
- Lee, S.H.; Golinska, M.; Griffiths, J.R. HIF-1-Independent Mechanisms Regulating Metabolic Adaptation in Hypoxic Cancer Cells. Cells 2021, 10, 2371. [Google Scholar] [CrossRef] [PubMed]
- Mathews, E.H.; Visagie, M.H.; Meyer, A.A.; Joubert, A.M.; Mathews, G.E. In vitro quantification: Long-term effect of glucose deprivation on various cancer cell lines. Nutrition 2020, 74, 110748. [Google Scholar] [CrossRef]
- Grasmann, G.; Mondal, A.; Leithner, K. Flexibility and Adaptation of Cancer Cells in a Heterogenous Metabolic Microenvironment. Int. J. Mol. Sci. 2021, 22, 1476. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef]
- Nunes, S.C. Tumor Microenvironment—Selective Pressures Boosting Cancer Progression. Adv. Exp. Med. Biol. 2020, 1219, 35–49. [Google Scholar] [CrossRef]
- Bustos, S.O.; Antunes, F.; Rangel, M.C.; Chammas, R. Emerging Autophagy Functions Shape the Tumor Microenvironment and Play a Role in Cancer Progression—Implications for Cancer Therapy. Front. Oncol. 2020, 10, 606436. [Google Scholar] [CrossRef]
- Wang, S.; Lv, Y.; Zhou, Y.; Ling, J.; Wang, H.; Gu, D.; Wang, C.; Qin, W.; Zheng, X.; Jin, H. Acidic extracellular pH induces autophagy to promote anoikis resistance of hepatocellular carcinoma cells via downregulation of miR-3663-3p. J. Cancer 2021, 12, 3418–3426. [Google Scholar] [CrossRef]
- Sadeghi, M.; Ordway, B.; Rafiei, I.; Borad, P.; Fang, B.; Koomen, J.L.; Zhang, C.; Yoder, S.; Johnson, J.; Damaghi, M. Integrative Analysis of Breast Cancer Cells Reveals an Epithelial-Mesenchymal Transition Role in Adaptation to Acidic Microenvironment. Front. Oncol. 2020, 10, 304. [Google Scholar] [CrossRef]
- Ibrahim-Hashim, A.; Estrella, V. Acidosis and cancer: From mechanism to neutralization. Cancer Metastasis Rev. 2019, 38, 149–155. [Google Scholar] [CrossRef]
- Emran, T.B.; Shahriar, A.; Mahmud, A.R.; Rahman, T.; Abir, M.H.; Siddiquee, M.F.; Ahmed, H.; Rahman, N.; Nainu, F.; Wahyudin, E.; et al. Multidrug Resistance in Cancer: Understanding Molecular Mechanisms, Immunoprevention and Therapeutic Approaches. Front. Oncol. 2022, 12, 891652. [Google Scholar] [CrossRef]
- Pillai, S.R.; Damaghi, M.; Marunaka, Y.; Spugnini, E.P.; Fais, S.; Gillies, R.J. Causes, consequences, and therapy of tumors acidosis. Cancer Metastasis Rev. 2019, 38, 205–222. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Goncalves, R.L.S.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta 2015, 1847, 171–181. [Google Scholar] [CrossRef]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Adam, C.; Paolini, L.; Gueguen, N.; Mabilleau, G.; Preisser, L.; Blanchard, S.; Pignon, P.; Manero, F.; Le Mao, M.; Morel, A.; et al. Acetoacetate protects macrophages from lactic acidosis-induced mitochondrial dysfunction by metabolic reprograming. Nat. Commun. 2021, 12, 7115. [Google Scholar] [CrossRef]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Piasentin, N.; Milotti, E.; Chignola, R. The control of acidity in tumor cells: A biophysical model. Sci. Rep. 2020, 10, 13613. [Google Scholar] [CrossRef]
- Wykoff, C.C.; Beasley, N.J.; Watson, P.H.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.D.; Turley, H.; Talks, K.L.; Maxwell, P.H.; et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar] [PubMed]
- Andreucci, E.; Peppicelli, S.; Ruzzolini, J.; Bianchini, F.; Biagioni, A.; Papucci, L.; Magnelli, L.; Mazzanti, B.; Stecca, B.; Calorini, L. The acidic tumor microenvironment drives a stem-like phenotype in melanoma cells. J. Mol. Med. 2020, 98, 1431–1446. [Google Scholar] [CrossRef]
- Wu, T.C.; Liao, C.Y.; Lu, W.C.; Chang, C.R.; Tsai, F.Y.; Jiang, S.S.; Chen, T.H.; Lin, K.M.; Chen, L.T.; Chang, W.W. Identification of distinct slow mode of reversible adaptation of pancreatic ductal adenocarcinoma to the prolonged acidic pH microenvironment. J. Exp. Clin. Cancer Res. CR 2022, 41, 137. [Google Scholar] [CrossRef]
- Rohani, N.; Hao, L.; Alexis, M.S.; Joughin, B.A.; Krismer, K.; Moufarrej, M.N.; Soltis, A.R.; Lauffenburger, D.A.; Yaffe, M.B.; Burge, C.B.; et al. Acidification of Tumor at Stromal Boundaries Drives Transcriptome Alterations Associated with Aggressive Phenotypes. Cancer Res. 2019, 79, 1952–1966. [Google Scholar] [CrossRef]
- Wojtkowiak, J.W.; Verduzco, D.; Schramm, K.J.; Gillies, R.J. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol. Pharm. 2011, 8, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wen, J.; Zhou, L.; Qin, L. Utilizing a high-throughput microfluidic platform to study hypoxia-driven mesenchymal-mode cell migration. Integr. Biol. 2015, 7, 672–680. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef]
- Quinn, W.J., 3rd; Jiao, J.; TeSlaa, T.; Stadanlick, J.; Wang, Z.; Wang, L.; Akimova, T.; Angelin, A.; Schafer, P.M.; Cully, M.D.; et al. Lactate Limits T Cell Proliferation via the NAD(H) Redox State. Cell Rep. 2020, 33, 108500. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Szent-Gyorgyi, A.; Hegyeli, A.; Mc, L.J. Cancer therapy: A possible new approach. Science 1963, 140, 1391–1392. [Google Scholar] [CrossRef]
- Szent-Gyorgyi, A. The living state and cancer. Physiol. Chem. Phys. 1980, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Szent-Gyorgyi, A. Bioelectronics and cancer. J. Bioenerg. 1973, 4, 533–562. [Google Scholar] [CrossRef]
- Weinhouse, S. On respiratory impairment in cancer cells. Science 1956, 124, 267–269. [Google Scholar] [CrossRef]
- Van den Brink, J.; Canelas, A.B.; van Gulik, W.M.; Pronk, J.T.; Heijnen, J.J.; de Winde, J.H.; Daran-Lapujade, P. Dynamics of glycolytic regulation during adaptation of Saccharomyces cerevisiae to fermentative metabolism. Appl. Environ. Microbiol. 2008, 74, 5710–5723. [Google Scholar] [CrossRef]
- Yuichiro, S.; Kenneth, Z.M.; Nijhout, H.F. 15—Regulation of Phenotypic Plasticity from the Perspective of Evolutionary Developmental Biology. In Phenotypic Switching: Implications in Biology and Medicine; Academic Press: Cambridge, MA, USA, 2020; pp. 403–442. [Google Scholar] [CrossRef]
- Ehrenreich, I.M.; Pfennig, D.W. Genetic assimilation: A review of its potential proximate causes and evolutionary consequences. Ann. Bot. 2016, 117, 769–779. [Google Scholar] [CrossRef]
- Paaby, A.B.; Testa, N.D. Developmental Plasticity and Evolution. In Evolutionary Developmental Biology; Nuño de la Rosa, L., Müller, G.B., Eds.; Springer: Cham, Switzerland, 2021. [Google Scholar]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L.; et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef]
- Gnocchi, D.; Sabba, C.; Massimi, M.; Mazzocca, A. Metabolism as a New Avenue for Hepatocellular Carcinoma Therapy. Int. J. Mol. Sci. 2023, 24, 3710. [Google Scholar] [CrossRef]
- Shegay, P.V.; Zabolotneva, A.A.; Shatova, O.P.; Shestopalov, A.V.; Kaprin, A.D. Evolutionary View on Lactate-Dependent Mechanisms of Maintaining Cancer Cell Stemness and Reprimitivization. Cancers 2022, 14, 4552. [Google Scholar] [CrossRef]
- Kocianova, E.; Piatrikova, V.; Golias, T. Revisiting the Warburg Effect with Focus on Lactate. Cancers 2022, 14, 6028. [Google Scholar] [CrossRef]
- Perez-Tomas, R.; Perez-Guillen, I. Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment. Cancers 2020, 12, 3244. [Google Scholar] [CrossRef]
- Dzeja, P.P.; Bortolon, R.; Perez-Terzic, C.; Holmuhamedov, E.L.; Terzic, A. Energetic communication between mitochondria and nucleus directed by catalyzed phosphotransfer. Proc. Natl. Acad. Sci. USA 2002, 99, 10156–10161. [Google Scholar] [CrossRef]
- De Boer, V.C.; Houten, S.M. A mitochondrial expatriate: Nuclear pyruvate dehydrogenase. Cell 2014, 158, 9–10. [Google Scholar] [CrossRef]
- Liu, X.; Si, W.; He, L.; Yang, J.; Peng, Y.; Ren, J.; Liu, X.; Jin, T.; Yu, H.; Zhang, Z.; et al. The existence of a nonclassical TCA cycle in the nucleus that wires the metabolic-epigenetic circuitry. Signal Transduct. Target. Ther. 2021, 6, 375. [Google Scholar] [CrossRef]
- Kafkia, E.; Andres-Pons, A.; Ganter, K.; Seiler, M.; Smith, T.S.; Andrejeva, A.; Jouhten, P.; Pereira, F.; Franco, C.; Kuroshchenkova, A.; et al. Operation of a TCA cycle subnetwork in the mammalian nucleus. Sci. Adv. 2022, 8, eabq5206. [Google Scholar] [CrossRef]
- Xu, D.; Shao, F.; Bian, X.; Meng, Y.; Liang, T.; Lu, Z. The Evolving Landscape of Noncanonical Functions of Metabolic Enzymes in Cancer and Other Pathologies. Cell Metab. 2021, 33, 33–50. [Google Scholar] [CrossRef]
- Shapiro, J.A. All living cells are cognitive. Biochem. Biophys. Res. Commun. 2021, 564, 134–149. [Google Scholar] [CrossRef]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e237. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. What Are the Reasons for Continuing Failures in Cancer Therapy? Are Misleading/Inappropriate Preclinical Assays to Be Blamed? Might Some Modern Therapies Cause More Harm than Benefit? Int. J. Mol. Sci. 2022, 23, 3217. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Brown, J.S. Integrating evolutionary dynamics into cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 675–686. [Google Scholar] [CrossRef]
- Gnocchi, D.; Sabba, C.; Mazzocca, A. The Edible Plant Crithmum maritimum Shows Nutraceutical Properties by Targeting Energy Metabolism in Hepatic Cancer. Plant Foods Hum. Nutr. 2022, 77, 481–483. [Google Scholar] [CrossRef]
- Gnocchi, D.; Del Coco, L.; Girelli, C.R.; Castellaneta, F.; Cesari, G.; Sabba, C.; Fanizzi, F.P.; Mazzocca, A. (1)H-NMR metabolomics reveals a multitarget action of Crithmum maritimum ethyl acetate extract in inhibiting hepatocellular carcinoma cell growth. Sci. Rep. 2021, 11, 1259. [Google Scholar] [CrossRef]
- Gnocchi, D.; Cesari, G.; Calabrese, G.J.; Capone, R.; Sabba, C.; Mazzocca, A. Inhibition of Hepatocellular Carcinoma Growth by Ethyl Acetate Extracts of Apulian Brassica oleracea L. and Crithmum maritimum L. Plant Foods Hum. Nutr. 2020, 75, 33–40. [Google Scholar] [CrossRef]
- Gnocchi, D.; Sabba, C.; Mazzocca, A. Crithmum maritimum Improves Sorafenib Sensitivity by Decreasing Lactic Acid Fermentation and Inducing a Pro-Hepatocyte Marker Profile in Hepatocellular Carcinoma. Plant Foods Hum. Nutr. 2023, 78, 230–232. [Google Scholar] [CrossRef]
- Gnocchi, D.; Castellaneta, F.; Cesari, G.; Fiore, G.; Sabba, C.; Mazzocca, A. Treatment of liver cancer cells with ethyl acetate extract of Crithmum maritimum permits reducing sorafenib dose and toxicity maintaining its efficacy. J. Pharm. Pharmacol. 2021, 73, 1369–1376. [Google Scholar] [CrossRef]
- Podpeskar, A.; Crazzolara, R.; Kropshofer, G.; Hetzer, B.; Meister, B.; Muller, T.; Salvador, C. Omega-3 Fatty Acids and Their Role in Pediatric Cancer. Nutrients 2021, 13, 1800. [Google Scholar] [CrossRef]
- Huang, C.C.; Hung, C.H.; Hung, T.W.; Lin, Y.C.; Wang, C.J.; Kao, S.H. Dietary delphinidin inhibits human colorectal cancer metastasis associating with upregulation of miR-204-3p and suppression of the integrin/FAK axis. Sci. Rep. 2019, 9, 18954. [Google Scholar] [CrossRef]
- Ksouri, R.; Ksouri, W.M.; Jallali, I.; Debez, A.; Magne, C.; Hiroko, I.; Abdelly, C. Medicinal halophytes: Potent source of health promoting biomolecules with medical, nutraceutical and food applications. Crit. Rev. Biotechnol. 2012, 32, 289–326. [Google Scholar] [CrossRef]
- Pilankar, A.; Singhavi, H.; Raghuram, G.V.; Siddiqui, S.; Khare, N.K.; Jadhav, V.; Tandel, H.; Pal, K.; Bhattacharjee, A.; Chaturvedi, P.; et al. A pro-oxidant combination of resveratrol and copper down-regulates hallmarks of cancer and immune checkpoints in patients with advanced oral cancer: Results of an exploratory study (RESCU 004). Front. Oncol. 2022, 12, 1000957. [Google Scholar] [CrossRef]
- Baronzio, G.; Parmar, G.; Baronzio, M. Overview of Methods for Overcoming Hindrance to Drug Delivery to Tumors, with Special Attention to Tumor Interstitial Fluid. Front. Oncol. 2015, 5, 165. [Google Scholar] [CrossRef] [PubMed]
- Herst, P.M.; Carson, G.M.; Eccles, D.A.; Berridge, M.V. Bioenergetic and Metabolic Adaptation in Tumor Progression and Metastasis. Front. Oncol. 2022, 12, 857686. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C. Stem Cell Metabolism in Cancer and Healthy Tissues: Pyruvate in the Limelight. Front. Pharmacol. 2017, 8, 958. [Google Scholar] [CrossRef] [PubMed]
- Mosier, J.A.; Schwager, S.C.; Boyajian, D.A.; Reinhart-King, C.A. Cancer cell metabolic plasticity in migration and metastasis. Clin. Exp. Metastasis 2021, 38, 343–359. [Google Scholar] [CrossRef]
- Fokas, E.; Engenhart-Cabillic, R.; Daniilidis, K.; Rose, F.; An, H.X. Metastasis: The seed and soil theory gains identity. Cancer Metastasis Rev. 2007, 26, 705–715. [Google Scholar] [CrossRef]
- Arena, G.O.; Arena, V.; Arena, M.; Abdouh, M. Transfer of malignant traits as opposed to migration of cells: A novel concept to explain metastatic disease. Med. Hypotheses 2017, 100, 82–86. [Google Scholar] [CrossRef]
- Abdouh, M.; Hamam, D.; Gao, Z.H.; Arena, V.; Arena, M.; Arena, G.O. Exosomes isolated from cancer patients’ sera transfer malignant traits and confer the same phenotype of primary tumors to oncosuppressor-mutated cells. J. Exp. Clin. Cancer Res. CR 2017, 36, 113. [Google Scholar] [CrossRef]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Vu, L.T.; Ismail, N.N.; Le, M.T.N.; Grimson, A. Landscape of extracellular vesicles in the tumour microenvironment: Interactions with stromal cells and with non-cell components, and impacts on metabolic reprogramming, horizontal transfer of neoplastic traits, and the emergence of therapeutic resistance. Semin. Cancer Biol. 2021, 74, 24–44. [Google Scholar] [CrossRef]
- Abdouh, M.; Tabah, R.; Arena, V.; Arena, M.; Gao, Z.H.; Lorico, A.; Arena, G.O. Oncosuppressor-Mutated Cell-Based Diagnostic Platform for Liquid Biopsy Diagnoses Benign Head and Neck Masses and Predicts Malignancy in Thyroid Nodules: Results from a Consecutive Cohort of Patients. Eur. Thyroid J. 2021, 10, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Milane, L.; Singh, A.; Mattheolabakis, G.; Suresh, M.; Amiji, M.M. Exosome mediated communication within the tumor microenvironment. J. Control Release 2015, 219, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.; Huang, Q.; Chen, Y.; Wang, Q.; Sang, R.; Wang, L.; Xie, Y.; Chen, W. Tumor-Derived Extracellular Vesicles Regulate Cancer Progression in the Tumor Microenvironment. Front. Mol. Biosci. 2021, 8, 796385. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Roy, G.; Budukh, A.; Dikshit, R.; Patil, V.M.; Joshi, A.; Noronha, V.; Prabash, K.; Roy, P. A competing risk analysis of death patterns in male genitourinary cancer. Cancer Rep. 2020, 3, e1174. [Google Scholar] [CrossRef]
- Kaliszewski, K.; Diakowska, D.; Wojtczak, B.; Forkasiewicz, Z.; Pupka, D.; Nowak, L.; Rudnicki, J. Which papillary thyroid microcarcinoma should be treated as “true cancer” and which as “precancer”? World J. Surg. Oncol. 2019, 17, 91. [Google Scholar] [CrossRef]
- Abdouh, M.; Hamam, D.; Arena, V.; Arena, M.; Alamri, H.; Arena, G.O. Novel blood test to predict neoplastic activity in healthy patients and metastatic recurrence after primary tumor resection. J. Circ. Biomark. 2016, 5, 1849454416663661. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Zhou, S.; Abdouh, M.; Arena, V.; Arena, M.; Arena, G.O. Reprogramming Malignant Cancer Cells toward a Benign Phenotype following Exposure to Human Embryonic Stem Cell Microenvironment. PLoS ONE 2017, 12, e0169899. [Google Scholar] [CrossRef]
- Cairns, R.A.; Papandreou, I.; Sutphin, P.D.; Denko, N.C. Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 2007, 104, 9445–9450. [Google Scholar] [CrossRef]
- Bhat, T.A.; Kumar, S.; Chaudhary, A.K.; Yadav, N.; Chandra, D. Restoration of mitochondria function as a target for cancer therapy. Drug Discov. Today 2015, 20, 635–643. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Bonnay, F.; Veloso, A.; Steinmann, V.; Kocher, T.; Abdusselamoglu, M.D.; Bajaj, S.; Rivelles, E.; Landskron, L.; Esterbauer, H.; Zinzen, R.P.; et al. Oxidative Metabolism Drives Immortalization of Neural Stem Cells during Tumorigenesis. Cell 2020, 182, 1490–1507.e1419. [Google Scholar] [CrossRef]
- Tsogtbaatar, E.; Landin, C.; Minter-Dykhouse, K.; Folmes, C.D.L. Energy Metabolism Regulates Stem Cell Pluripotency. Front. Cell Dev. Biol. 2020, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Tomkova, V.; Sandoval-Acuna, C.; Torrealba, N.; Truksa, J. Mitochondrial fragmentation, elevated mitochondrial superoxide and respiratory supercomplexes disassembly is connected with the tamoxifen-resistant phenotype of breast cancer cells. Free Radic. Biol. Med. 2019, 143, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Gnocchi, D.; Kurzyk, A.; Mintrone, A.; Lentini, G.; Sabba, C.; Mazzocca, A. Inhibition of LPAR6 overcomes sorafenib resistance by switching glycolysis into oxidative phosphorylation in hepatocellular carcinoma. Biochimie 2022, 202, 180–189. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Z.; Bunker, E.; Ramirez, A.; Lee, S.; Peng, Y.; Tan, A.C.; Eckhardt, S.G.; Chapnick, D.A.; Liu, X. Sorafenib targets the mitochondrial electron transport chain complexes and ATP synthase to activate the PINK1-Parkin pathway and modulate cellular drug response. J. Biol. Chem. 2017, 292, 15105–15120. [Google Scholar] [CrossRef]
- Rao, S.; Mondragon, L.; Pranjic, B.; Hanada, T.; Stoll, G.; Kocher, T.; Zhang, P.; Jais, A.; Lercher, A.; Bergthaler, A.; et al. AIF-regulated oxidative phosphorylation supports lung cancer development. Cell Res. 2019, 29, 579–591. [Google Scholar] [CrossRef]
- Yan, B.; Stantic, M.; Zobalova, R.; Bezawork-Geleta, A.; Stapelberg, M.; Stursa, J.; Prokopova, K.; Dong, L.; Neuzil, J. Mitochondrially targeted vitamin E succinate efficiently kills breast tumour-initiating cells in a complex II-dependent manner. BMC Cancer 2015, 15, 401. [Google Scholar] [CrossRef]
- An, S.T.; James, W.B.; Michael, V.B. The role of mitochondrial electron transport in tumorigenesis and metastasis. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2014, 1840, 1454–1463. [Google Scholar] [CrossRef]
- Pranzini, E.; Pardella, E.; Paoli, P.; Fendt, S.M.; Taddei, M.L. Metabolic Reprogramming in Anticancer Drug Resistance: A Focus on Amino Acids. Trends Cancer 2021, 7, 682–699. [Google Scholar] [CrossRef]
- Loong, J.H.; Wong, T.L.; Tong, M.; Sharma, R.; Zhou, L.; Ng, K.Y.; Yu, H.J.; Li, C.H.; Man, K.; Lo, C.M.; et al. Glucose deprivation-induced aberrant FUT1-mediated fucosylation drives cancer stemness in hepatocellular carcinoma. J. Clin. Investig. 2021, 131, e143377. [Google Scholar] [CrossRef] [PubMed]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yoshimura, K.; Goto, K.; Lee, C.; Hamura, K.; Kwon, O.; Tamanoi, F. Nanoformulation of Geranylgeranyltransferase-I Inhibitors for Cancer Therapy: Liposomal Encapsulation and pH-Dependent Delivery to Cancer Cells. PLoS ONE 2015, 10, e0137595. [Google Scholar] [CrossRef]
- Luo, L.; Wu, W.; Sun, D.; Dai, H.B.; Wang, Y.; Zhong, Y.; Wang, J.X.; Maruf, A.; Nurhidayah, D.; Zhang, X.J.; et al. Acid-Activated Melittin for Targeted and Safe Antitumor Therapy. Bioconjug. Chem. 2018, 29, 2936–2944. [Google Scholar] [CrossRef]
- Tu, V.Y.; Ayari, A.; O’Connor, R.S. Beyond the Lactate Paradox: How Lactate and Acidity Impact T Cell Therapies against Cancer. Antibodies 2021, 10, 25. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Shayoub, M.E.; Muddathir, A.K.; Elhassan, G.O.; Bashir, A.H. Evolution of Tumor Metabolism might Reflect Carcinogenesis as a Reverse Evolution process (Dismantling of Multicellularity). Cancers 2011, 3, 3002–3017. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef]
- Ban, J.J.; Lee, M.; Im, W.; Kim, M. Low pH increases the yield of exosome isolation. Biochem. Biophys. Res. Commun. 2015, 461, 76–79. [Google Scholar] [CrossRef]
- Vaupel, P.; Harrison, L. Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 2004, 9 Suppl. 5, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Blaszczak, W.; Swietach, P. What do cellular responses to acidity tell us about cancer? Cancer Metastasis Rev. 2021, 40, 1159–1176. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.; Cheng, Q.; Tian, Y.; Teng, C.; Xie, K.; Yan, L. Double pH-sensitive nanotheranostics of polypeptide nanoparticle encapsulated BODIPY with both NIR activated fluorescence and enhanced photodynamic therapy. J. Mater. Chem. B 2021, 9, 8871–8881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xue, R.; Lyu, J.; Gao, A.; Sun, C. Tumor acidity/redox hierarchical-activable nanoparticles for precise combination of X-ray-induced photodynamic therapy and hypoxia-activated chemotherapy. J. Mater. Chem. B 2022, 10, 3849–3860. [Google Scholar] [CrossRef]
- Santos, M.F.; Rappa, G.; Karbanova, J.; Fontana, S.; Bella, M.A.D.; Pope, M.R.; Parrino, B.; Cascioferro, S.M.; Vistoli, G.; Diana, P.; et al. Itraconazole inhibits nuclear delivery of extracellular vesicle cargo by disrupting the entry of late endosomes into the nucleoplasmic reticulum. J. Extracell. Vesicles 2021, 10, e12132. [Google Scholar] [CrossRef]
- Campanella, C.; Caruso Bavisotto, C.; Logozzi, M.; Marino Gammazza, A.; Mizzoni, D.; Cappello, F.; Fais, S. On the Choice of the Extracellular Vesicles for Therapeutic Purposes. Int. J. Mol. Sci. 2019, 20, 236. [Google Scholar] [CrossRef]
- Uthamacumaran, A.; Abdouh, M.; Sengupta, K.; Gao, Z.-h.; Forte, S.; Tsering, T.; Burnier, J.V.; Arena, G. Machine intelligence-driven classification of cancer patients-derived extracellular vesicles using fluorescence correlation spectroscopy: Results from a pilot study. Neural Comput. Appl. 2023, 35, 8407–8422. [Google Scholar] [CrossRef]
- McCann, C.; Kerr, E.M. Metabolic Reprogramming: A Friend or Foe to Cancer Therapy? Cancers 2021, 13, 3351. [Google Scholar] [CrossRef]
- Marchetti, P.; Fovez, Q.; Germain, N.; Khamari, R.; Kluza, J. Mitochondrial spare respiratory capacity: Mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J. 2020, 34, 13106–13124. [Google Scholar] [CrossRef]
- Kwan, Y.P.; Saito, T.; Ibrahim, D.; Al-Hassan, F.M.; Ein Oon, C.; Chen, Y.; Jothy, S.L.; Kanwar, J.R.; Sasidharan, S. Evaluation of the cytotoxicity, cell-cycle arrest, and apoptotic induction by Euphorbia hirta in MCF-7 breast cancer cells. Pharm. Biol. 2016, 54, 1223–1236. [Google Scholar] [CrossRef]
- Karna, P.; Chagani, S.; Gundala, S.R.; Rida, P.C.; Asif, G.; Sharma, V.; Gupta, M.V.; Aneja, R. Benefits of whole ginger extract in prostate cancer. Br. J. Nutr. 2012, 107, 473–484. [Google Scholar] [CrossRef]
- Jo, K.J.; Cha, M.R.; Lee, M.R.; Yoon, M.Y.; Park, H.R. Methanolic extracts of Uncaria rhynchophylla induce cytotoxicity and apoptosis in HT-29 human colon carcinoma cells. Plant Foods Hum. Nutr. 2008, 63, 77–82. [Google Scholar] [CrossRef]
- Gill, M.S.A.; Saleem, H.; Ahemad, N. Plant Extracts and their Secondary Metabolites as Modulators of Kinases. Curr. Top. Med. Chem. 2020, 20, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Encalada, M.A.; Hoyos, K.M.; Rehecho, S.; Berasategi, I.; de Ciriano, M.G.; Ansorena, D.; Astiasaran, I.; Navarro-Blasco, I.; Cavero, R.Y.; Calvo, M.I. Anti-proliferative effect of Melissa officinalis on human colon cancer cell line. Plant Foods Hum. Nutr. 2011, 66, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Gnocchi, D.; Afonso, M.B.; Cavalluzzi, M.M.; Lentini, G.; Ingravallo, G.; Sabba, C.; Rodrigues, C.M.P.; Mazzocca, A. Inhibition of lysophosphatidic acid receptor 6 upregulated by the choline-deficient l-amino acid-defined diet prevents hepatocarcinogenesis in mice. Mol. Carcinog. 2023, 62, 577–582. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, M.A.; de la Cruz-Ojeda, P.; Lopez-Grueso, M.J.; Navarro-Villaran, E.; Requejo-Aguilar, R.; Castejon-Vega, B.; Negrete, M.; Gallego, P.; Vega-Ochoa, A.; Victor, V.M.; et al. Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer. Redox Biol. 2020, 36, 101510. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernandez, M.A.; de la Cruz-Ojeda, P.; Gallego, P.; Navarro-Villaran, E.; Stankova, P.; Del Campo, J.A.; Kucera, O.; Elkalaf, M.; Maseko, T.E.; Cervinkova, Z.; et al. Dose-dependent regulation of mitochondrial function and cell death pathway by sorafenib in liver cancer cells. Biochem. Pharmacol. 2020, 176, 113902. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, M.A.; Chapresto-Garzon, R.; Cadenas, M.; Navarro-Villaran, E.; Negrete, M.; Gomez-Bravo, M.A.; Victor, V.M.; Padillo, F.J.; Muntane, J. Differential effectiveness of tyrosine kinase inhibitors in 2D/3D culture according to cell differentiation, p53 status and mitochondrial respiration in liver cancer cells. Cell Death Dis. 2020, 11, 339. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Pioronska, W.; Battello, N.; Zimmer, A.D.; Dewidar, B.; Han, M.; Pereira, S.; Blagojevic, B.; Castven, D.; Charlestin, V.; et al. Severe metabolic alterations in liver cancer lead to ERK pathway activation and drug resistance. EBioMedicine 2020, 54, 102699. [Google Scholar] [CrossRef]
- Damaghi, M.; West, J.; Robertson-Tessi, M.; Xu, L.; Ferrall-Fairbanks, M.C.; Stewart, P.A.; Persi, E.; Fridley, B.L.; Altrock, P.M.; Gatenby, R.A.; et al. The harsh microenvironment in early breast cancer selects for a Warburg phenotype. Proc. Natl. Acad. Sci. USA 2021, 118, e2011342118. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gnocchi, D.; Nikolic, D.; Paparella, R.R.; Sabbà, C.; Mazzocca, A. Cellular Adaptation Takes Advantage of Atavistic Regression Programs during Carcinogenesis. Cancers 2023, 15, 3942. https://doi.org/10.3390/cancers15153942
Gnocchi D, Nikolic D, Paparella RR, Sabbà C, Mazzocca A. Cellular Adaptation Takes Advantage of Atavistic Regression Programs during Carcinogenesis. Cancers. 2023; 15(15):3942. https://doi.org/10.3390/cancers15153942
Chicago/Turabian StyleGnocchi, Davide, Dragana Nikolic, Rosa Rita Paparella, Carlo Sabbà, and Antonio Mazzocca. 2023. "Cellular Adaptation Takes Advantage of Atavistic Regression Programs during Carcinogenesis" Cancers 15, no. 15: 3942. https://doi.org/10.3390/cancers15153942
APA StyleGnocchi, D., Nikolic, D., Paparella, R. R., Sabbà, C., & Mazzocca, A. (2023). Cellular Adaptation Takes Advantage of Atavistic Regression Programs during Carcinogenesis. Cancers, 15(15), 3942. https://doi.org/10.3390/cancers15153942