

A Blood-Based Immune Gene Signature with Prognostic Significance in Localized Prostate Cancer


 , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Study Individuals and Sample Collection
2.2. Ethics Approval
2.3. RNA Isolation
2.4. Library Preparation and Next-Generation Sequencing
2.5. Gene Expression Analysis from Blood Samples
2.6. TCGA Data Analysis
3. Results
3.1. Demographics of the Study Cohorts
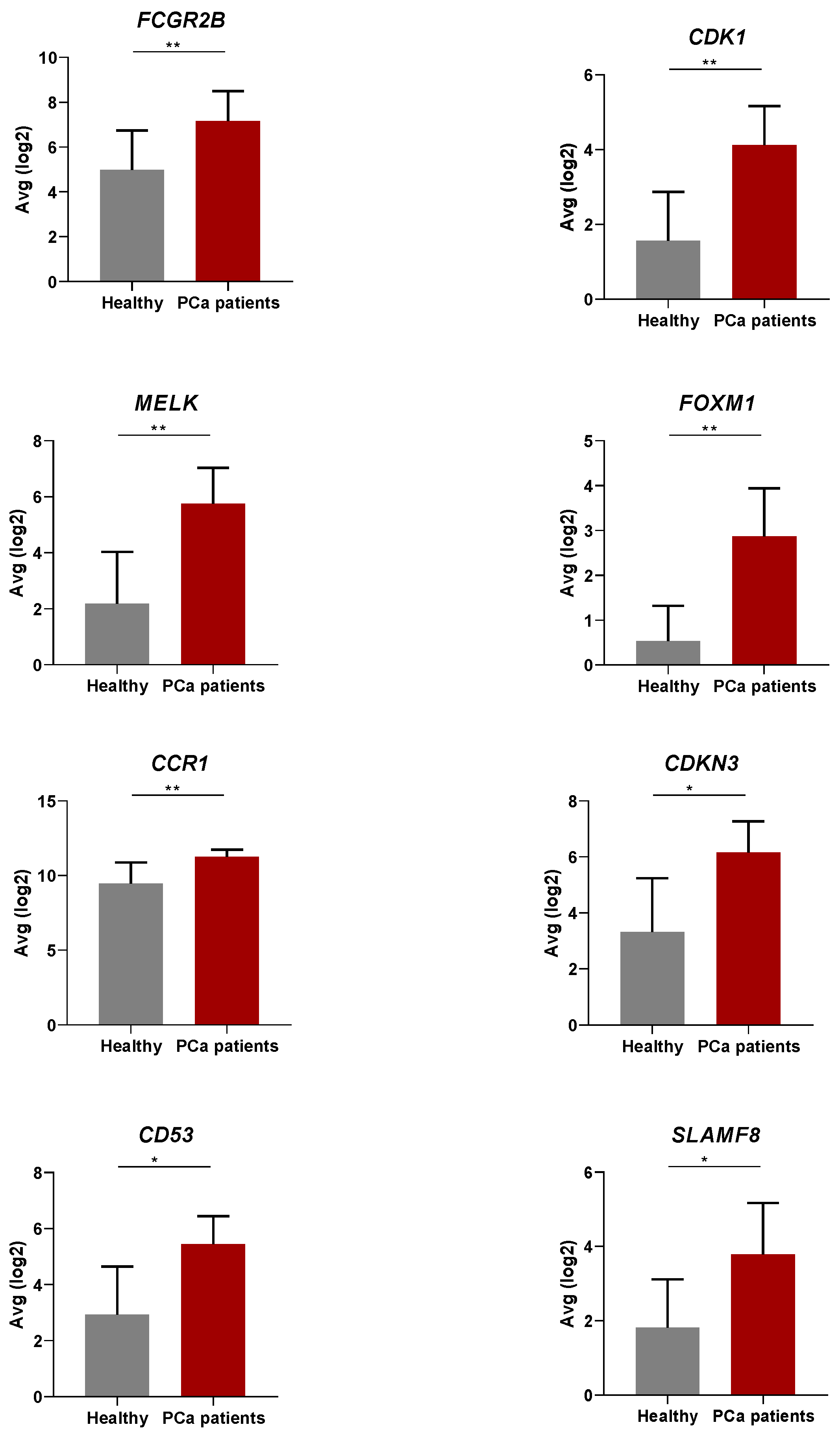
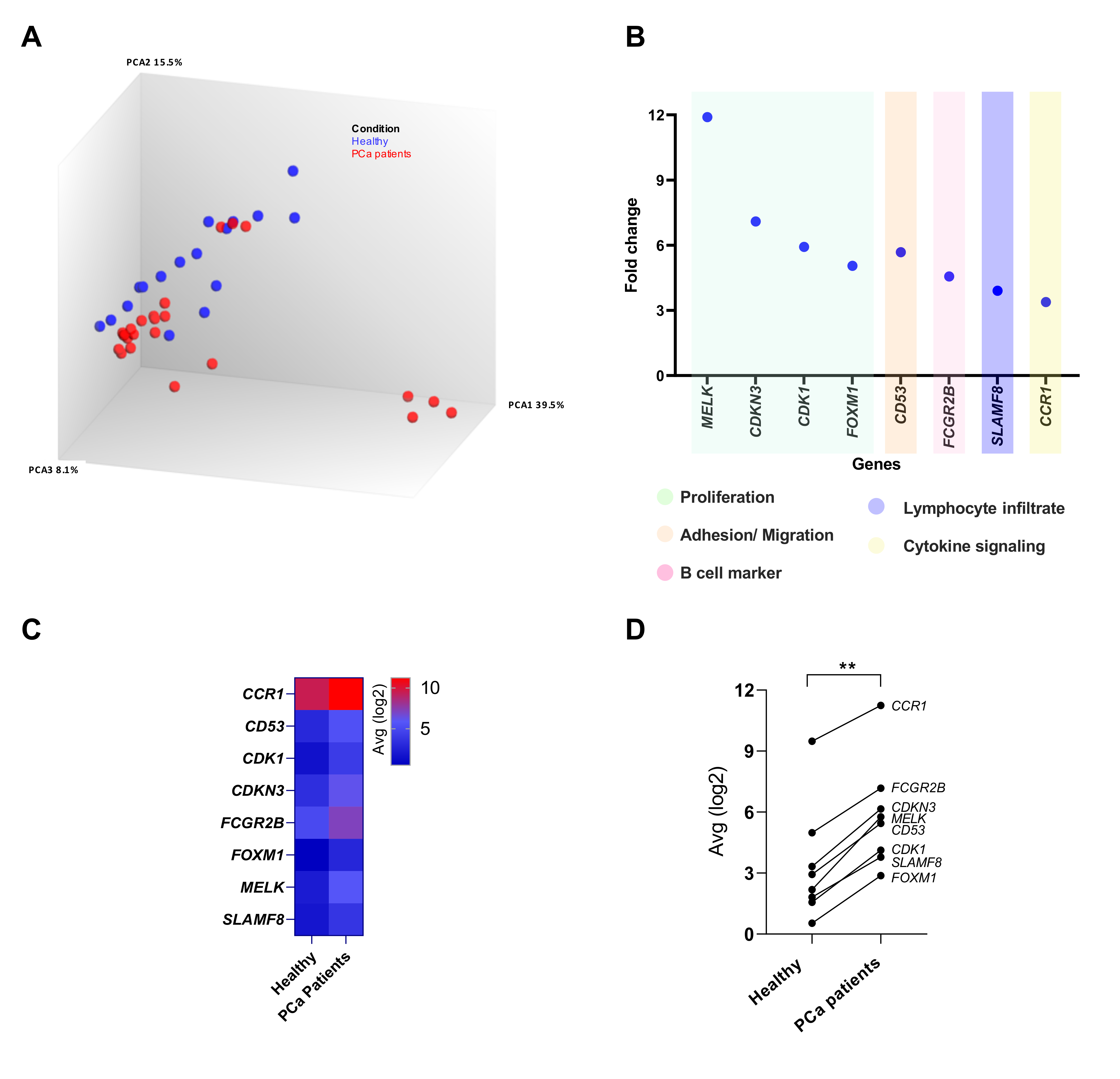
3.2. Differential Gene Expression in the Peripheral Blood of PCa Patients Compared to Healthy Donors
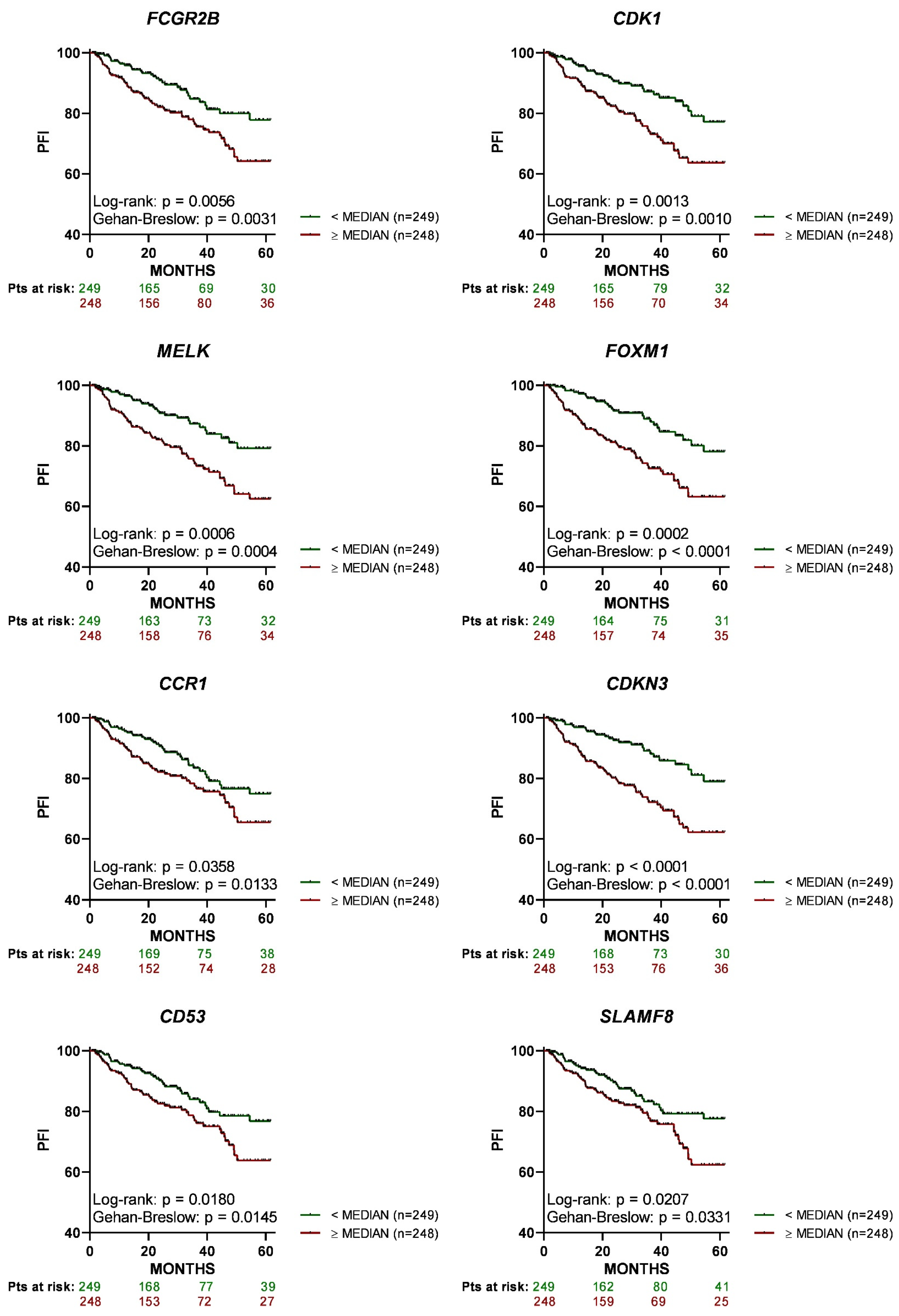
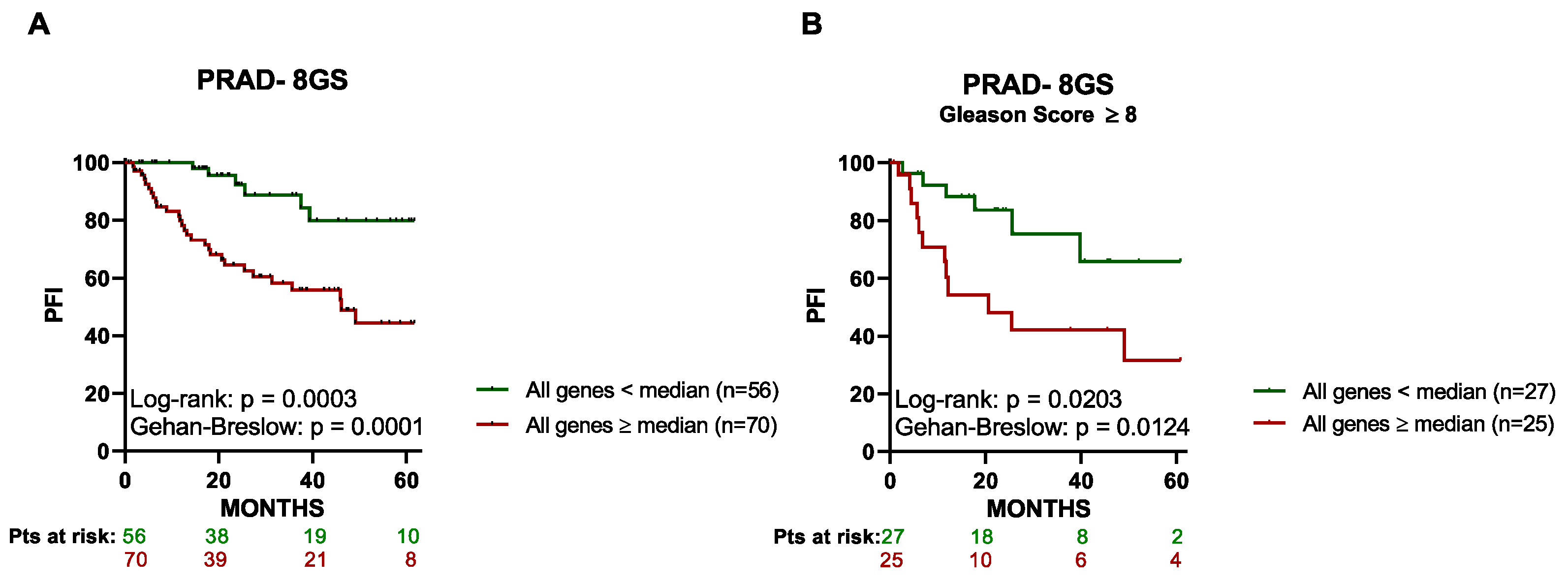
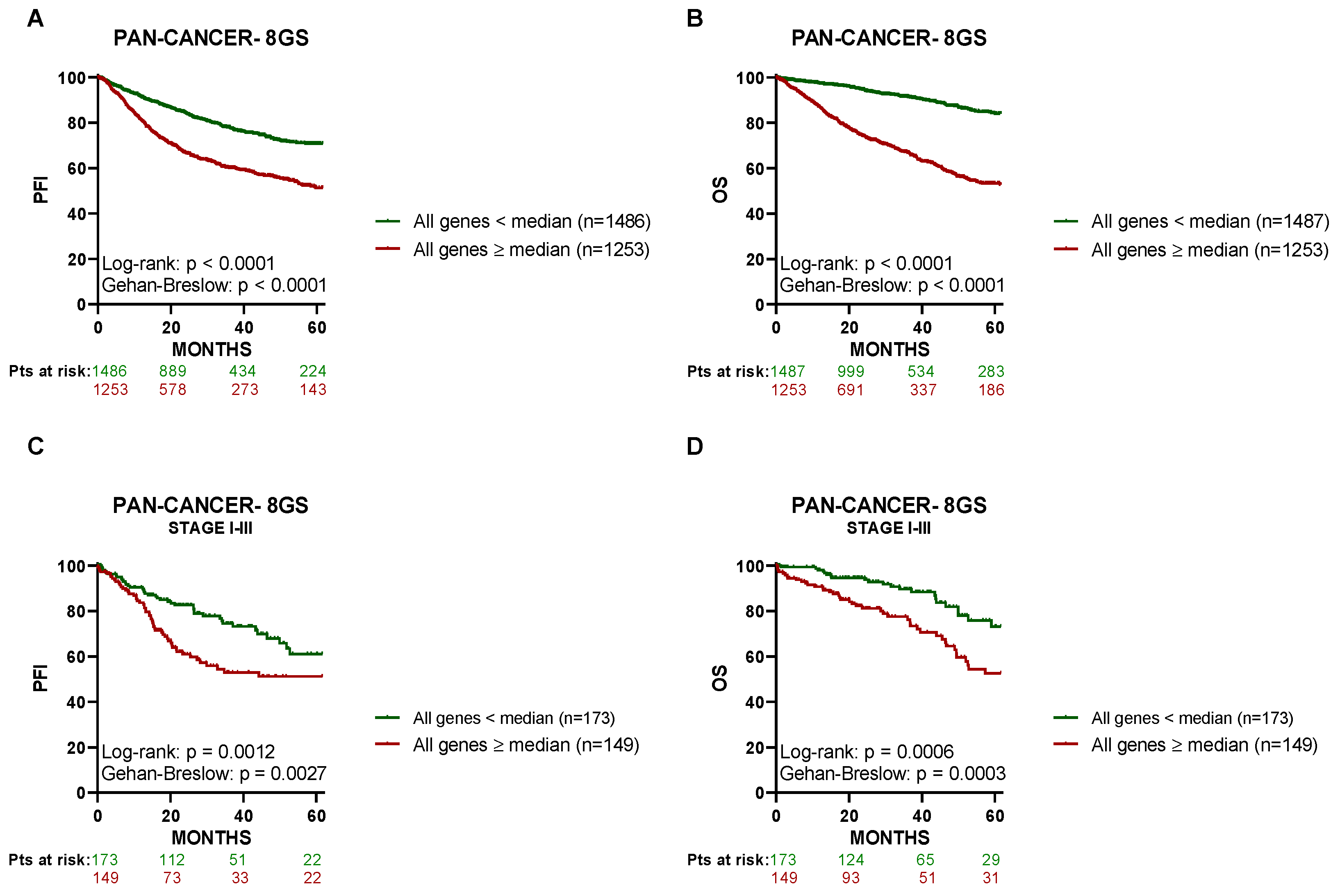
3.3. High Expression of the 8 Genes in Primary Tumor Tissue Associates with a Lower PFI in PCa Patients
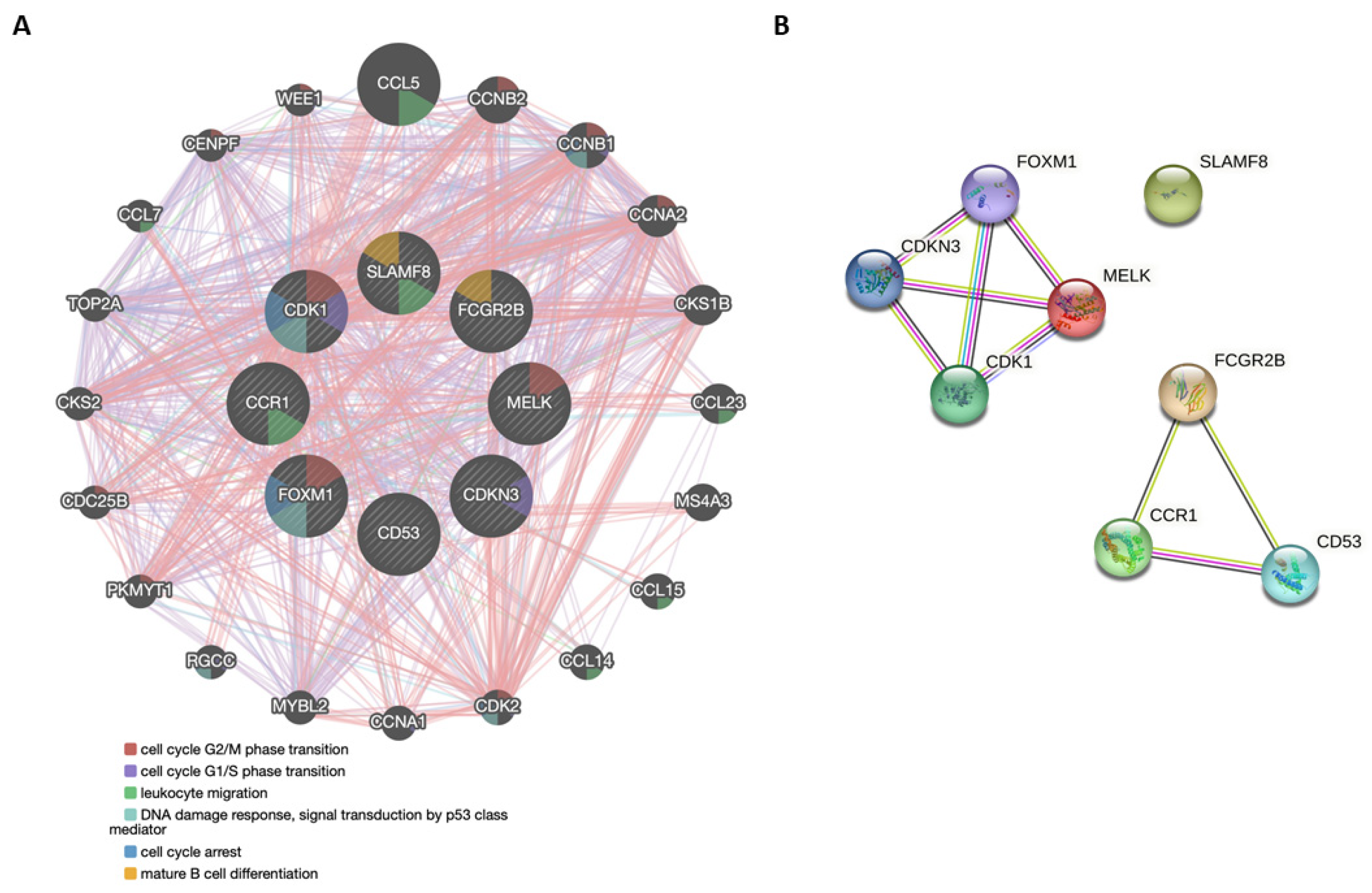
3.4. Functional Genetic and Protein Networks Structured by the Genes Comprising the 8-GS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Prim. 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, D.; Spring, D.J.; DePinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef] [PubMed]
- Sekhoacha, M.; Riet, K.; Motloung, P.; Gumenku, L.; Adegoke, A.; Mashele, S. Prostate Cancer Review: Genetics, Diagnosis, Treatment Options, and Alternative Approaches. Molecules 2022, 27, 5730. [Google Scholar] [CrossRef]
- Narla, G.; DiFeo, A.; Fernandez, Y.; Dhanasekaran, S.; Huang, F.; Sangodkar, J.; Hod, E.; Leake, D.; Friedman, S.L.; Hall, S.J.; et al. KLF6-SV1 overexpression accelerates human and mouse prostate cancer progression and metastasis. J. Clin. Invest. 2008, 118, 2711–2721. [Google Scholar] [CrossRef]
- Gurel, B.; Ali, T.Z.; Montgomery, E.A.; Begum, S.; Hicks, J.; Goggins, M.; Eberhart, C.G.; Clark, D.P.; Bieberich, C.J.; Epstein, J.I.; et al. NKX3.1 as a marker of prostatic origin in metastatic tumors. Am. J. Surg. Pathol. 2010, 34, 1097–1105. [Google Scholar] [CrossRef]
- Porzycki, P.; Ciszkowicz, E. Modern biomarkers in prostate cancer diagnosis. Cent. Eur. J. Urol. 2020, 73, 300–306. [Google Scholar] [CrossRef]
- Eickelschulte, S.; Riediger, A.L.; Angeles, A.K.; Janke, F.; Duensing, S.; Sültmann, H.; Görtz, M. Biomarkers for the Detection and Risk Stratification of Aggressive Prostate Cancer. Cancers 2022, 14, 6094. [Google Scholar] [CrossRef]
- Eggener, S.E.; Rumble, R.B.; Armstrong, A.J.; Morgan, T.M.; Crispino, T.; Cornford, P.; van der Kwast, T.; Grignon, D.J.; Rai, A.J.; Agarwal, N.; et al. Molecular Biomarkers in Localized Prostate Cancer: ASCO Guideline. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1474–1494. [Google Scholar] [CrossRef]
- Borley, N.; Feneley, M.R. Prostate cancer: Diagnosis and staging. Asian J. Androl. 2009, 11, 74–80. [Google Scholar] [CrossRef]
- Liu, B.; Wang, Y.; Weitz, P.; Lindberg, J.; Hartman, J.; Wang, W.; Egevad, L.; Grönberg, H.; Eklund, M.; Rantalainen, M. Using deep learning to detect patients at risk for prostate cancer despite benign biopsies. iScience 2022, 25, 104663. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Localio, A.R.; Armstrong, K.; Malkowicz, S.B.; Schwartz, J.S. A meta-analysis of the performance characteristics of the free prostate-specific antigen test. Urology 2006, 67, 762–768. [Google Scholar] [CrossRef]
- Maldonado, L.; Brait, M.; Loyo, M.; Sullenberger, L.; Wang, K.; Peskoe, S.B.; Rosenbaum, E.; Howard, R.; Toubaji, A.; Albadine, R.; et al. GSTP1 promoter methylation is associated with recurrence in early stage prostate cancer. J. Urol. 2014, 192, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Cullen, J.; Rosner, I.L.; Brand, T.C.; Zhang, N.; Tsiatis, A.C.; Moncur, J.; Ali, A.; Chen, Y.; Knezevic, D.; Maddala, T.; et al. A Biopsy-based 17-gene Genomic Prostate Score Predicts Recurrence After Radical Prostatectomy and Adverse Surgical Pathology in a Racially Diverse Population of Men with Clinically Low- and Intermediate-risk Prostate Cancer. Eur. Urol. 2015, 68, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Fortis, S.P.; Goulielmaki, M.; Aubert, N.; Batsaki, P.; Ouzounis, S.; Cavouras, D.; Marodon, G.; Stokidis, S.; Gritzapis, A.D.; Baxevanis, C.N. Radiotherapy-Related Gene Signature in Prostate Cancer. Cancers 2022, 14, 5032. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Jurmeister, S.; Ramos-Montoya, A.; Sandi, C.; Pértega-Gomes, N.; Wadhwa, K.; Lamb, A.D.; Dunning, M.J.; Attig, J.; Carroll, J.S.; Fryer, L.G.D.; et al. Identification of potential therapeutic targets in prostate cancer through a cross-species approach. EMBO Mol. Med. 2018, 10, e8274. [Google Scholar] [CrossRef]
- Oshi, M.; Gandhi, S.; Huyser, M.R.; Tokumaru, Y.; Yan, L.; Yamada, A.; Matsuyama, R.; Endo, I.; Takabe, K. MELK expression in breast cancer is associated with infiltration of immune cell and pathological compete response (pCR) after neoadjuvant chemotherapy. Am. J. Cancer Res. 2021, 11, 4421–4437. [Google Scholar] [CrossRef] [PubMed]
- Madhi, H.; Lee, J.-S.; Choi, Y.E.; Li, Y.; Kim, M.H.; Choi, Y.; Goh, S.-H. FOXM1 Inhibition Enhances the Therapeutic Outcome of Lung Cancer Immunotherapy by Modulating PD-L1 Expression and Cell Proliferation. Adv. Sci. 2022, 9, 2202702. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Q.; Guo, X.; Yuan, Q.; Nian, S.; Kang, P.; Xu, Z.; Li, L.; Ye, Y. Systematic Pan-Cancer Analysis Identifies CDK1 as an Immunological and Prognostic Biomarker. J. Oncol. 2022, 2022, 8115474. [Google Scholar] [CrossRef] [PubMed]
- Hedblom, A.; Laursen, K.B.; Miftakhova, R.; Sarwar, M.; Anagnostaki, L.; Bredberg, A.; Mongan, N.P.; Gudas, L.J.; Persson, J.L. CDK1 interacts with RARγ and plays an important role in treatment response of acute myeloid leukemia. Cell Cycle 2013, 12, 1251–1266. [Google Scholar] [CrossRef]
- Zhu, Y.; Bian, Y.; Zhang, Q.; Hu, J.; Li, L.; Yang, M.; Qian, H.; Yu, L.; Liu, B.; Qian, X. Construction and analysis of dysregulated lncRNA-associated ceRNA network in colorectal cancer. J. Cell. Biochem. 2019, 120, 9250–9263. [Google Scholar] [CrossRef]
- Zhu, Y.; Bian, Y.; Zhang, Q.; Hu, J.; Li, L.; Yang, M.; Qian, H.; Yu, L.; Liu, B.; Qian, X. LINC00365 promotes colorectal cancer cell progression through the Wnt/β-catenin signaling pathway. J. Cell. Biochem. 2020, 121, 1260–1272. [Google Scholar] [CrossRef]
- Li, S.; Xue, P.; Diao, X.; Fan, Q.-Y.; Ye, K.; Tang, X.-M.; Liu, J.; Huang, Z.-Y.; Tang, Q.-H.; Jia, C.-Y.; et al. Identification and validation of functional roles for three MYC-associated genes in hepatocellular carcinoma. J. Adv. Res. 2023. [Google Scholar] [CrossRef]
- Wu, L.; Xu, Y.; Zhao, H.; Zhou, Y.; Chen, Y.; Yang, S.; Lei, J.; Zhang, J.; Wang, J.; Wu, Y.; et al. FcγRIIB potentiates differentiation of myeloid-derived suppressor cells to mediate tumor immunoescape. Theranostics 2022, 12, 842–858. [Google Scholar] [CrossRef]
- Hussain, K.; Liu, R.; Smith, R.C.G.; Müller, K.T.J.; Ghorbani, M.; Macari, S.; Cleary, K.L.S.; Oldham, R.J.; Foxall, R.B.; James, S.; et al. HIF activation enhances FcγRIIb expression on mononuclear phagocytes impeding tumor targeting antibody immunotherapy. J. Exp. Clin. Cancer Res. 2022, 41, 131. [Google Scholar] [CrossRef]
- Dunlock, V.E. Tetraspanin CD53: An overlooked regulator of immune cell function. Med. Microbiol. Immunol. 2020, 209, 545–552. [Google Scholar] [CrossRef]
- Todros-Dawda, I.; Kveberg, L.; Vaage, J.T.; Inngjerdingen, M. The tetraspanin CD53 modulates responses from activating NK cell receptors, promoting LFA-1 activation and dampening NK cell effector functions. PLoS ONE 2014, 9, e97844. [Google Scholar] [CrossRef] [PubMed]
- Alarcón-Zendejas, A.P.; Scavuzzo, A.; Jiménez-Ríos, M.A.; Álvarez-Gómez, R.M.; Montiel-Manríquez, R.; Castro-Hernández, C.; Jiménez-Dávila, M.A.; Pérez-Montiel, D.; González-Barrios, R.; Jiménez-Trejo, F.; et al. The promising role of new molecular biomarkers in prostate cancer: From coding and non-coding genes to artificial intelligence approaches. Prostate Cancer Prostatic Dis. 2022, 25, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Lentzsch, S.; Gries, M.; Janz, M.; Bargou, R.; Dörken, B.; Mapara, M.Y. Macrophage inflammatory protein 1-alpha (MIP-1α) triggers migration and signaling cascades mediating survival and proliferation in multiple myeloma (MM) cells. Blood 2003, 101, 3568–3573. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Fujita, Y.; Nakane, K.; Mizutani, K.; Terazawa, R.; Ehara, H.; Kanimoto, Y.; Kojima, T.; Nozawa, Y.; Deguchi, T.; et al. CCR1/CCL5 interaction promotes invasion of taxane-resistant PC3 prostate cancer cells by increasing secretion of MMPs 2/9 and by activating ERK and Rac signaling. Cytokine 2013, 64, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Fujishita, T.; Loetscher, P.; Revesz, L.; Hashida, H.; Kizaka-Kondoh, S.; Aoki, M.; Taketo, M.M. Inactivation of chemokine (C-C motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proc. Natl. Acad. Sci. USA. 2010, 107, 13063–13068. [Google Scholar] [CrossRef]
- Sugimoto, A.; Kataoka, T.R.; Ueshima, C.; Takei, Y.; Kitamura, K.; Hirata, M.; Nomura, T.; Haga, H. SLAM family member 8 is involved in oncogenic KIT-mediated signalling in human mastocytosis. Exp. Dermatol. 2018, 27, 641–646. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, P.; Ma, J.; Xu, J.; Yang, L.; Xu, W.; Que, H.; Chen, M.; Xu, H. Serum biomarker panels for the diagnosis of gastric cancer. Cancer Med. 2019, 8, 1576–1583. [Google Scholar] [CrossRef]
- Zhang, Q.; Cheng, L.; Qin, Y.; Kong, L.; Shi, X.; Hu, J.; Li, L.; Ding, Z.; Wang, T.; Shen, J.; et al. SLAMF8 expression predicts the efficacy of anti-PD1 immunotherapy in gastrointestinal cancers. Clin. Transl. Immunol. 2021, 10, e1347. [Google Scholar] [CrossRef]
- McDonald, I.M.; Graves, L.M. Enigmatic MELK: The controversy surrounding its complex role in cancer. J. Biol. Chem. 2020, 295, 8195–8203. [Google Scholar] [CrossRef]
- Choi, S.; Ku, J.-L. Resistance of colorectal cancer cells to radiation and 5-FU is associated with MELK expression. Biochem. Biophys. Res. Commun. 2011, 412, 207–213. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, C.; Li, C.; Jiao, X.; Griffin, B.B.; Dongol, S.; Wu, H.; Zhang, C.; Cao, W.; Dong, R.; et al. Upregulated MELK Leads to Doxorubicin Chemoresistance and M2 Macrophage Polarization via the miR-34a/JAK2/STAT3 Pathway in Uterine Leiomyosarcoma. Front. Oncol. 2020, 10, 453. [Google Scholar] [CrossRef]
- Li, L.; Wu, D.; Yu, Q.; Li, L.; Wu, P. Prognostic value of FOXM1 in solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 32298–32308. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Khoshbakht, T.; Hussen, B.M.; Dong, P.; Gassler, N.; Taheri, M.; Baniahmad, A.; Dilmaghani, N.A. A review on the role of cyclin dependent kinases in cancers. Cancer Cell Int. 2022, 22, 325. [Google Scholar] [CrossRef]
- Li, T.; Xue, H.; Guo, Y.; Guo, K. CDKN3 is an independent prognostic factor and promotes ovarian carcinoma cell proliferation in ovarian cancer. Oncol. Rep. 2014, 31, 1825–1831. [Google Scholar] [CrossRef]
- Jin, X.; Kang, J.; Lu, Q.; Guo, S.-L.; Liu, M.; Zhang, Y.; Cui, C.; Liu, H.-L.; Xu, X.; Yin, J. Fc gamma receptor IIb in tumor-associated macrophages and dendritic cells drives poor prognosis of recurrent glioblastoma through immune-associated signaling pathways. Front. Genet. 2022, 13, 1046008. [Google Scholar] [CrossRef]
- Zeissig, M.N.; Hewett, D.R.; Panagopoulos, V.; Mrozik, K.M.; To, L.B.; Croucher, P.I.; Zannettino, A.C.W.; Vandyke, K. Expression of the chemokine receptor CCR1 promotes the dissemination of multiple myeloma plasma cells in vivo. Haematologica 2020, 106, 3176–3187. [Google Scholar] [CrossRef]
- Zou, C.-Y.; Guan, G.-F.; Zhu, C.; Liu, T.-Q.; Guo, Q.; Cheng, W.; Wu, A.-H. Costimulatory checkpoint SLAMF8 is an independent prognosis factor in glioma. CNS Neurosci. Ther. 2019, 25, 333–342. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Q.; Han, X.; Han, L.; Wang, T.; Hu, J.; Li, L.; Ding, Z.; Shi, X.; Qian, X. SLAMF8, a potential novel immune checkpoint molecule, is associated with the prognosis of colorectal cancer. Transl. Oncol. 2022, 31, 101654. [Google Scholar] [CrossRef]
- Marchetti, P.; Antonov, A.; Anemona, L.; Vangapandou, C.; Montanaro, M.; Botticelli, A.; Mauriello, A.; Melino, G.; Catani, M.V. New immunological potential markers for triple negative breast cancer: IL18R1, CD53, TRIM, Jaw1, LTB, PTPRCAP. Discov. Oncol. 2021, 12, 6. [Google Scholar] [CrossRef]
- Pabla, S.; Conroy, J.M.; Nesline, M.K.; Glenn, S.T.; Papanicolau-Sengos, A.; Burgher, B.; Hagen, J.; Giamo, V.; Andreas, J.; Lenzo, F.L.; et al. Proliferative potential and resistance to immune checkpoint blockade in lung cancer patients. J. Immunother. Cancer 2019, 7, 27. [Google Scholar] [CrossRef]
- Glenn, S.T.; Pabla, S.; Zhu, J.; Labriola, M.; Gupta, R.; George, D.J.; McCall, S.; Yau, E.; Conroy, J.M.; Nesline, M.; et al. Cell proliferation as a biomarker for response to immune checkpoint inhibitors in PD-L1 negative renal cell carcinoma. J. Clin. Oncol. 2019, 37, 62. [Google Scholar] [CrossRef]
- Casarrubios, M.; Provencio, M.; Nadal, E.; Insa, A.; del Rosario García-Campelo, M.; Lázaro-Quintela, M.; Dómine, M.; Majem, M.; Rodriguez-Abreu, D.; Martinez-Marti, A.; et al. Tumor microenvironment gene expression profiles associated to complete pathological response and disease progression in resectable NSCLC patients treated with neoadjuvant chemoimmunotherapy. J. Immunother. Cancer 2022, 10, e005320. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Liu, H.; Xu, J.; Mo, Y.; Dai, F. Integrated analysis of tumor-associated macrophage infiltration and prognosis in ovarian cancer. Aging 2021, 13, 23210–23232. [Google Scholar] [CrossRef] [PubMed]
- Chifman, J.; Pullikuth, A.; Chou, J.W.; Bedognetti, D.; Miller, L.D. Conservation of immune gene signatures in solid tumors and prognostic implications. BMC Cancer 2016, 16, 911. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Daza, J.; Itzel, T.; Betge, J.; Zhan, T.; Marmé, F.; Teufel, A. Prognostic Cancer Gene Expression Signatures: Current Status and Challenges. Cells 2021, 10, 648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, X.; Peng, K.; Jia, Z.; Yang, J. Identification of biomarkers for immunotherapy response in prostate cancer and potential drugs to alleviate immunosuppression. Aging 2022, 14, 4839–4857. [Google Scholar] [CrossRef]
- Boettcher, A.N.; Usman, A.; Morgans, A.; VanderWeele, D.J.; Sosman, J.; Wu, J.D. Past, Current, and Future of Immunotherapies for Prostate Cancer. Front. Oncol. 2019, 9, 884. [Google Scholar] [CrossRef]
- Bou-Dargham, M.J.; Sha, L.; Sang, Q.-X.A.; Zhang, J. Immune landscape of human prostate cancer: Immune evasion mechanisms and biomarkers for personalized immunotherapy. BMC Cancer 2020, 20, 572. [Google Scholar] [CrossRef]
- Nair, S.S.; Weil, R.; Dovey, Z.; Davis, A.; Tewari, A.K. The Tumor Microenvironment and Immunotherapy in Prostate and Bladder Cancer. Urol. Clin. North Am. 2020, 47, e17–e54. [Google Scholar] [CrossRef]
- Xu, P.; Wasielewski, L.J.; Yang, J.C.; Cai, D.; Evans, C.P.; Murphy, W.J.; Liu, C. The Immunotherapy and Immunosuppressive Signaling in Therapy-Resistant Prostate Cancer. Biomedicines 2022, 10, 1778. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics of Patients with Localized PCa (n = 23) | |
|---|---|
| Age at diagnosis (years) | |
| Median | 73 |
| Range | 53–81 |
| PSA (ng/mL) | |
| Mean | 18.74 |
| Standard Deviation | 20.85 |
| Range | 5.51–100.00 |
| Gleason Score | |
| 6 | 4 (17.4%) |
| 7 | 11 (47.8%) |
| 8 | 4 (17.4%) |
| 9 | 4 (17.4%) |
| Mean (range) | 7 (1) |
| T stage | |
| T1c | 3 (13.0%) |
| T2a, T2b, T2c | 12 (52.2%) |
| T3a, T3b | 8 (34.8%) |
| Type of therapy received up to the time of blood sampling | |
| ADT | 17 (73.9%) |
| RP + ADT | 6 (26.1%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fortis, S.P.; Batsaki, P.; Stokidis, S.; Moschandreou, D.; Grouzi, E.; Baxevanis, C.N.; Gritzapis, A.D.; Goulielmaki, M. A Blood-Based Immune Gene Signature with Prognostic Significance in Localized Prostate Cancer. Cancers 2023, 15, 3697. https://doi.org/10.3390/cancers15143697
Fortis SP, Batsaki P, Stokidis S, Moschandreou D, Grouzi E, Baxevanis CN, Gritzapis AD, Goulielmaki M. A Blood-Based Immune Gene Signature with Prognostic Significance in Localized Prostate Cancer. Cancers. 2023; 15(14):3697. https://doi.org/10.3390/cancers15143697
Chicago/Turabian StyleFortis, Sotirios P., Panagiota Batsaki, Savvas Stokidis, Dimitra Moschandreou, Elisavet Grouzi, Constantin N. Baxevanis, Angelos D. Gritzapis, and Maria Goulielmaki. 2023. "A Blood-Based Immune Gene Signature with Prognostic Significance in Localized Prostate Cancer" Cancers 15, no. 14: 3697. https://doi.org/10.3390/cancers15143697
APA StyleFortis, S. P., Batsaki, P., Stokidis, S., Moschandreou, D., Grouzi, E., Baxevanis, C. N., Gritzapis, A. D., & Goulielmaki, M. (2023). A Blood-Based Immune Gene Signature with Prognostic Significance in Localized Prostate Cancer. Cancers, 15(14), 3697. https://doi.org/10.3390/cancers15143697