
Comprehensive Immune Profiling Unveils a Subset of Leiomyosarcoma with “Hot” Tumor Immune Microenvironment

 , , , , ,
, , , , ,  and
and 
Abstract
Simple Summary
Abstract
1. Background
2. Materials and Methods
2.1. Patient Samples
2.2. Whole Transcriptome Sequencing (WTS)
2.3. Bioinformatics and Statistical Analysis
2.4. Data Availability
3. Results
3.1. Patient/Tumor Characterization in the ICGC/TCGA Cohorts
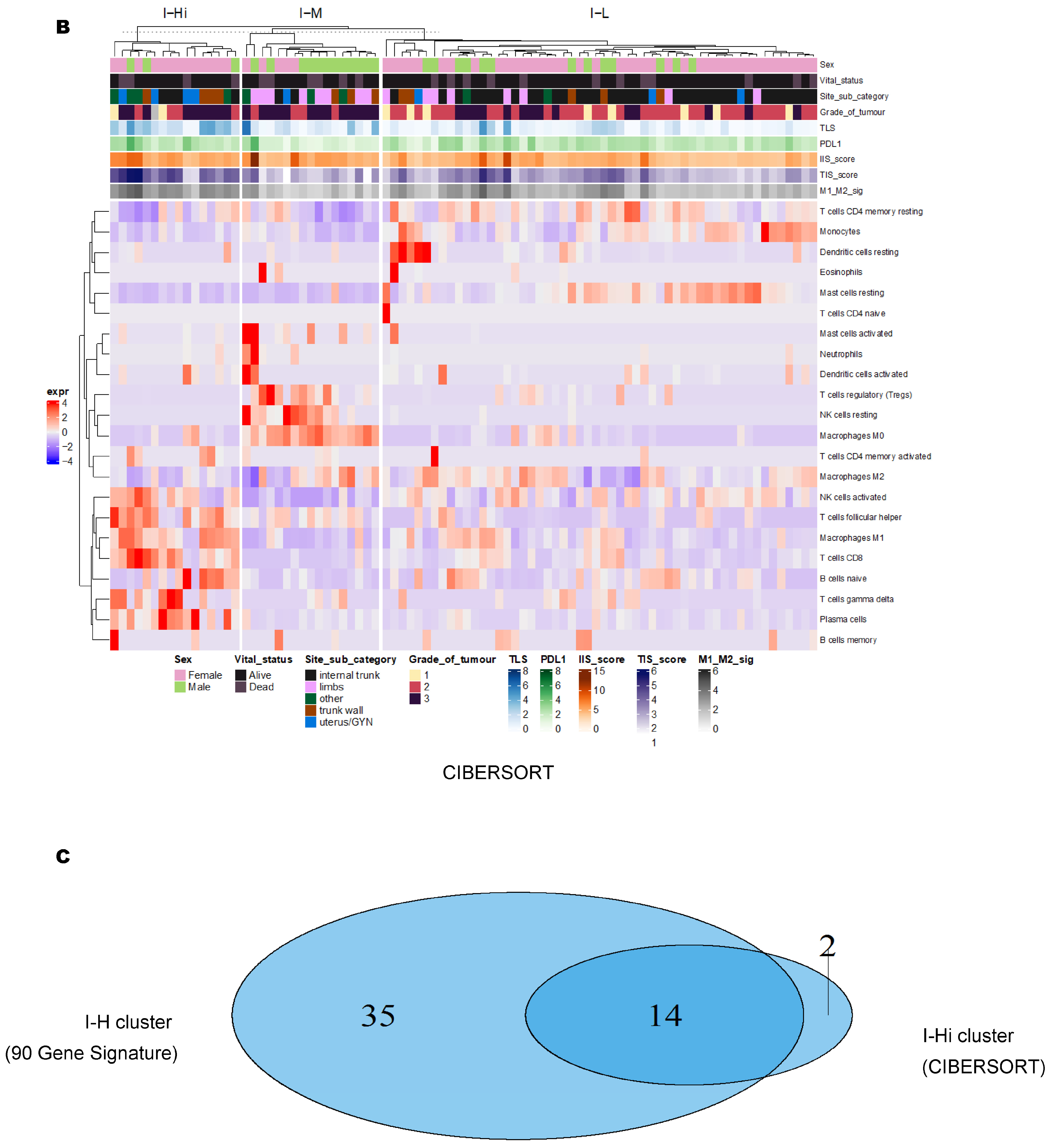
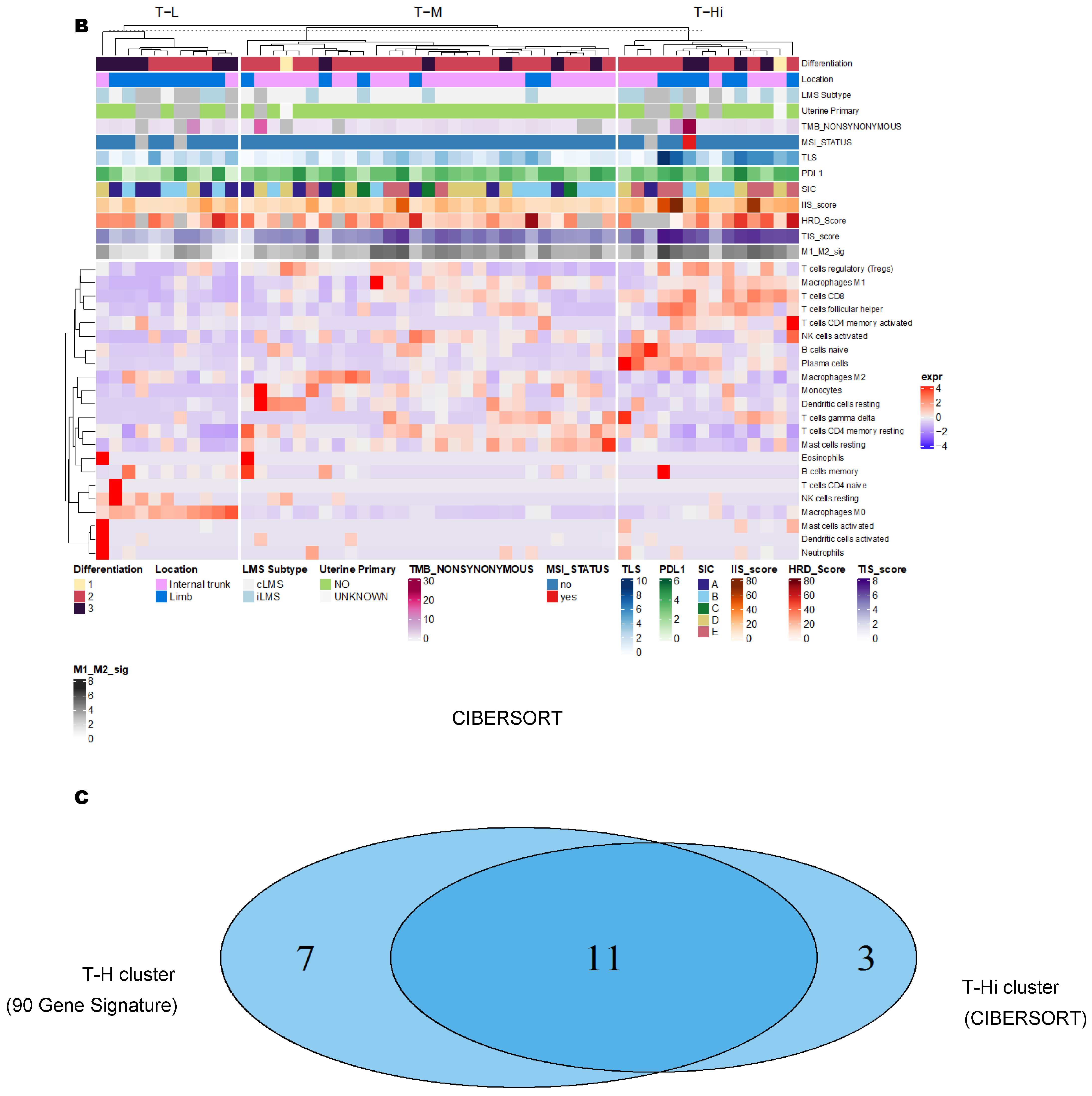
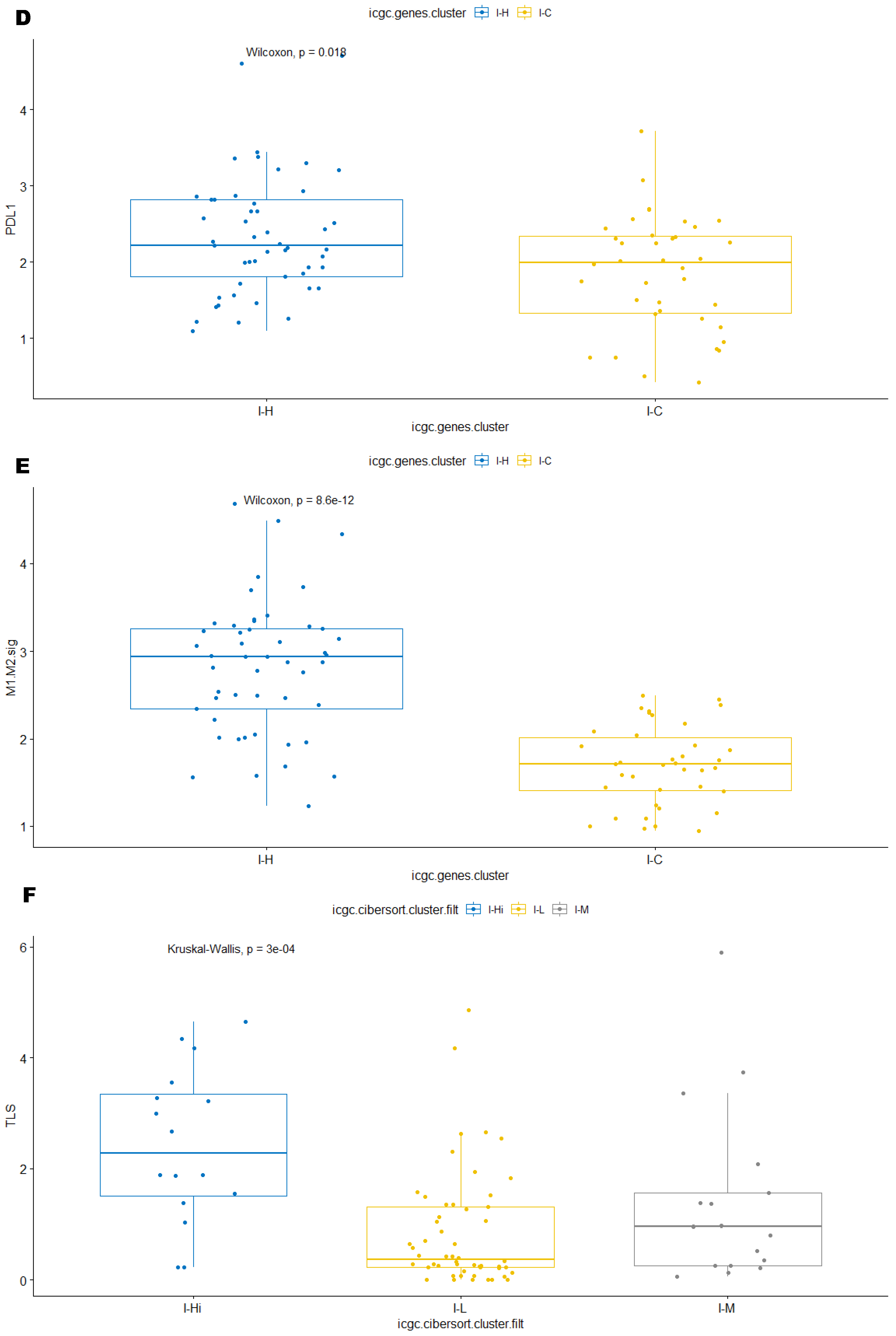
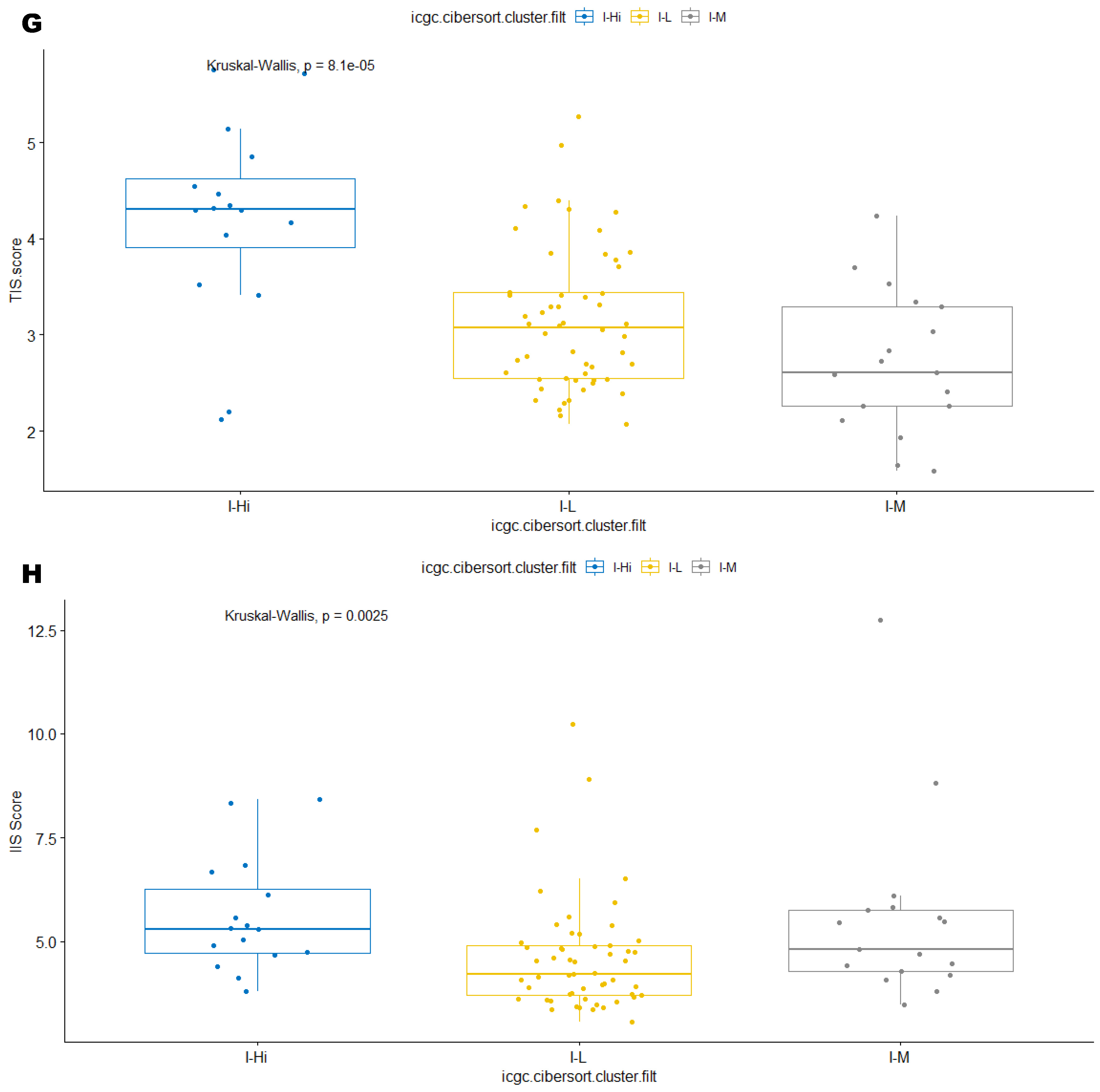
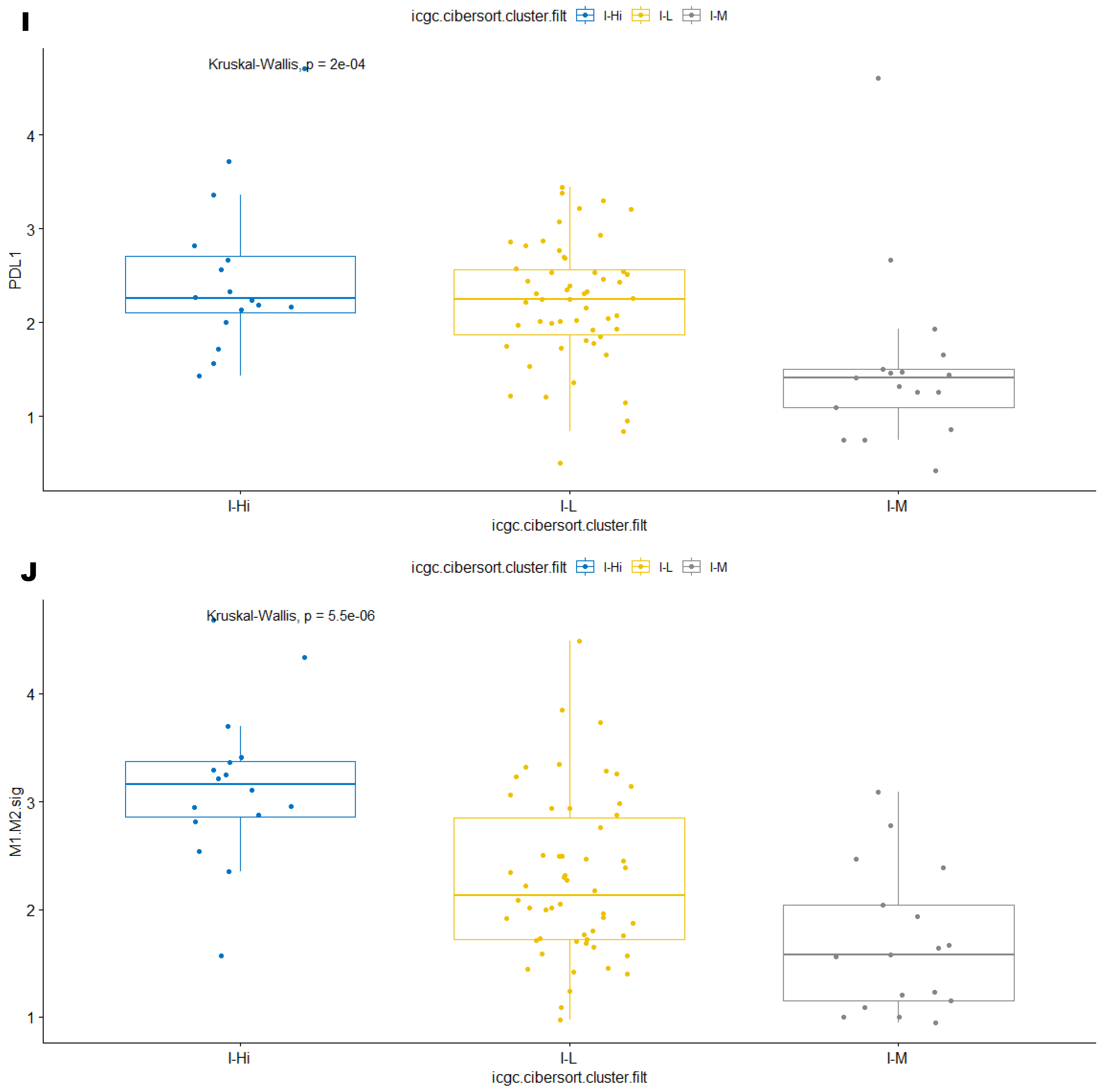
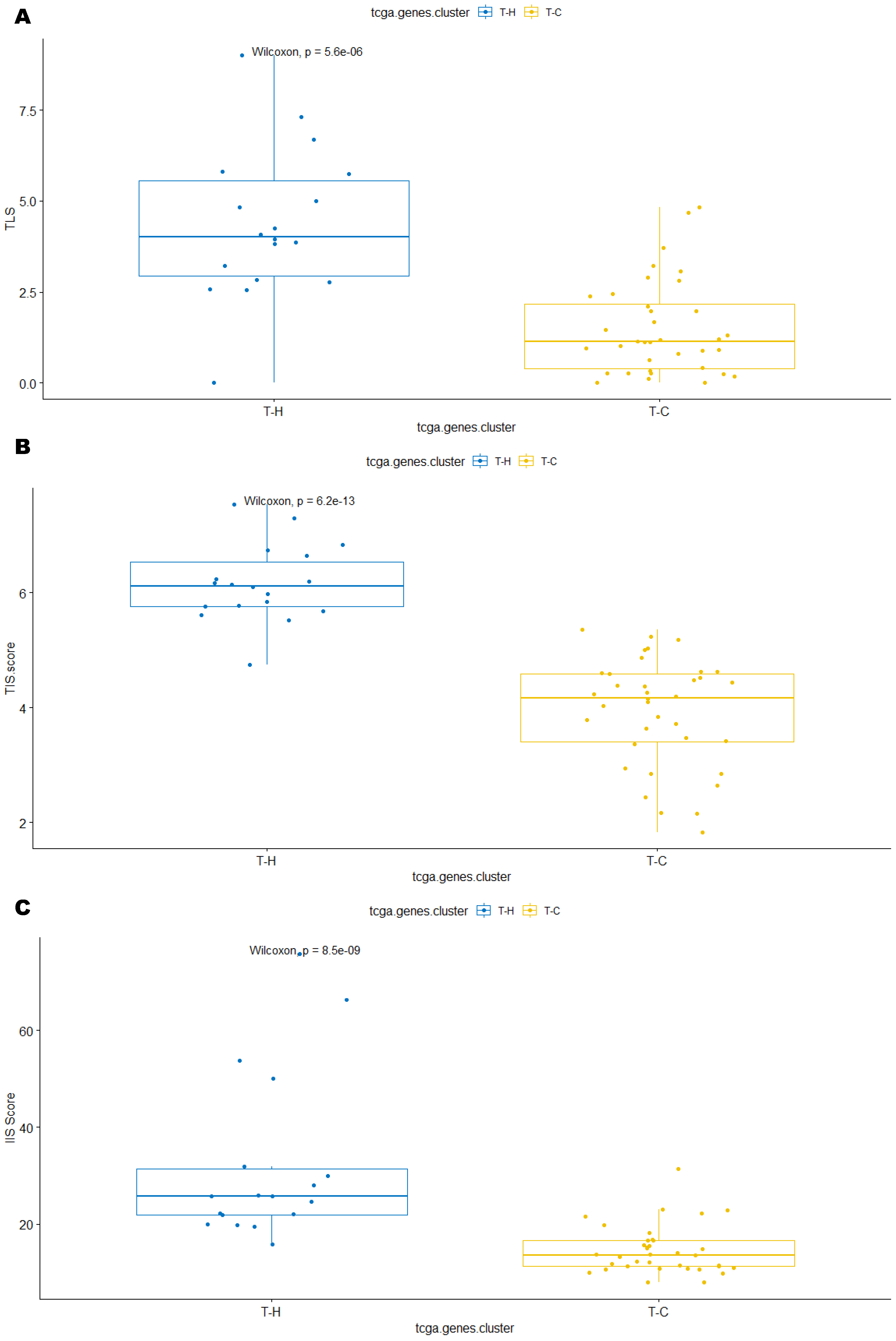
3.2. Unsupervised Clustering Revealed a Small Subset (~15%) of LMS with Active (“Hot”) TIME with Combined 90-Gene Signature and CIBERSORT Methods
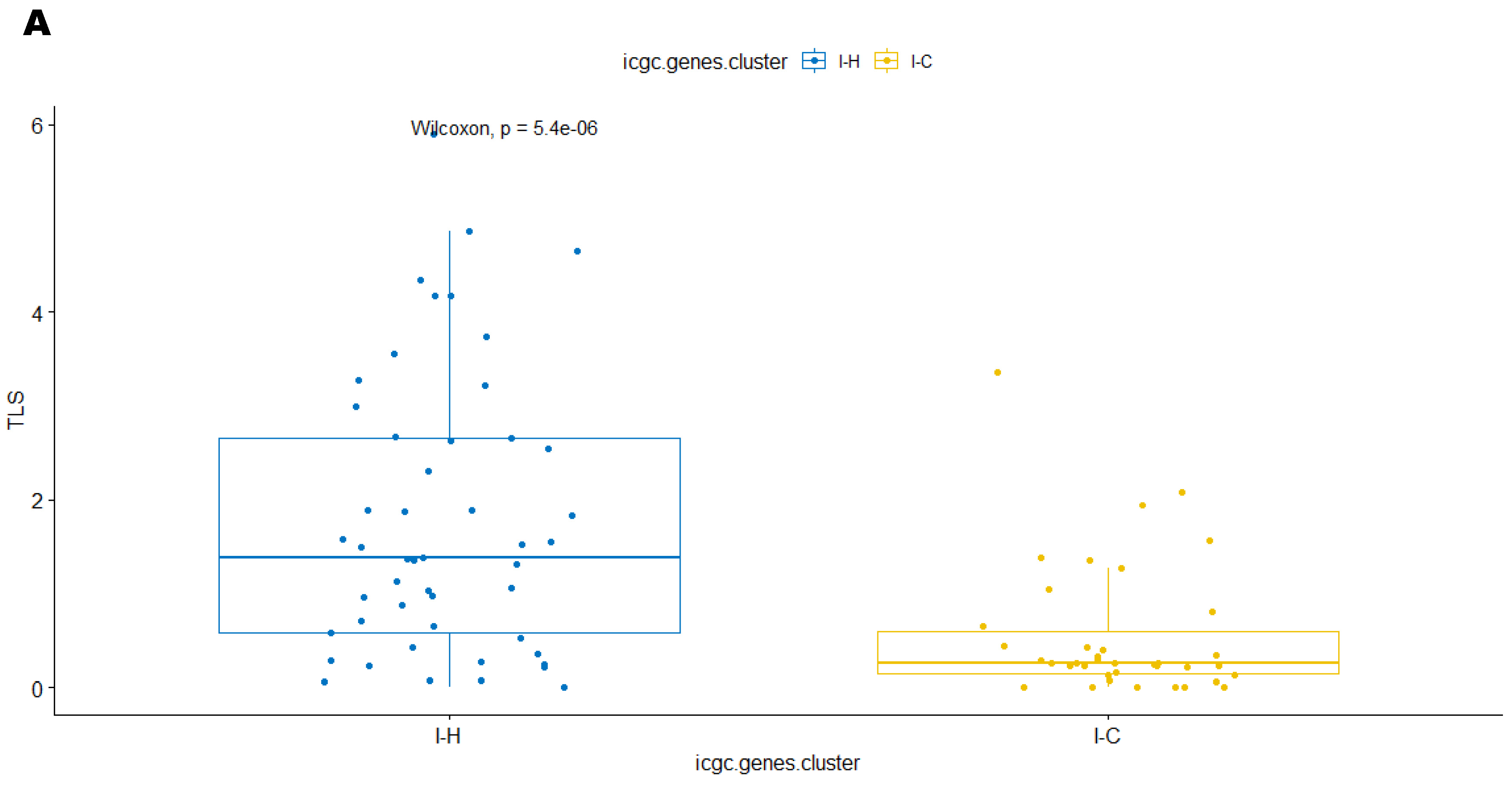
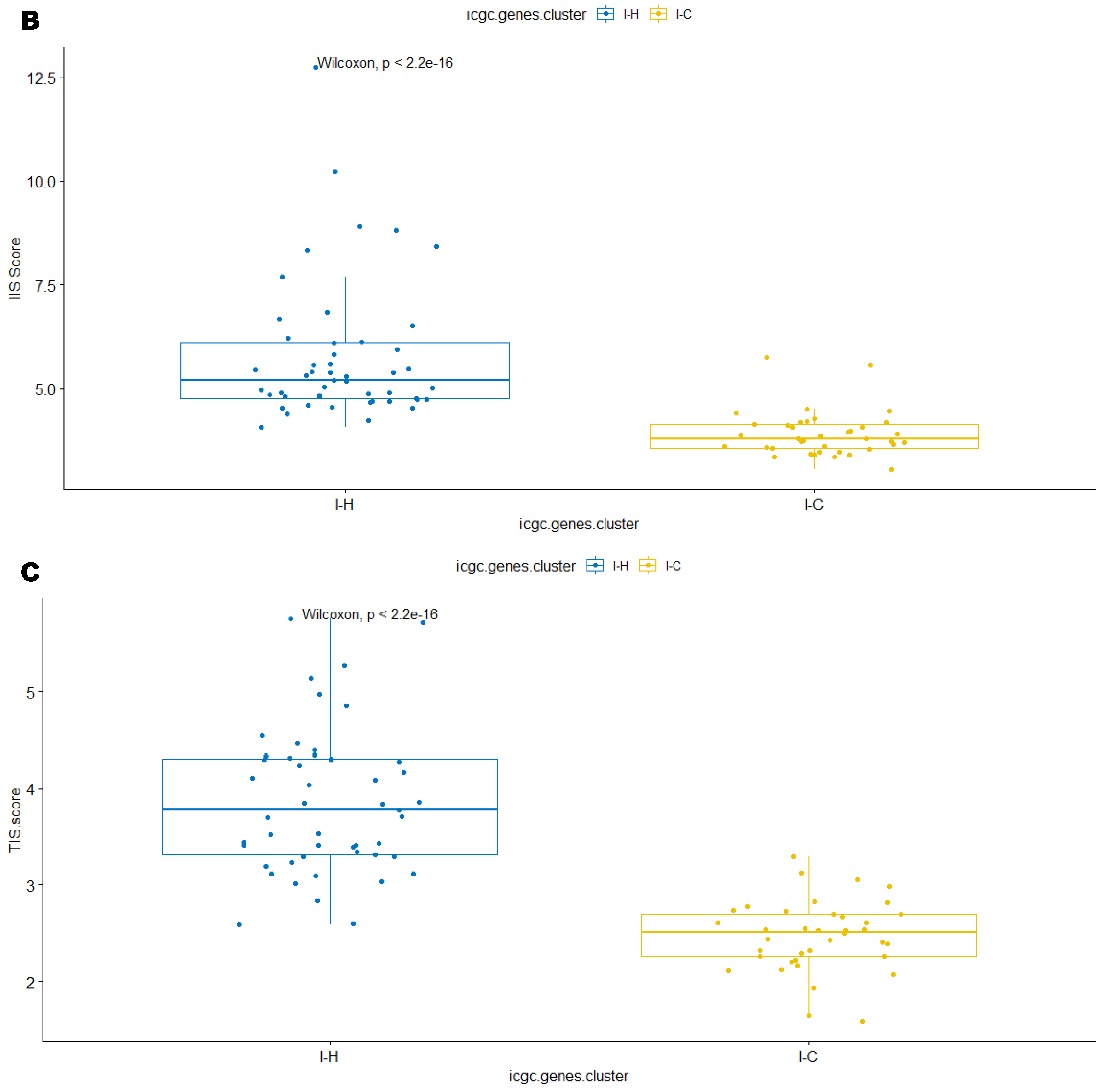
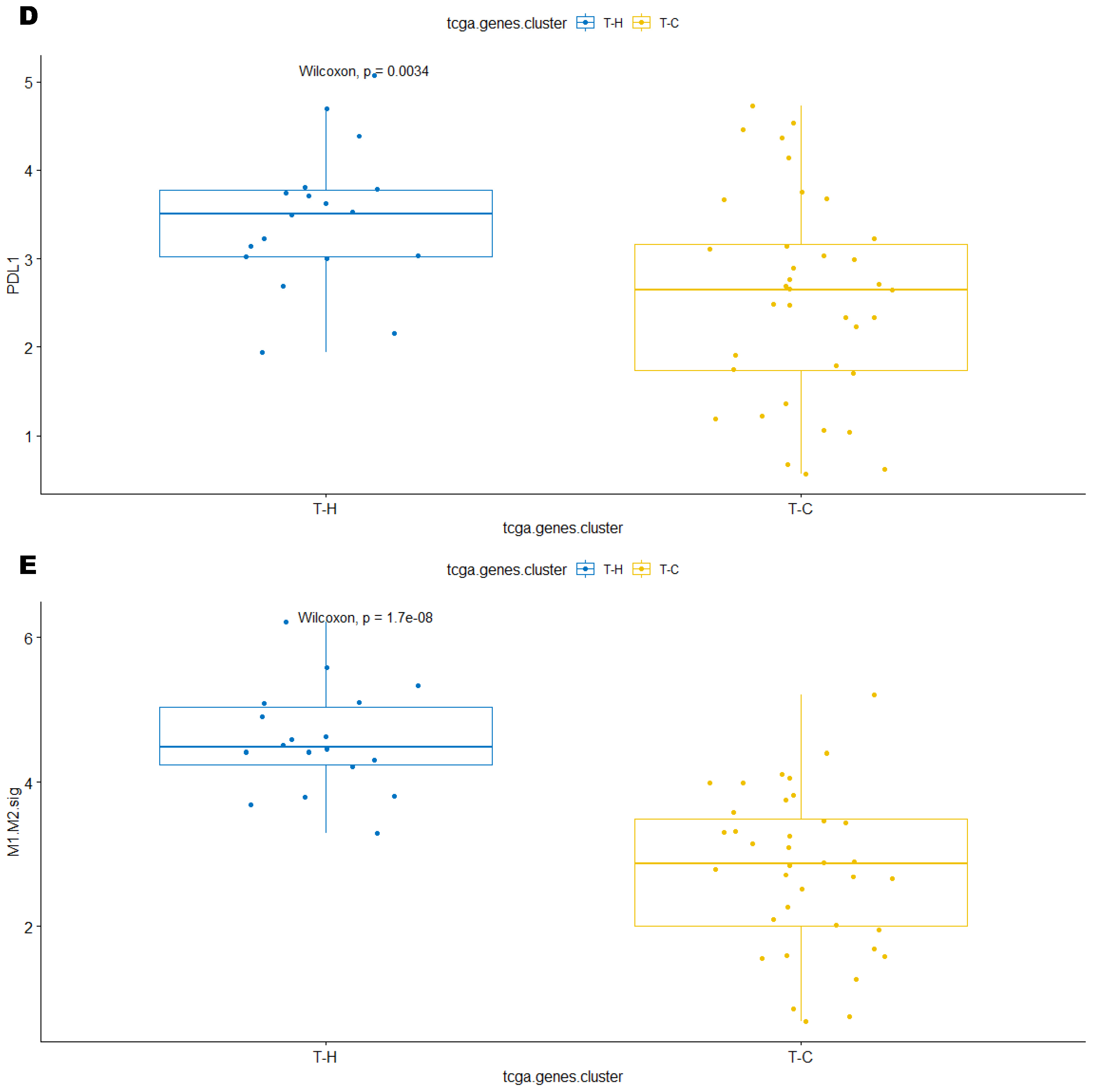
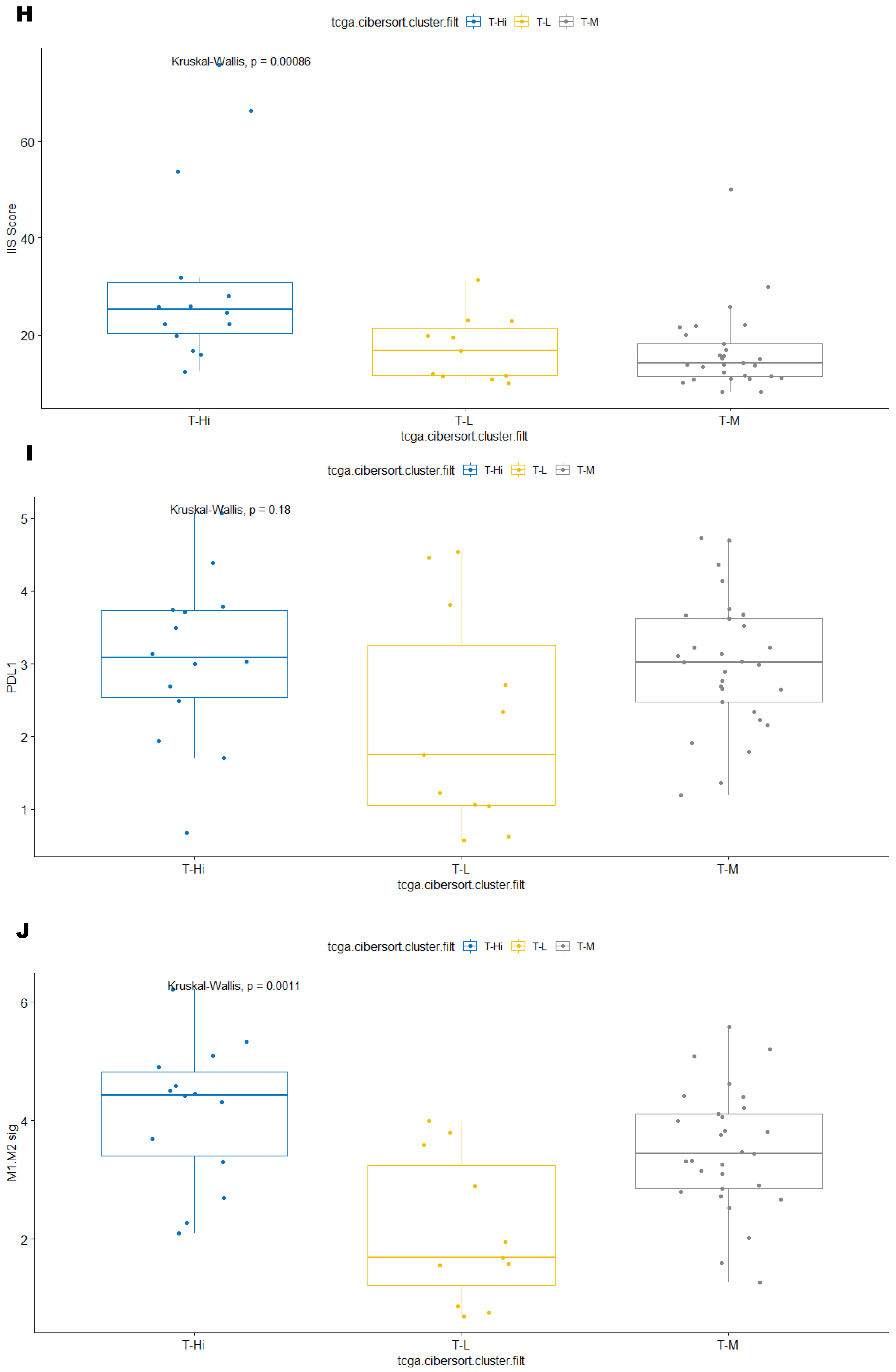
3.3. Immune Clusters Identified through 90-Gene Signature and CIBERSORT Methods Were Associated with Other Immune Signatures
3.4. Intra-Tumor Homogeneity of TIME Signatures
3.5. Correlation of Immune Clusters with Clinical Factors, Molecular Classifiers and Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Classification of Tumours of Soft Tissue and Bone Tumours: WHO Classification of Tumours, 5th ed.; International Agency for Research on Cancer Publications: Lyon, France, 2020; Volume 3.
- de Pinieux, G.; Karanian, M.; Le Loarer, F.; Le Guellec, S.; Chabaud, S.; Terrier, P.; Bouvier, C.; Batistella, M.; Neuville, A.; Robin, Y.; et al. Nationwide incidence of sarcomas and connective tissue tumors of intermediate malignancy over four years using an expert pathology review network. PLoS ONE 2021, 16, e0246958. [Google Scholar] [CrossRef]
- Italiano, A.; Mathoulin-Pelissier, S.; Cesne, A.L.; Terrier, P.; Bonvalot, S.; Collin, F.; Michels, J.; Blay, J.; Coindre, J.; Bui, B.; et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer 2011, 117, 1049–1054. [Google Scholar] [CrossRef]
- Leitao, M.M.; Hensley, M.L.; Barakat, R.R.; Aghajanian, C.; Gardner, G.J.; Jewell, E.L.; O’Cearbhaill, R.; Soslow, R.A. Immunohistochemical expression of estrogen and progesterone receptors and outcomes in patients with newly diagnosed uterine leiomyosarcoma. Gynecol. Oncol. 2012, 124, 558–562. [Google Scholar] [CrossRef]
- Chudasama, P.; Mughal, S.S.; Sanders, M.A.; Hübschmann, D.; Chung, I.; Deeg, K.I.; Wong, S.; Rabe, S.; Hlevnjak, M.; Zapatka, M.; et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat. Commun. 2018, 9, 144. [Google Scholar] [CrossRef]
- Oza, J.; Doshi, S.D.; Hao, L.; Musi, E.; Schwartz, G.K.; Ingham, M. Homologous recombination repair deficiency as a therapeutic target in sarcoma. Semin. Oncol. 2020, 47, 380–389. [Google Scholar] [CrossRef]
- Hemming, M.L.; Fan, C.; Raut, C.P.; Demetri, G.D.; Armstrong, S.A.; Sicinska, E.; George, S. Oncogenic Gene-Expression Programs in Leiomyosarcoma and Characterization of Conventional, Inflammatory, and Uterogenic Subtypes. Mol. Cancer Res. 2020, 18, 1302–1314. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar]
- Saerens, M.; Brusselaers, N.; Rottey, S.; Decruyenaere, A.; Creytens, D.; Lapeire, L. Immune checkpoint inhibitors in treatment of soft-tissue sarcoma: A systematic review and meta-analysis. Eur. J. Cancer 2021, 152, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Wilky, B.A.; Trucco, M.M.; Subhawong, T.K.; Florou, V.; Park, W.; Kwon, D.; Wieder, E.D.; Kolonias, D.; Rosenberg, A.E.; Kerr, D.A.; et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: A single-centre, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 837–848. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Lazar, A.J.; McLellan, M.D.; Bailey, M.H.; Miller, C.A.; Appelbaum, E.L.; Cordes, M.G.; Fronick, C.C.; Fulton, L.A.; Fulton, R.S.; Mardis, E.R. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar]
- Cohen, J.E.; Eleyan, F.; Zick, A.; Peretz, T.; Katz, D. Intratumoral immune-biomarkers and mismatch repair status in leiyomyosarcoma -potential predictive markers for adjuvant treatment: A pilot study. Oncotarget 2018, 9, 30847–30854. [Google Scholar] [CrossRef] [PubMed]
- Shanes, E.D.; Friedman, L.A.; Mills, A.M. PD-L1 Expression and Tumor-infiltrating Lymphocytes in Uterine Smooth Muscle Tumors: Implications for Immunotherapy. Am. J. Surg. Pathol. 2019, 43, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Pollack, S.M.; Ingham, M.; Spraker, M.B.; Schwartz, G.K. Emerging Targeted and Immune-Based Therapies in Sarcoma. J. Clin. Oncol. 2018, 36, 125–135. [Google Scholar] [CrossRef]
- Keung, E.Z.; Burgess, M.; Salazar, R.; Parra, E.R.; Rodrigues-Canales, J.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Attia, S.; Riedel, R.F.; et al. Correlative Analyses of the SARC028 Trial Reveal an Association Between Sarcoma-Associated Immune Infiltrate and Response to Pembrolizumab. Clin. Cancer Res. 2020, 26, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Cote, G.M.; He, J.; Choy, E. Next-Generation Sequencing for Patients with Sarcoma: A Single Center Experience. Oncologist 2018, 23, 234–242. [Google Scholar] [CrossRef]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 1, 2017. [Google Scholar] [CrossRef] [PubMed]
- Dancsok, A.R.; Gao, D.; Lee, A.F.; Steigen, S.E.; Blay, J.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology 2020, 9, 1747340. [Google Scholar] [CrossRef]
- Pollack, S.M.; He, Q.; Yearley, J.H.; Emerson, R.; Vignali, M.; Zhang, Y.; Redman, M.W.; Baker, K.K.; Cooper, S.; Donahue, B.; et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer 2017, 123, 3291–3304. [Google Scholar] [CrossRef]
- Wunder, J.S.; Lee, M.J.; Nam, J.; Lau, B.Y.; Dickson, B.C.; Pinnaduwage, D.; Bull, S.B.; Ferguson, P.C.; Seto, A.; Gokgoz, N.; et al. Osteosarcoma and soft-tissue sarcomas with an immune infiltrate express PD-L1: Relation to clinical outcome and Th1 pathway activation. Oncoimmunology 2020, 9, 1737385. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.T.J.; Chew, W.; Wilding, C.P.; Guljar, N.; Smith, M.J.; Strauss, D.C.; Fisher, C.; Hayes, A.J.; Judson, I.; Thway, K.; et al. The adequacy of tissue microarrays in the assessment of inter- and intra-tumoural heterogeneity of infiltrating lymphocyte burden in leiomyosarcoma. Sci. Rep. 2019, 9, 14602. [Google Scholar] [CrossRef]
- Zhu, M.M.T.; Shenasa, E.; Nielsen, T.O. Sarcomas: Immune biomarker expression and checkpoint inhibitor trials. Cancer Treat. Rev. 2020, 91, 102115. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.; Sun, C.; Calderaro, J.; Jeng, Y.; Hsiao, L.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Bessede, A.; Bompas, E.; Piperno-Neumann, S.; Chevreau, C.; Penel, N.; Bertucci, F.; Toulmonde, M.; Bellera, C.A.; Guegan, J.; et al. PD1 inhibition in soft-tissue sarcomas with tertiary lymphoid structures: A multicenter phase II trial. JCO 2021, 39, 11507. [Google Scholar] [CrossRef]
- Vanhersecke, L.; Brunet, M.; Guégan, J.; Rey, C.; Bougouin, A.; Cousin, S.; Le Moulec, S.; Besse, B.; Loriot, Y.; Larroquette, M.; et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat. Cancer 2021, 2, 794–802. [Google Scholar] [CrossRef]
- Ben-Ami, E.; Barysauskas, C.M.; Solomon, S.; Tahlil, K.; Malley, R.; Hohos, M.; Polson, K.; Loucks, M.; Severgnini, M.; Patel, T.; et al. Immunotherapy with single agent nivolumab for advanced leiomyosarcoma of the uterus: Results of a phase 2 study. Cancer 2017, 123, 3285–3290. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Danaher, P.; Warren, S.; Lu, R.; Samayoa, J.; Sullivan, A.; Pekker, I.; Wallden, B.; Marincola, F.M.; Cesano, A. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): Results from The Cancer Genome Atlas (TCGA). J. Immunother. Cancer 2018, 6, 63. [Google Scholar] [CrossRef]
- Pantaleo, M.A.; Tarantino, G.; Agostinelli, C.; Urbini, M.; Nannini, M.; Saponara, M.; Castelli, C.; Stacchiotti, S.; Fumagalli, E.; Gatto, L.; et al. Immune microenvironment profiling of gastrointestinal stromal tumors (GIST) shows gene expression patterns associated to immune checkpoint inhibitors response. Oncoimmunology 2019, 8, e1617588. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Pleasance, E.; Zhao, E.Y.; Ng, T.; Grewal, J.K.; Mohammad, N.; Taylor, S.K.; Simmons, C.; Srikanthan, A.; Rassekh, S.R.; et al. Therapeutic Implication of Genomic Landscape of Adult Metastatic Sarcoma. JCO Precis. Oncol. 2019, 3, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Trojani, M.; Contesso, G.; Coindre, J.M.; Rouesse, J.; Bui, N.B.; de Mascarel, A.; Goussot, J.F.; David, M.; Bonichon, F.; Lagarde, C. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int. J. Cancer 1984, 33, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Lesluyes, T.; Pérot, G.; Largeau, M.R.; Brulard, C.; Lagarde, P.; Dapremont, V.; Lucchesi, C.; Neuville, A.; Terrier, P.; Vince-Ranchère, D.; et al. RNA sequencing validation of the Complexity INdex in SARComas prognostic signature. Eur. J. Cancer 2016, 57, 104–111. [Google Scholar] [CrossRef]
- Silva, T.C.; Colaprico, A.; Olsen, C.; D’Angelo, F.; Bontempi, G.; Ceccarelli, M.; Noushmehr, H. TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Research 2016, 5, 1542. [Google Scholar] [CrossRef]
- Dufresne, A.; Lesluyes, T.; Ménétrier-Caux, C.; Brahmi, M.; Darbo, E.; Toulmonde, M.; Italiano, A.; Mir, O.; Le Cesne, A.; Le Guellec, S.; et al. Specific immune landscapes and immune checkpoint expressions in histotypes and molecular subtypes of sarcoma. Oncoimmunology 2020, 9, 1792036. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Pender, A.; Titmuss, E.; Pleasance, E.D.; Fan, K.Y.; Pearson, H.; Brown, S.D.; Grisdale, C.J.; Topham, J.T.; Shen, Y.; Bonakdar, M.; et al. Genome and Transcriptome Biomarkers of Response to Immune Checkpoint Inhibitors in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 202–212. [Google Scholar] [CrossRef]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Wang, F.; Yu, T.; Ma, C.; Yuan, H.; Zhang, H.; Zhang, Z. Prognostic Value of Programmed Cell Death 1 Ligand-1 in Patients with Bone and Soft Tissue Sarcomas: A Systemic and Comprehensive Meta-Analysis Based on 3,680 Patients. Front. Oncol. 2020, 10, 749. [Google Scholar] [CrossRef]
- Toulmonde, M.; Penel, N.; Adam, J.; Chevreau, C.; Blay, J.; Le Cesne, A.; Bompas, E.; Piperno-Neumann, S.; Cousin, S.; Grellety, T.; et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 93–97. [Google Scholar] [CrossRef]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Tanaka, T.; Yamamoto, Y.; Akasaki, Y.; Sasaki, H. Dual role of macrophage in tumor immunity. Immunotherapy 2018, 10, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Espinosa, I.; Vrijaldenhoven, S.; Subramanian, S.; Montgomery, K.D.; Zhu, S.; Marinelli, R.J.; Peterse, J.L.; Poulin, N.; Nielsen, T.O.; et al. Prognostic significance of macrophage infiltration in leiomyosarcomas. Clin. Cancer Res. 2008, 14, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Ismaila, N.; Allison, K.H.; Barlow, W.E.; Collyar, D.E.; Damodaran, S.; Henry, N.L.; Jhaveri, K.; Kalinsky, K.; Kuderer, N.M.; et al. Biomarkers for Adjuvant Endocrine and Chemotherapy in Early-Stage Breast Cancer: ASCO Guideline Update. J. Clin. Oncol. 2022, 40, 1816–1837. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient/Tumor Characteristics | ICGC Cohort (N = 111) | TCGA Cohort (N = 74) |
|---|---|---|
| Age | ||
| Median (min–max) | 64 (22–85) | 60 (33–90) |
| Gender | ||
| Female | 84 (75.7%) | 40 (54.1%) |
| Male | 27 (24.3%) | 34 (45.9%) |
| Tumor Size (cm) | Not available | |
| ≤5 | 27 (24.3%) | |
| >5 and ≤10 | 43 (38.7%) | |
| >10 | 40 (36%) | |
| Unknown | 1 (0.9%) | |
| Tumor Grade (/FRSCC) | ||
| 1 | 14 (12.6%) | 3 (4.1%) |
| 2 | 38 (34.2%) | 54 (73%) |
| 3 | 52 (46.8%) | 17 (23%) |
| Unknown | 7 (6.3%) | 0 (0%) |
| Tumor Location | 24 (32.4%) 50 (67.6%) | |
| Limb | 20 (18%) | |
| Internal trunk | 56 (50.5%) | |
| Trunk wall | 10 (9%) | |
| Head and neck | 5 (4.5%) | |
| Gynecological | 16 (14.4%) | |
| Others | 4 (3.6%) | |
| Tumor Depth | Not available | |
| Superficial | 8 (7.2%) | |
| Deep | 103 (92.8%) | |
| Tumor Multifocality | Not available | |
| No | 108 (97.3%) | |
| Yes | 3 (2.7%) | |
| Surgery | Not available | |
| Yes | 110 (99.1%) | |
| NA | 1 (0.9%) | |
| Re-resection | Not available | |
| No | 96 (86.5%) | |
| Yes | 9 (8.1%) | |
| Unknown | 6 (5.4%) | |
| Surgical Margin | Not available | |
| R0 | 68 (61.3%) | |
| R1 | 32 (28.8%) | |
| R2 | 1 (0.9%) | |
| Not evaluable/Unknown | 10 (9%) | |
| (Neo)Adjuvant chemotherapy | Not available | |
| No | 92 (82.9%) | |
| Yes * | 19 (17.1%) * | |
| (Neo)Adjuvant Radiotherapy | Not available | |
| No | 73 (65.8%) | |
| Yes ** | 38 (34.2%) ** | |
| Local Recurrence | ||
| No | 99 (89.2%) | |
| Yes | 12 (10.8%) | |
| Metastatic Recurrence | ||
| No | 49 (44.1%) | 40 (54.15) |
| Yes | 62 (55.5%) | 34 (45.9%) |
| Metastatic Site (first) | Not available | |
| Lung only | 27 (43.5%) | |
| Liver only | 10 (16.1%) | |
| Lung, liver, and others | 7 (11.3%) | |
| Peritoneum | 5 (8.1%) | |
| Bone | 3 (4.8%) | |
| Skin/Soft tissue/Lymph node | 9 (14.5%) | |
| Brain only | 1 (1.6%) | |
| Locoregional treatment for metastatic disease | Not available | |
| No | 41 (66.1%) | |
| Yes *** | 21 (33.9%) *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, X.; Tonon, L.; Li, H.; Darbo, E.; Pleasance, E.; Macagno, N.; Dufresne, A.; Brahmi, M.; Bollard, J.; Ducimetière, F.; et al. Comprehensive Immune Profiling Unveils a Subset of Leiomyosarcoma with “Hot” Tumor Immune Microenvironment. Cancers 2023, 15, 3705. https://doi.org/10.3390/cancers15143705
Feng X, Tonon L, Li H, Darbo E, Pleasance E, Macagno N, Dufresne A, Brahmi M, Bollard J, Ducimetière F, et al. Comprehensive Immune Profiling Unveils a Subset of Leiomyosarcoma with “Hot” Tumor Immune Microenvironment. Cancers. 2023; 15(14):3705. https://doi.org/10.3390/cancers15143705
Chicago/Turabian StyleFeng, Xiaolan, Laurie Tonon, Haocheng Li, Elodie Darbo, Erin Pleasance, Nicolas Macagno, Armelle Dufresne, Mehdi Brahmi, Julien Bollard, Francoise Ducimetière, and et al. 2023. "Comprehensive Immune Profiling Unveils a Subset of Leiomyosarcoma with “Hot” Tumor Immune Microenvironment" Cancers 15, no. 14: 3705. https://doi.org/10.3390/cancers15143705
APA StyleFeng, X., Tonon, L., Li, H., Darbo, E., Pleasance, E., Macagno, N., Dufresne, A., Brahmi, M., Bollard, J., Ducimetière, F., Karanian, M., Meurgey, A., Pérot, G., Valentin, T., Chibon, F., & Blay, J.-Y. (2023). Comprehensive Immune Profiling Unveils a Subset of Leiomyosarcoma with “Hot” Tumor Immune Microenvironment. Cancers, 15(14), 3705. https://doi.org/10.3390/cancers15143705