

A Systematic Review of Mesenchymal Epithelial Transition Factor (MET) and Its Impact in the Development and Treatment of Non-Small-Cell Lung Cancer



Abstract
:Simple Summary
Abstract
1. Introduction
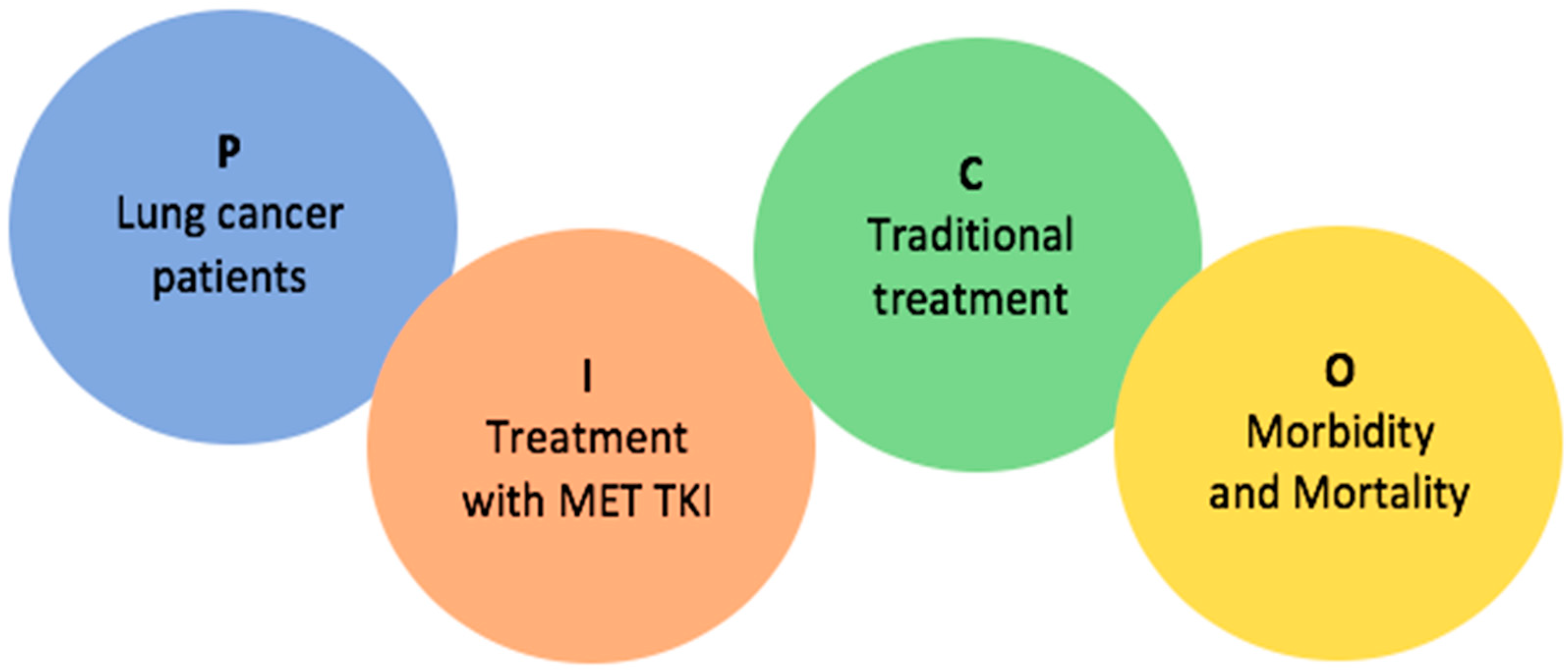
2. Methods
2.1. Search Strategy
2.2. Exclusion and Inclusion Criteria
2.3. Data Extraction and Processing
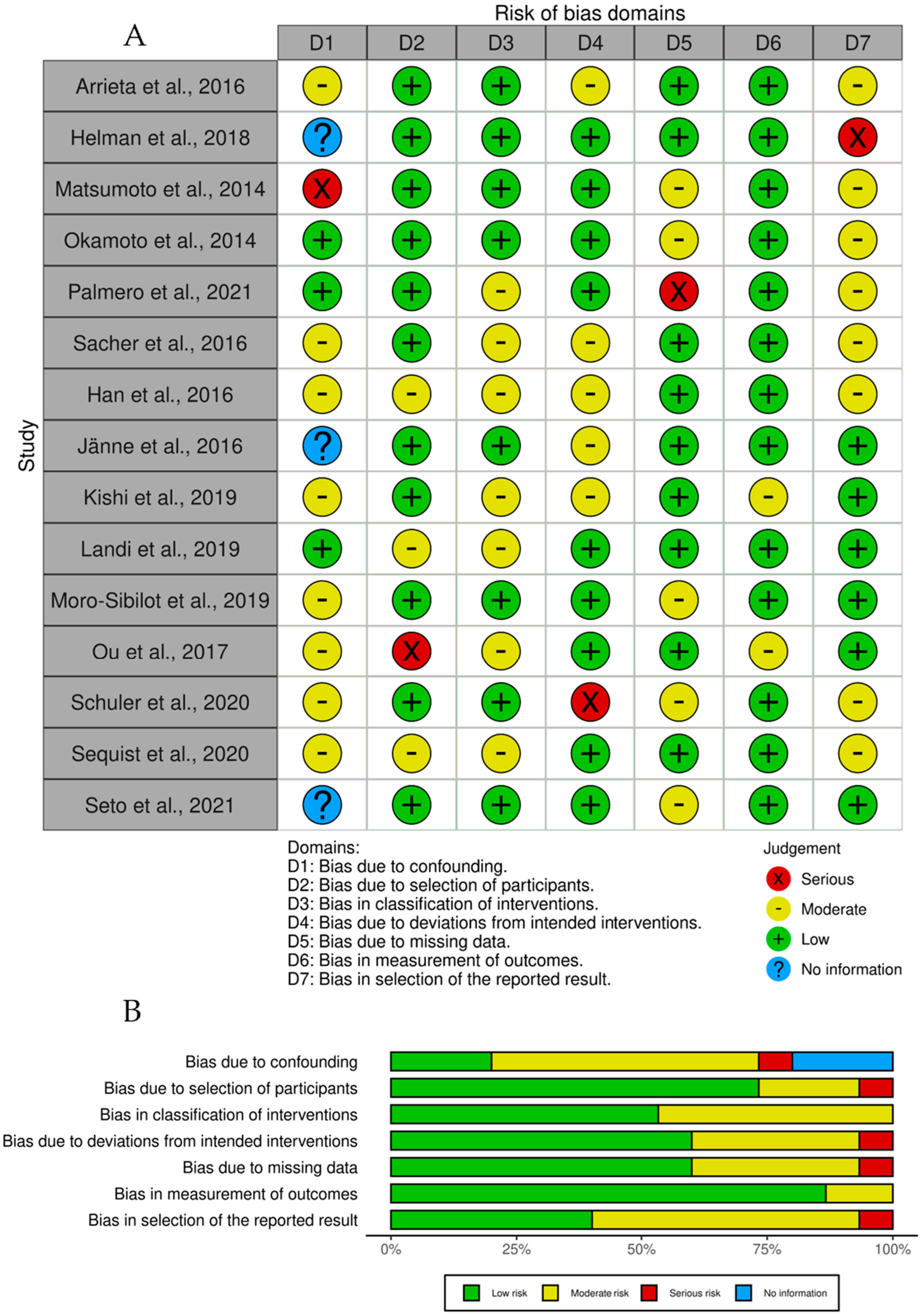
3. Bias
4. Results
4.1. Epidemiology
4.2. Prevalence of Aberrant MET Expression
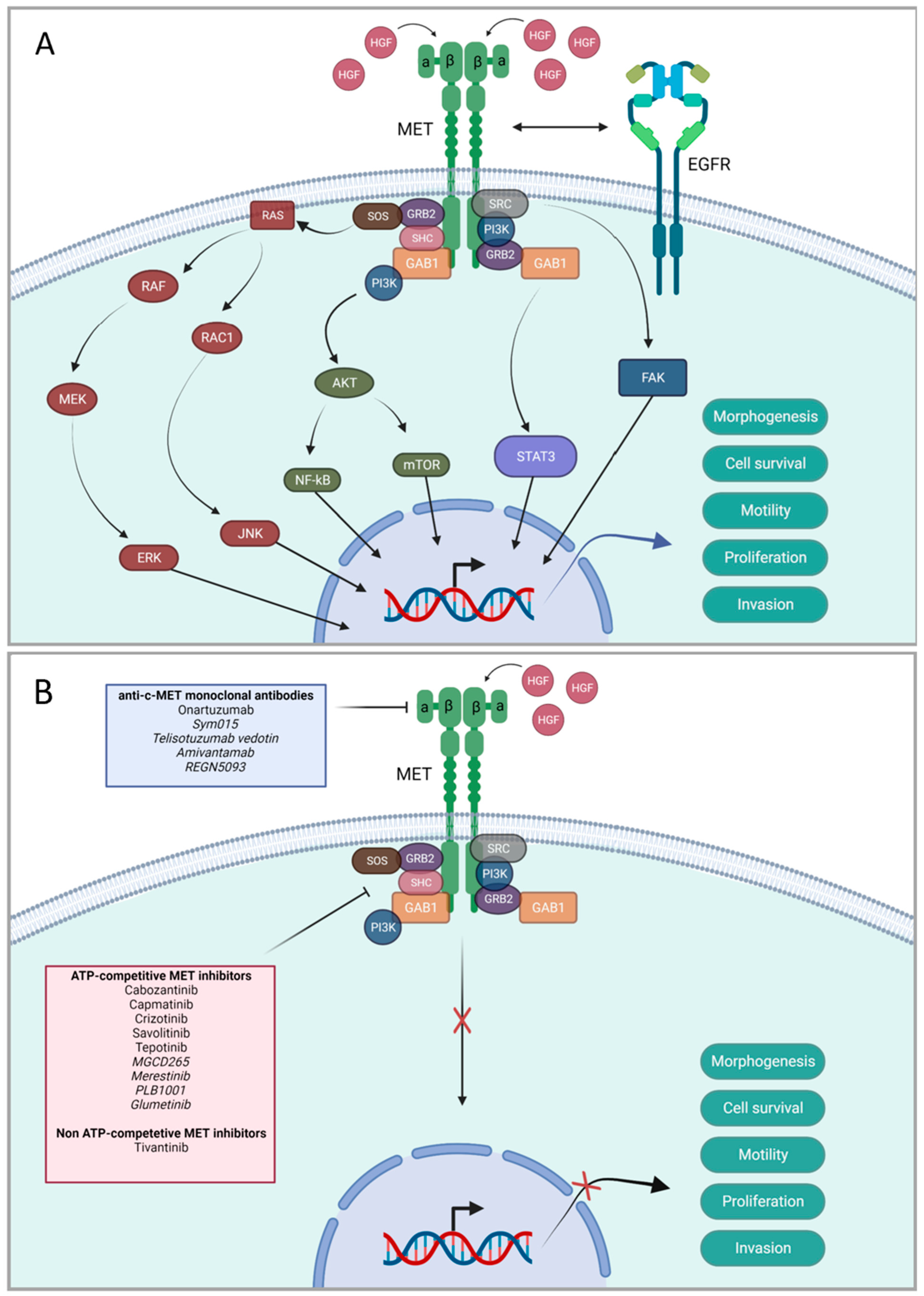
4.3. Targeted Therapies
5. Discussion
6. Strengths and Limitations
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
Search Queries
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Section and Topic | Item # | Checklist Item | Location Where Item Is Reported |
|---|---|---|---|
| TITLE | |||
| Title | 1 | Identify the report as a systematic review. | Page 1 |
| ABSTRACT | |||
| Abstract | 2 | See the PRISMA 2020 for Abstracts checklist. | Page 1, paragraph 2 |
| INTRODUCTION | |||
| Rationale | 3 | Describe the rationale for the review in the context of existing knowledge. | Page 2, paragraph 2 |
| Objectives | 4 | Provide an explicit statement of the objective(s) or question(s) the review addresses. | Page 4, paragraph 2 |
| METHODS | |||
| Eligibility criteria | 5 | Specify the inclusion and exclusion criteria for the review and how studies were grouped for the syntheses. | Page 5, paragraph 2 |
| Information sources | 6 | Specify all databases, registers, websites, organisations, reference lists and other sources searched or consulted to identify studies. Specify the date when each source was last searched or consulted. | Page 5, paragraph 1 |
| Search strategy | 7 | Present the full search strategies for all databases, registers and websites, including any filters and limits used. | Page 22 |
| Selection process | 8 | Specify the methods used to decide whether a study met the inclusion criteria of the review, including how many reviewers screened each record and each report retrieved, whether they worked independently, and if applicable, details of automation tools used in the process. | Page 5, paragraph 2 |
| Data collection process | 9 | Specify the methods used to collect data from reports, including how many reviewers collected data from each report, whether they worked independently, any processes for obtaining or confirming data from study investigators, and if applicable, details of automation tools used in the process. | Page 5, paragraph 2 |
| Data items | 10a | List and define all outcomes for which data were sought. Specify whether all results that were compatible with each outcome domain in each study were sought (e.g., for all measures, time points, analyses), and if not, the methods used to decide which results to collect. | Page 6, paragraph 1 |
| 10b | List and define all other variables for which data were sought (e.g., participant and intervention characteristics, funding sources). Describe any assumptions made about any missing or unclear information. | Page 6, paragraph 1 | |
| Study risk of bias assessment | 11 | Specify the methods used to assess risk of bias in the included studies, including details of the tool(s) used, how many reviewers assessed each study and whether they worked independently, and if applicable, details of automation tools used in the process. | Page 11, paragraph 1 |
| Effect measures | 12 | Specify for each outcome the effect measure(s) (e.g., risk ratio, mean difference) used in the synthesis or presentation of results. | N/A |
| Synthesis methods | 13a | Describe the processes used to decide which studies were eligible for each synthesis (e.g., tabulating the study intervention characteristics and comparing against the planned groups for each synthesis (item #5)). | Page 8, Table 3 |
| 13b | Describe any methods required to prepare the data for presentation or synthesis, such as handling of missing summary statistics, or data conversions. | N/A | |
| 13c | Describe any methods used to tabulate or visually display results of individual studies and syntheses. | Page 8, Table 3 | |
| 13d | Describe any methods used to synthesize results and provide a rationale for the choice(s). If meta-analysis was performed, describe the model(s), method(s) to identify the presence and extent of statistical heterogeneity, and software package(s) used. | N/A | |
| 13e | Describe any methods used to explore possible causes of heterogeneity among study results (e.g., subgroup analysis, meta-regression). | N/A | |
| 13f | Describe any sensitivity analyses conducted to assess robustness of the synthesized results. | N/A | |
| Reporting bias assessment | 14 | Describe any methods used to assess risk of bias due to missing results in a synthesis (arising from reporting biases). | Page 11, paragraph 1 |
| Certainty assessment | 15 | Describe any methods used to assess certainty (or confidence) in the body of evidence for an outcome. | N/A |
| RESULTS | |||
| Study selection | 16a | Describe the results of the search and selection process, from the number of records identified in the search to the number of studies included in the review, ideally using a flow diagram. | Page 6, Figure 3 |
| 16b | Cite studies that might appear to meet the inclusion criteria, but which were excluded, and explain why they were excluded. | N/A | |
| Study characteristics | 17 | Cite each included study and present its characteristics. | Page 11, paragraph 4 |
| Risk of bias in studies | 18 | Present assessments of risk of bias for each included study. | Pages 27–28 |
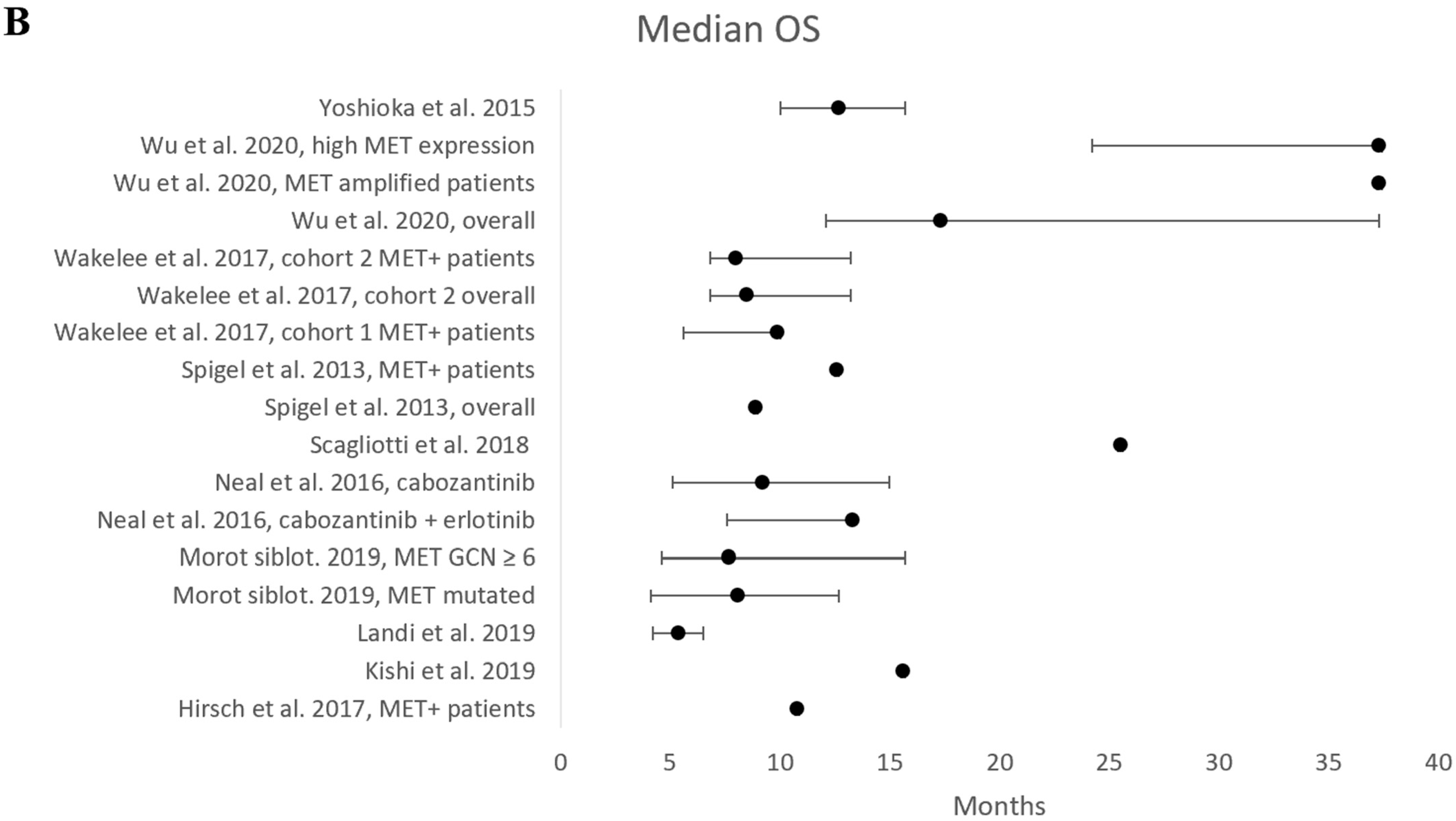
| Results of individual studies | 19 | For all outcomes, present, for each study: (a) summary statistics for each group (where appropriate) and (b) an effect estimate and its precision (e.g., confidence/credible interval), ideally using structured tables or plots. | Pages 29–30 |
| Results of syntheses | 20a | For each synthesis, briefly summarise the characteristics and risk of bias among contributing studies. | Page 21, paragraph 2 |
| 20b | Present results of all statistical syntheses conducted. If meta-analysis was done, present for each the summary estimate and its precision (e.g., confidence/credible interval) and measures of statistical heterogeneity. If comparing groups, describe the direction of the effect. | N/A | |
| 20c | Present results of all investigations of possible causes of heterogeneity among study results. | Page 21, paragraph 2 | |
| 20d | Present results of all sensitivity analyses conducted to assess the robustness of the synthesized results. | N/A | |
| Reporting biases | 21 | Present assessments of risk of bias due to missing results (arising from reporting biases) for each synthesis assessed. | Pages 27–28 |
| Certainty of evidence | 22 | Present assessments of certainty (or confidence) in the body of evidence for each outcome assessed. | N/A |
| DISCUSSION | |||
| Discussion | 23a | Provide a general interpretation of the results in the context of other evidence. | Page 18, paragraph 1 |
| 23b | Discuss any limitations of the evidence included in the review. | Page 21, paragraph 2 | |
| 23c | Discuss any limitations of the review processes used. | Page 21, paragraph 2 | |
| 23d | Discuss implications of the results for practice, policy, and future research. | Page 18, paragraph 3 | |
| OTHER INFORMATION | |||
| Registration and protocol | 24a | Provide registration information for the review, including register name and registration number, or state that the review was not registered. | Page 5, paragraph 1 |
| 24b | Indicate where the review protocol can be accessed, or state that a protocol was not prepared. | Page 5, paragraph 1 | |
| 24c | Describe and explain any amendments to information provided at registration or in the protocol. | N/A | |
| Support | 25 | Describe sources of financial or non-financial support for the review, and the role of the funders or sponsors in the review. | Page 22 |
| Competing interests | 26 | Declare any competing interests of review authors. | Page 22 |
| Availability of data, code and other materials | 27 | Report which of the following are publicly available and where they can be found: template data collection forms; data extracted from included studies; data used for all analyses; analytic code; any other materials used in the review. | Page 22 |
| Topic | No. | Item | Reported? |
|---|---|---|---|
| TITLE | |||
| Title | 1 | Identify the report as a systematic review. | Yes |
| BACKGROUND | |||
| Objectives | 2 | Provide an explicit statement of the main objective(s) or question(s) the review addresses. | Yes |
| METHODS | |||
| Eligibility criteria | 3 | Specify the inclusion and exclusion criteria for the review. | No |
| Information sources | 4 | Specify the information sources (e.g., databases, registers) used to identify studies and the date when each was last searched. | Yes |
| Risk of bias | 5 | Specify the methods used to assess risk of bias in the included studies. | No |
| Synthesis of results | 6 | Specify the methods used to present and synthesize results. | No |
| RESULTS | |||
| Included studies | 7 | Give the total number of included studies and participants and summarise relevant characteristics of studies. | Yes |
| Synthesis of results | 8 | Present results for main outcomes, preferably indicating the number of included studies and participants for each. If meta-analysis was done, report the summary estimate and confidence/credible interval. If comparing groups, indicate the direction of the effect (i.e., which group is favoured). | Yes |
| DISCUSSION | |||
| Limitations of evidence | 9 | Provide a brief summary of the limitations of the evidence included in the review (e.g., study risk of bias, inconsistency and imprecision). | No |
| Interpretation | 10 | Provide a general interpretation of the results and important implications. | Yes |
| OTHER | |||
| Funding | 11 | Specify the primary source of funding for the review. | No |
| Registration | 12 | Provide the register name and registration number. | Yes |
Appendix C


| Mutational Status | Median Age (Years) | Male Gender (%) | History of No Smoking (%) | |
|---|---|---|---|---|
| Arrieta et al., 2016 [1] | EGFR+/WT | 60.1 | 33.3% | 39.4% |
| Han et al., 2017 [14] | EGFR+/WT, MET+/− | Treatment: 63.0 Placebo: 63.0 | Treatment: 56.0% Placebo: 57.0% | Treatment: 18.5% Placebo: 15.8% |
| Helman et al., 2018 [25] | EGFR+ | 61.0 | 28.6% | NS |
| Hirsch et al., 2017 [17] | MET+/− | Treatment: 68.0 Placebo: 66.0 | Treatment: 70.9% Placebo: 74.1% | Treatment: 3.6% Placebo: 5.6% |
| Jänne et al., 2016 [21] | EGFR+/WT | 59.5 | 39.0% | 51.0% |
| Kishi et al., 2019 [15] | EGFR+ MET+ | 67.0 | 43% | NS |
| Landi et al., 2019 [18] | EGFR WT MET+ | 56.0 | 65% | 23% |
| Matsumoto et al., 2014 [19] | EGFR WT | 64.0 | 19.7% | 89.1% |
| Moro-Sibilot et al., 2019 [29] | EGFR+/WT MET+/− | MET amplified: 59.0 MET mutated: 72.0 | MET amplified: 56.0% MET mutated: 32.0% | MET amplified: 24.0% MET mutation: 48.0% |
| Neal et al., 2016 [2] | EGFR WT | 65.3 | 45.0% | 15% |
| Okamoto et al., 2014 [3] | NS | 64.0 | 77.0% | 18.0% |
| Ou et al., 2017 [24] | NS | 60.0 | 30.0% | 52.0% |
| Palmero et al., 2021 [11] | NS | 64.3 | 65.0% | 27.0% |
| Sacher et al., 2016 [28] | EGFR+/WT | 64.0 | 45.0% | 18.0% |
| Scagliotti et al., 2018 [30] | EGFR+ MET+/− | Treatment: 59.5 Placebo: 65.0 | Treatment: 42.9% Placebo: 47.2% | Treatment: 48.2% Placebo: 60.4% |
| Schuler et al., 2020 [20] | EGFR WT MET+ | 60.0 | 60.0% | NS |
| Sequist et al., 2020 [22] | EGFR+ MET+ | Cohort B: 59.0 Cohort D: 62.0 | Cohort B: 59% Cohort D: 60% | NS |
| Seto et al., 2021 [31] | EGFR WT MET+ | 68.0 | 66.7% | 42.2% |
| Spigel et al., 2013 [6] | EGFR+/WT MET+/− | Treatment MET-: 63.0 Placebo MET-: 61.0 Treatment MET+: 66.0 Placebo MET+: 64.0 | Treatment MET-: 65.0% Placebo MET-: 55.0% Treatment MET+: 51.0% Placebo MET+: 65.0% | Treatment MET -: 7.0% Placebo MET -: 3.0% Treatment MET+: 20.0% Placebo MET+: 19.0% |
| Wakelee et al., 2017 [32] | EGFR+/WT MET+/− | Cohort I treatment: 60.0 Cohort I placebo: 60.5 Cohort II treatment: 66.0 Cohort II placebo: 63.0 | Cohort I treatment: 68.1% Cohort I placebo: 48.6% Cohort II treatment: 55.9% Cohort II placebo: 42.6% | Cohort I treatment: 21.7% Cohort I placebo: 25.7% Cohort II treatment: 27.1% Cohort II placebo: 13.1% |
| Wu et al., 2020 [23] | EGFR+ MET+ | Phase Ib: 65.5 Phase II treatment: 61.0 Phase II placebo: 58.3 | Phase Ib: 44.0% Phase II treatment: 35.5% Phase II placebo: 50.0% | Phase Ib: 72.0% Phase II treatment: 68.0% Phase II placebo: 67.0% |
| Yoshioka et al., 2015 [5] | EGFR WT MET+/− | Treatment: 63.0 Placebo: 63.0 | Treatment: 66.7% Placebo: 70.8% | Treatment: 25.5% Placebo: 26.2% |
References
- Arrieta, O.; Cruz-Rico, G.; Soto-Perez-de-Celis, E.; Ramírez-Tirado, L.A.; Caballe-Perez, E.; Martínez-Hernández, J.N.; Martinez-Alvarez, I.; Soca-Chafre, G.; Macedo-Pérez, E.O.; Astudillo-de la Vega, H.; et al. Reduction in Hepatocyte Growth Factor Serum Levels is Associated with Improved Prognosis in Advanced Lung Adenocarcinoma Patients Treated with Afatinib: A Phase II Trial. Target Oncol. 2016, 11, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.W.; Dahlberg, S.E.; Wakelee, H.A.; Aisner, S.C.; Bowden, M.; Huang, Y.; Carbone, D.P.; Gerstner, G.J.; Lerner, R.E.; Rubin, J.L.; et al. Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second-line or third-line treatment of patients with EGFR wild-type advanced non-small-cell lung cancer (ECOG-ACRIN 1512): A randomised, controlled, open-label, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 1661–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, I.; Sakai, K.; Morita, S.; Yoshioka, H.; Kaneda, H.; Takeda, K.; Hirashima, T.; Kogure, Y.; Kimura, T.; Takahashi, T.; et al. Multiplex genomic profiling of non-small cell lung cancers from the LETS phase III trial of first-line S-1/carboplatin versus paclitaxel/carboplatin: Results of a West Japan Oncology Group study. Oncotarget 2014, 5, 2293–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, H.; Azuma, K.; Yamamoto, N.; Takahashi, T.; Nishio, M.; Katakami, N.; Ahn, M.J.; Hirashima, T.; Maemondo, M.; Kim, S.W.; et al. A randomized, double-blind, placebo-controlled, phase III trial of erlotinib with or without a c-Met inhibitor tivantinib (ARQ 197) in Asian patients with previously treated stage IIIB/IV nonsquamous nonsmall-cell lung cancer harboring wild-type epidermal growth factor receptor (ATTENTION study). Ann. Oncol. 2015, 26, 2066–2072. [Google Scholar] [PubMed]
- Spigel, D.R.; Ervin, T.J.; Ramlau, R.A.; Daniel, D.B.; Goldschmidt, J.H., Jr.; Blumenschein, G.R., Jr.; Krzakowski, M.J.; Robinet, G.; Godbert, B.; Barlesi, F.; et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 4105–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urvay, S.E.; Yucel, B.; Erdis, E.; Turan, N. Prognostic Factors in Stage III Non-Small-Cell Lung Cancer Patients. Asian. Pac. J. Cancer Prev. 2016, 17, 4693–4697. [Google Scholar]
- Önal, Ö.; Koçer, M.; Eroğlu, H.N.; Yilmaz, S.D.; Eroğlu, I.; Karadoğan, D. Survival analysis and factors affecting survival in patients who presented to the medical oncology unit with non-small cell lung cancer. Turk. J. Med. Sci. 2020, 50, 1838–1850. [Google Scholar] [CrossRef]
- Shields, M.D.; Marin-Acevedo, J.A.; Pellini, B. Immunotherapy for Advanced Non-Small Cell Lung Cancer: A Decade of Progress; American Society of Clinical Oncology Educational Book: Alexandria, VA, USA, 2021; pp. 105–127. [Google Scholar]
- Bodén, E.; Andreasson, J.; Hirdman, G.; Malmsjö, M.; Lindstedt, S. Quantitative Proteomics Indicate Radical Removal of Non-Small Cell Lung Cancer and Predict Outcome. Biomedicines 2022, 10, 2738. [Google Scholar] [CrossRef]
- Palmero, R.; Taus, A.; Viteri, S.; Majem, M.; Carcereny, E.; Garde-Noguera, J.; Felip, E.; Nadal, E.; Malfettone, A.; Sampayo, M.; et al. Biomarker Discovery and Outcomes for Comprehensive Cell-Free Circulating Tumor DNA Versus Standard-of-Care Tissue Testing in Advanced Non-Small-Cell Lung Cancer. JCO Precis. Oncol. 2021, 5, 93–102. [Google Scholar] [CrossRef]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar] [PubMed]
- Coleman, N.; Hong, L.; Zhang, J.; Heymach, J.; Hong, D.; Le, X. Beyond epidermal growth factor receptor: MET amplification as a general resistance driver to targeted therapy in oncogene-driven non-small-cell lung cancer. ESMO Open 2021, 6, 100319. [Google Scholar] [CrossRef]
- Han, K.; Chanu, P.; Jonsson, F.; Winter, H.; Bruno, R.; Jin, J.; Stroh, M. Exposure-Response and Tumor Growth Inhibition Analyses of the Monovalent Anti-c-MET Antibody Onartuzumab (MetMAb) in the Second- and Third-Line Non-Small Cell Lung Cancer. AAPS J. 2017, 19, 527–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishi, K.; Sakai, H.; Seto, T.; Kozuki, T.; Nishio, M.; Imamura, F.; Hiroshi, N.; Miyako, S.; Shintaro, N.; Takashi, T.; et al. First-line onartuzumab plus erlotinib treatment for patients with MET-positive and EGFR mutation-positive non-small-cell lung cancer. Cancer Treat Res. Commun. 2019, 18, 100113. [Google Scholar] [CrossRef] [PubMed]
- Organ, S.L.; Tsao, M. S An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, F.R.; Govindan, R.; Zvirbule, Z.; Braiteh, F.; Rittmeyer, A.; Belda-Iniesta, C.; Isla, D.; Cosgriff, T.; Boyer, M.; Ueda, M.; et al. Efficacy and Safety Results From a Phase II, Placebo-Controlled Study of Onartuzumab Plus First-Line Platinum-Doublet Chemotherapy for Advanced Squamous Cell Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2017, 18, 43–49. [Google Scholar] [CrossRef]
- Landi, L.; Chiari, R.; Tiseo, M.; D’Incà, F.; Dazzi, C.; Chella, A.; Delmonte, A.; Bonanno, L.; Giannarelli, D.; Cortinovis, D.L.; et al. Crizotinib in MET-Deregulated or ROS1-Rearranged Pretreated Non-Small Cell Lung Cancer (METROS): A Phase II, Prospective, Multicenter, Two-Arms Trial. Clin. Cancer Res. 2019, 25, 7312–7319. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Maemondo, M.; Ishii, Y.; Okudera, K.; Demura, Y.; Takamura, K.; Kobayashi, K.; Morikawa, N.; Gemm, A.; Ishimoto, O.; et al. A phase II study of erlotinib monotherapy in pre-treated non-small cell lung cancer without EGFR gene mutation who have never/light smoking history: Re-evaluation of EGFR gene status (NEJ006/TCOG0903). Lung Cancer 2014, 86, 195–200. [Google Scholar] [CrossRef]
- Schuler, M.; Berardi, R.; Lim, W.T.; de Jonge, M.; Bauer, T.M.; Azaro, A.; Gottfried, M.; Han, J.-Y.; Lee, D.; Wollner, M.; et al. Molecular correlates of response to capmatinib in advanced non-small-cell lung cancer: Clinical and biomarker results from a phase I trial. Ann. Oncol. 2020, 31, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Shaw, A.T.; Camidge, D.R.; Giaccone, G.; Shreeve, S.M.; Tang, Y.; Goldberg, Z.; Martini, J.-F.; Xu, H.; James, L.P.; et al. Combined Pan-HER and ALK/ROS1/MET Inhibition with Dacomitinib and Crizotinib in Advanced Non-Small Cell Lung Cancer: Results of a Phase I Study. J. Thorac. Oncol. 2016, 11, 737–747. [Google Scholar] [CrossRef]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.-H.; Lee, J.S.; Su, W.-C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Cheng, Y.; Zhou, J.; Lu, S.; Zhang, Y.; Zhao, J.; Kim, D.-W.; Soo, R.A.; Kim, S.-W.; Pan, H.; et al. Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): An open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir. Med. 2020, 8, 1132–1143. [Google Scholar] [CrossRef]
- Ou, S.I.; Govindan, R.; Eaton, K.D.; Otterson, G.A.; Gutierrez, M.E.; Mita, A.C.; Argiris, A.; Brega, N.M.; Usari, T.; Tan, W.; et al. Phase I Results from a Study of Crizotinib in Combination with Erlotinib in Patients with Advanced Nonsquamous Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 145–151. [Google Scholar] [CrossRef]
- Helman, E.; Nguyen, M.; Karlovich, C.A.; Despain, D.; Choquette, A.K.; Spira, A.I.; Yu, H.A.; Camidge, D.R.; Harding, T.C.; Lanman, R.B.; et al. Cell-Free DNA Next-Generation Sequencing Prediction of Response and Resistance to Third-Generation EGFR Inhibitor. Clin. Lung Cancer 2018, 19, 518–530. [Google Scholar] [CrossRef] [Green Version]
- Morganti, S.; Tarantino, P.; Ferraro, E.; D’Amico, P.; Duso, B.A.; Curigliano, G. Next Generation Sequencing (NGS): A Revolutionary Technology in Pharmacogenomics and Personalized Medicine in Cancer; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; pp. 9–30. [Google Scholar]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Jennifer, M.; Elie, A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Syst. Rev. 2021, 88, 105906. [Google Scholar]
- Sacher, A.G.; Le, L.W.; Lara-Guerra, H.; Waddell, T.K.; Sakashita, S.; Chen, Z.; Kim, L.; Zhang, T.; Kamel-Reid, S.; Salvarrey, A.; et al. A window of opportunity study of potential tumor and soluble biomarkers of response to preoperative erlotinib in early stage non-small cell lung cancer. Oncotarget 2016, 7, 25632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro-Sibilot, D.; Cozic, N.; Pérol, M.; Mazières, J.; Otto, J.; Souquet, P.J.; Bahleda, R.; Wislez, M.; Zalcman, G.; Guibert, S.D.; et al. Crizotinib in c-MET- or ROS1-positive NSCLC: Results of the AcSé phase II trial. Ann. Oncol. 2019, 30, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.V.; Shuster, D.; Orlov, S.; von Pawel, J.; Shepherd, F.A.; Ross, J.S.; Wang, Q.; Schwartz, B.; Akerley, W. Tivantinib in Combination with Erlotinib versus Erlotinib Alone for EGFR-Mutant NSCLC: An Exploratory Analysis of the Phase 3 MARQUEE Study. J. Thorac. Oncol. 2018, 13, 849–854. [Google Scholar] [CrossRef] [Green Version]
- Seto, T.; Ohashi, K.; Sugawara, S.; Nishio, M.; Takeda, M.; Aoe, K.; Moizumi, S.; Nomura, S.; Tajima, T.; Hida, T.; et al. Capmatinib in Japanese patients with MET exon 14 skipping-mutated or MET-amplified advanced NSCLC: GEOMETRY mono-1 study. Cancer Sci. 2021, 112, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Wakelee, H.; Zvirbule, Z.; De Braud, F.; Kingsley, C.D.; Mekhail, T.; Lowe, T.; Schütte, W.; Lena, H.; Lawler, W.; Braiteh, F.; et al. Efficacy and Safety of Onartuzumab in Combination With First-Line Bevacizumab- or Pemetrexed-Based Chemotherapy Regimens in Advanced Non-Squamous Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2017, 18, 50–59. [Google Scholar] [CrossRef]
- McGuinness, L.A.; Higgins, J.P.T. Risk-of-bias VISualization (robvis): An R package and Shiny web app for visualizing risk-of-bias assessments. Res. Synth. Methods 2021, 12, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, J.; Bodén, E.; Fakhro, M.; von Wachter, C.; Olm, F.; Malmsjö, M.; Hallgren, O.; Lindstedt, S. Exhaled phospholipid transfer protein and hepatocyte growth factor receptor in lung adenocarcinoma. Respir. Res. 2022, 23, 369. [Google Scholar] [CrossRef] [PubMed]
- Liguori, N.R.; Lee, Y.; Borges, W.; Zhou, L.; Azzoli, C.; El-Deiry, W.S. Absence of Biomarker-Driven Treatment Options in Small Cell Lung Cancer, and Selected Preclinical Candidates for Next Generation Combination Therapies. Front. Pharmacol. 2021, 12, 747180. [Google Scholar] [CrossRef] [PubMed]
- Toumazis, I.; Bastani, M.; Han, S.S.; Plevritis, S.K. Risk-Based lung cancer screening: A systematic review. Lung Cancer 2020, 147, 154–186. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.; Salgia, R. Biomarkers in lung cancer: From early detection to novel therapeutics and decision making. Biomark. Med. 2008, 2, 577–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, E.C.; Sclafani, F.; Cunningham, D. Emerging molecular targets in oncology: Clinical potential of MET/hepatocyte growth-factor inhibitors. OncoTargets Ther. 2014, 7, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Beau-Faller, M.; Ruppert, A.M.; Voegeli, A.C.; Neuville, A.; Meyer, N.; Guerin, E.; Legrain, M.; Mennecier, B.; Wihlm, J.-M.; Massard, G.; et al. MET gene copy number in non-small cell lung cancer: Molecular analysis in a targeted tyrosine kinase inhibitor naïve cohort. J. Thorac. Oncol. 2008, 3, 331–339. [Google Scholar] [CrossRef]
- Onozato, R.; Kosaka, T.; Kuwano, H.; Sekido, Y.; Yatabe, Y.; Mitsudomi, T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J. Thorac. Oncol. 2009, 4, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Kent, D.G.; Green, A.R. Order Matters: The Order of Somatic Mutations Influences Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a027060. [Google Scholar] [CrossRef] [Green Version]
- Jolly, C.; Van Loo, P. Timing somatic events in the evolution of cancer. Genome Biol. 2018, 19, 95. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Xu, J.; Sun, B.; Wang, J.; Wang, Z. MET-Targeted Therapies and Clinical Outcomes: A Systematic Literature Review. Mol. Diagn. Ther. 2022, 26, 203–227. [Google Scholar] [CrossRef] [PubMed]
- Kollmannsberger, C.; Hurwitz, H.; Bazhenova, L.; Cho, B.C.; Hong, D.; Park, K.; Sharma, S.; Der-Torossian, H.; Christensen, J.G.; Faltaos, D.; et al. Phase I Study Evaluating Glesatinib (MGCD265), An Inhibitor of MET and AXL, in Patients with Non-small Cell Lung Cancer and Other Advanced Solid Tumors. Target Oncol. 2023, 18, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Janku, F.; Bueno, A.M.; Catenacci, D.V.; Lee, J.; Lee, S.H.; Chung, H.C.; Dowlati, A.; Rohrberg, K.S.; Font, F.E.; et al. 1490PD—A phase Ia/IIa trial of Sym015, a MET antibody mixture, in patients with advanced solid tumours. Ann. Oncol. 2019, 30, V610–V611. [Google Scholar] [CrossRef]

| Drug Name | Effect | References |
|---|---|---|
| Afatinib | Binds covalently and irreversibly to the kinase domain of EGFR. | Arrieta et al. [1] |
| Cabozantinib | A small molecule TKI that targets MET, VEGFR2, RET, ROS1, KIT, TIE-2, and AXL. Binds intracellularly to MET. | Neal et al. [2] Landi et al. [18] |
| Capmatinib | A highly selective intracellular MET inhibitor. | Schuler et al. [20] Sequist et al. [22] |
| Crizotinib | An intracellular MET/ALK/ROS1 RTK inhibitor with high specificity for MET. | Landi et al. [18] |
| Dacomitinib | A small irreversible pan-human EGFR inhibitor. | Jänne et al. [21] |
| Erlotinib | A reversible, small-molecule EGFR TKI. | Spigel et al. [6] |
| Gefitinib | A reversible EGFR TKI. | Arrieta et al. [1] Wu et al. [23] |
| Onartuzumab | A recombinant, fully humanized, one-armed anti-MET monovalent monoclonal antibody. Binds to the extracellular domain of MET without activating it and without dimerizing. | Hirsch et al. [17] Kishi et al. [15] |
| Osimertinib | A CNS-active, irreversible EGFR TKI. | Sequist et al. [22] |
| Rociletinib | An irreversible EGFR TKI targeting mutated form of the EGFR gene. | Arrieta et al. [1] |
| Savolitinib | A small molecule, ATP competitive, selective MET TKI. | Sequist et al. [22] |
| Tepotinib | A highly selective, ATP competitive MET inhibitor. | Wu et al. [23] |
| Tivantinib | A selective, non-ATP-competitive MET inhibitor metabolized by CYP2C19. | Yoshioka et al. [5] |
| Total Participants | Cancer Type | Mutational Status | Active Drug | Results | |
|---|---|---|---|---|---|
| Arrieta et al., 2016 [1] | n = 66 | NSCLC | EGFR WT EGFR+ | Afatinib (EGFR TKI) | Reduced levels of HGF led to improved ORR, PFS, and OS. |
| Helman et al., 2018 [25] | n = 77 | NSCLC | EGFR+ | Rociletinib (EGFR TKI) | Prevalence of MET alteration was 15.0%. MET amplification was seen in 7.6% of these, 4.5% had focal amplification, and 3.1% had aneuploidy. |
| Matsumoto et al., 2014 [19] | n = 47 | NSCLC | EGFR WT | Erlotinib (EGFR TKI) | Expression of HGF resulted in poor response to erlotinib with shorter PFS. MET mutational status did not correlate to response to erlotinib or PFS. |
| Okamoto et al., 2014 [3] | n = 295 | NSCLC | NS | Chemotherapy | Totally, 21 patients (7.6%) had MET mutations. MET amplifications were present in 9 (3.9%) cases. Median GCN was 8.8 among MET amplified patients. |
| Palmero et al., 2021 [11] | n = 186 | NSCLC | NS | None | Totally, 22.0% of patients had MET amplifications and 11.0% had METex14 mutations. |
| Sacher et al., 2016 [28] | n = 22 | NSCLC | EGFR WT EGFR+ | Erlotinib | Totally, 45.0% of subjects harbored a MET alteration. MET amplification was present in 9.0% of the patients. |
| Study Design | Total Participants | Cancer Type | Mutational Status | Definition of MET+ | Active Drug | Results | |
|---|---|---|---|---|---|---|---|
| Han et al., 2017 [14] | Phase III | n = 636 | NSCLC | EGFR+/WT, MET+/− | Not defined | Onartuzumab (MET TKI) + erlotinib (EGFR TKI) vs. erlotinib | High dose onartuzumab resulted in longer PFS (4.4 months) compared to low dose (2.5 months) and erlotinib (2.5 months). No significant difference in OS. |
| Hirsch et al., 2017 [17] | Phase II | n = 106 | NSCLC | MET+/− | MET IHC2+ or IHC3+ | Onartuzumab (MET TKI) + chemotherapy vs. chemotherapy + placebo | Median PFS was 5 months for onartuzumab and 5.2 months for placebo (ns). Median OS was 10.8 months for onartuzumab and 7.9 months for placebo (ns). |
| Jänne et al., 2016 [21] | Phase I | n = 67 | NSCLC | EGFR+/WT | MET IHC2+ or IHC3+, MET GCN ≥ 2.1 | Crizotinib (MET TKI) + dacomitinib (EGFR TKI) | Median PFS was 3 months with 61.0% stable disease in the escalation phase. Median PFS was 2.1 months with 32.0% stable disease in the expansion phase. |
| Kishi et al., 2019 [15] | Phase II | n = 61 | NSCLC | EGFR+ MET+ | MET IHC2+ or IHC3+, the total number of MET genes in 20 cancer cells ≥ 90 | Onartuzumab (MET TKI) + erlotinib (EGFR TKI) | Median PFS was 8.5 months, median OS 15.6 months, and ORR 68.9%. |
| Landi et al., 2019 [18] | Phase II | n = 26 | NSCLC | EGFR WT MET+ | MET-CEP7/ratio ≥ 2.2, METex14 mutation | Crizotinib (MET TKI) | ORR of 27.0%, median PFS 4.4 months, and median OS 5.4 months. |
| Moro-Sibilot et al., 2019 [29] | Phase II | n = 53 | NSCLC | EGFR WT EGFR+ MET+/− | MET IHC2+ or IHC3+, MET GCN ≥ 6, MET exon skipping mutations in exon 14, 16–19 determined by NGS | Crizotinib (MET TKI) | ORR of 16.0%, median PFS of 3.2 months, and median OS of 7.7 months in the MET GCN ≥ 6 cohort. ORR of 10.7%, median PFS of 2.4 months, and median OS of 8.1 months in the MET mutated cohort. |
| Neal et al., 2016 [2] | Phase II | n = 111 | NSCLC | EGFR WT | Tested through IHC, positive if MET was expressed in either membrane or cytoplasm | Cabozantinib (MET TKI) + erlotinib (EGFR TKI) vs. cabozantinib vs. erlotinib | Median PFS of 1.8 months and OS of 5.1 months for erlotinib. PFS was 4.7 months and OS was 12.3 months for erlotinib + cabozantinib. PFS was 4.3 months and OS was 9.2 months (ns) for cabozantinib. No association between MET IHC+ and PFS in the cabozantinib group. |
| Ou et al., 2017 [24] | Phase I | n = 26 | NSCLC | NS | Mutational status not mentioned | Crizotinib (MET TKI) + erlotinib (EGFR TKI) | Two patients had partial response, 8 had stable disease, and 10 had progressive disease. |
| Scagliotti et al., 2018 [30] | Phase III | n = 109 | NSCLC | EGFR+ MET+/− | MET IHC2+ or IHC3+, MET GCN ≥ 4 | Tivantinib (MET TKI) + erlotinib (EGFR TKI) vs. erlotinib + placebo | Greater overall response rate (60.7%) and median PFS (13.0 months) for tivantinib + erlotinib compared to erlotinib + placebo (43.4%, 7.5 months). Similar median OS between groups (25.5 months for tivantinib and 20.3 months for placebo). |
| Schuler et al., 2020 [20] | Phase I | n = 55 | NSCLC | EGFR WT MET+ | MET H-score ≥ 150, MET/CEP7 ≥ 2.0, MET GCN ≥ 5, MET IHC 2+ or IHC3+ | Capmatinib (MET TKI) | Median PFS was 3.7 months. In patients with MET GCN ≥ 6 median PFS was 9.3 months. |
| Sequist et al., 2020 [22] | Phase Ib | n = 180 | NSCLC | EGFR+ MET+ | MET GCN ≥ 5, MET/CEP7 ratio ≥ 2, MET IHC3+ or ≥20.0% tumor cells in NGS | Savolitinib (MET TKI) + osimertinib (EGFR TKI) | Partial response in 89 patients treated with savolitinib and osimertinib. PFS was 7.6 months and 9.1 months in two different subgroups. |
| Seto et al., 2021 [31] | Phase II | n = 45 | NSCLC | EGFR WT MET+ | METex14 mutation, MET amplification | Capmatinib (MET TKI) | In a subcohort with METex14 mutations receiving second or third-line (2/3L) treatment with capmatinib, overall response rate was 36.4%. In a second cohort with MET GCN ≥ 10, overall response rate to 2/3L capmatinib was 45.5%. |
| Spigel et al., 2013 [6] | Phase II | n = 136 | NSCLC | EGFR+/WT MET+/− | MET IHC2+ or IHC3+ | Onartuzumab (MET TKI) + erlotinib (EGFR TKI) vs. erlotinib | No difference in PFS and OS between groups. In a MET+ subgroup, PFS was significantly longer for onartuzumab compared to erlotinib + placebo (2.9 vs. 1.5 months). Longer OS in the MET+ subgroup receiving onartuzumab (12.6 vs. 3.8 months). |
| Wakelee et al., 2017 [32] | Phase II | n = 259 | NSCLC | EGFR+/WT MET+/− | MET IHC2+ or ICH3+ | Cohort 1: onartuzumab (MET TKI) + bevacizumab (VEGF-A TKI) + chemotherapy vs. placebo + bevacizumab + chemotherapy. Cohort 2: onartuzumab + chemotherapy vs. placebo + chemotherapy | Cohort 1: longer median PFS on placebo (6.8 months) compared to onartuzumab (5.0 months). A MET+ subgroup had a median PFS of 4.8 months and OS of 9.9 months on onartuzumab vs. 6.9 months and 16.5 months on placebo. Cohort 2: median PFS of 4.9 months and median OS of 8.5 months on onartuzumab vs. 5.1 months and 13.7 months on placebo. In a MET+ subgroup, median PFS was 5.0 months and OS was 8.0 months on onartuzumab vs. 5.0 months and 7.6 months on placebo. |
| Wu et al., 2020 [23] | Phase Ib/II | n = 55 | NSCLC | EGFR+ MET+ | MET IHC2+ or IHC3+, MET GCN ≥ 5 | Tepotinib (MET TKI) + gefitinib (EGFR TKI) vs. chemotherapy | Significantly longer OS (37.3 months vs. 13.1 months) and PFS (16.6 months vs. 4.2 months) for tepotinib + gefitinib in patients with MET IHC3+ or MET GCN ≥ 5. |
| Yoshioka et al., 2015 [5] | Phase III | n = 303 | NSCLC | EGFR WT MET+/− | IHC with moderate/strong intensity ≥ 50.0% of tumor cells, MET GCN ≥ 4 | Tivantinib (MET TKI) + erlotinib (EGFR TKI) vs. erlotinib + placebo | Significantly longer PFS for tivantinib + erlotinib (2.9 months) compared to erlotinib + placebo (2 months). No effect on OS. |
| Clinical Trial ID | Study Design | Study Type | Total Participants | Cancer Type | Mutational Status | Active Drug and Effect |
|---|---|---|---|---|---|---|
| NCT02544633 | Phase II | Non-randomized | n = 68 | NSCLC | MET activating mutation MET amplification | MGCD265: oral RTK inhibitor targeting MET |
| NCT02920996 | Phase II | Single arm | n = 12 | NSCLC | METex14 mutation | Merestinib: reversible type II ATP-competitive MET inhibitor |
| NCT02896231 | Phase I | Dose escalation | n = 37 | NSCLC | MET+ | PLB1001: selective MET inhibitor |
| NCT04270591 | Phase Ib/II | Single arm | n = 183 | NSCLC | METex14 mutation MET amplification MET overexpression | Glumetinib: selective MET inhibitor |
| NCT02648724 | Phase I/II | Non-randomized | n = 57 | NSCLC | MET amplification METex14 deletion | Sym015: monoclonal antibody mixture targeting MET |
| NCT03539536 | Phase II | Single arm | n = 275 | NSCLC | MET+ | Telisotuzumab vedotin: antibody-drug conjugate targeting MET |
| NCT02609776 | Phase I | Non-randomized | n = 780 | Advanced NSCLC | Varying | Amivantamab: human bispecific antibody targeting EGFR and MET |
| NCT04077099 | Phase I/II | Single arm | n = 82 | NSCLC | Any MET alteration | REGN5093: human bispecific antibody targeting MET, inducing internalization and degradation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bodén, E.; Sveréus, F.; Olm, F.; Lindstedt, S. A Systematic Review of Mesenchymal Epithelial Transition Factor (MET) and Its Impact in the Development and Treatment of Non-Small-Cell Lung Cancer. Cancers 2023, 15, 3827. https://doi.org/10.3390/cancers15153827
Bodén E, Sveréus F, Olm F, Lindstedt S. A Systematic Review of Mesenchymal Epithelial Transition Factor (MET) and Its Impact in the Development and Treatment of Non-Small-Cell Lung Cancer. Cancers. 2023; 15(15):3827. https://doi.org/10.3390/cancers15153827
Chicago/Turabian StyleBodén, Embla, Fanny Sveréus, Franziska Olm, and Sandra Lindstedt. 2023. "A Systematic Review of Mesenchymal Epithelial Transition Factor (MET) and Its Impact in the Development and Treatment of Non-Small-Cell Lung Cancer" Cancers 15, no. 15: 3827. https://doi.org/10.3390/cancers15153827
APA StyleBodén, E., Sveréus, F., Olm, F., & Lindstedt, S. (2023). A Systematic Review of Mesenchymal Epithelial Transition Factor (MET) and Its Impact in the Development and Treatment of Non-Small-Cell Lung Cancer. Cancers, 15(15), 3827. https://doi.org/10.3390/cancers15153827