
CDC20 Is Regulated by the Histone Methyltransferase, KMT5A, in Castration-Resistant Prostate Cancer
 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Compounds
2.3. Cell Culture
2.4. siRNA
2.5. Western Blotting and Quantitative Polymerase Chain Reaction
2.6. Microarray
2.7. RNA-Seq Analysis
2.8. Chromatin Immunoprecipitation Assays
2.9. Sulforhodamine B Growth Analysis
2.10. Gamma H2AX Assay
3. Results
3.1. Identification of KMT5A-Regulated Genes in Androgen-Independent Prostate Cancer
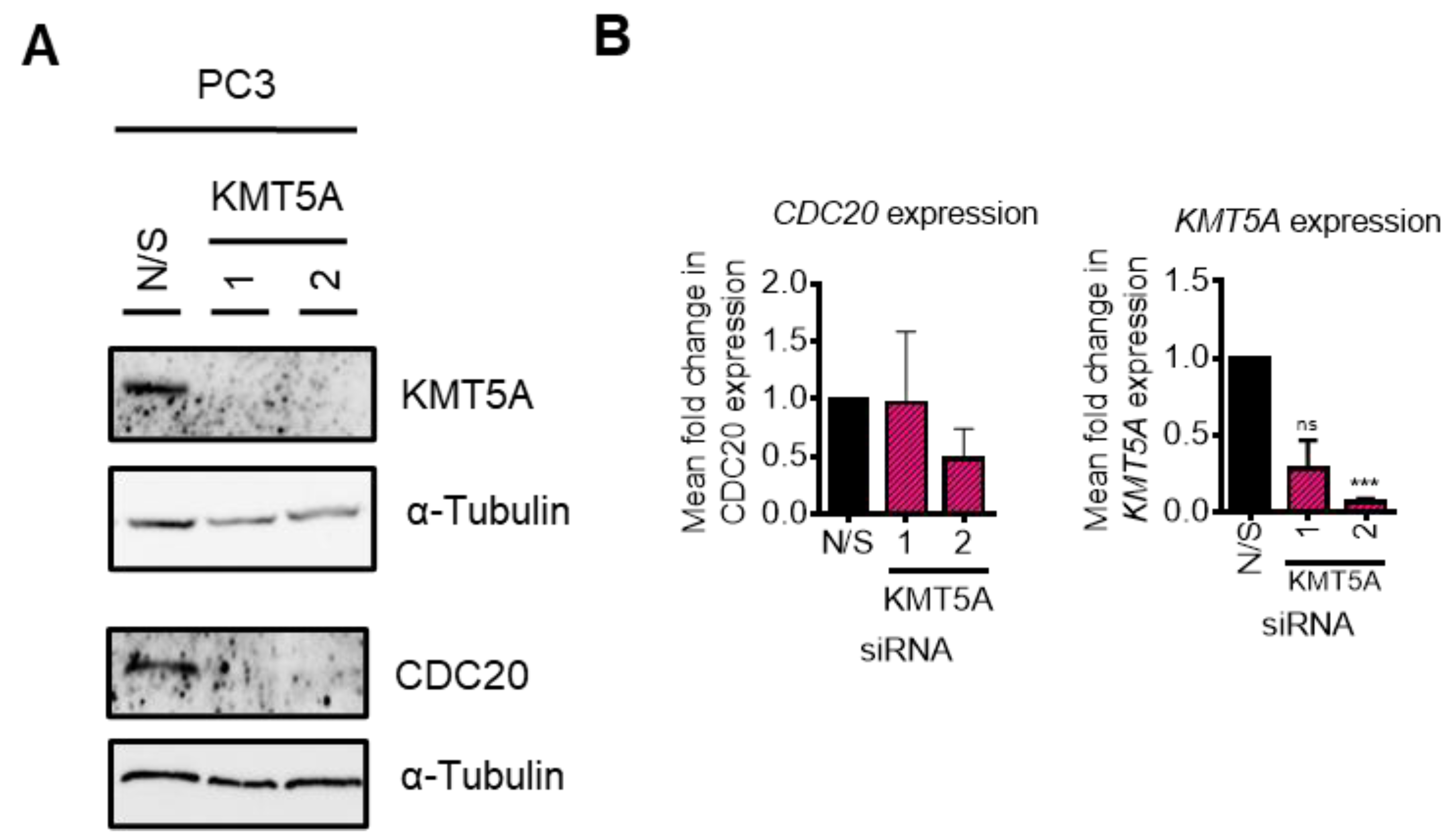
3.2. KMT5A Depletion Reduces CDC20 Expression
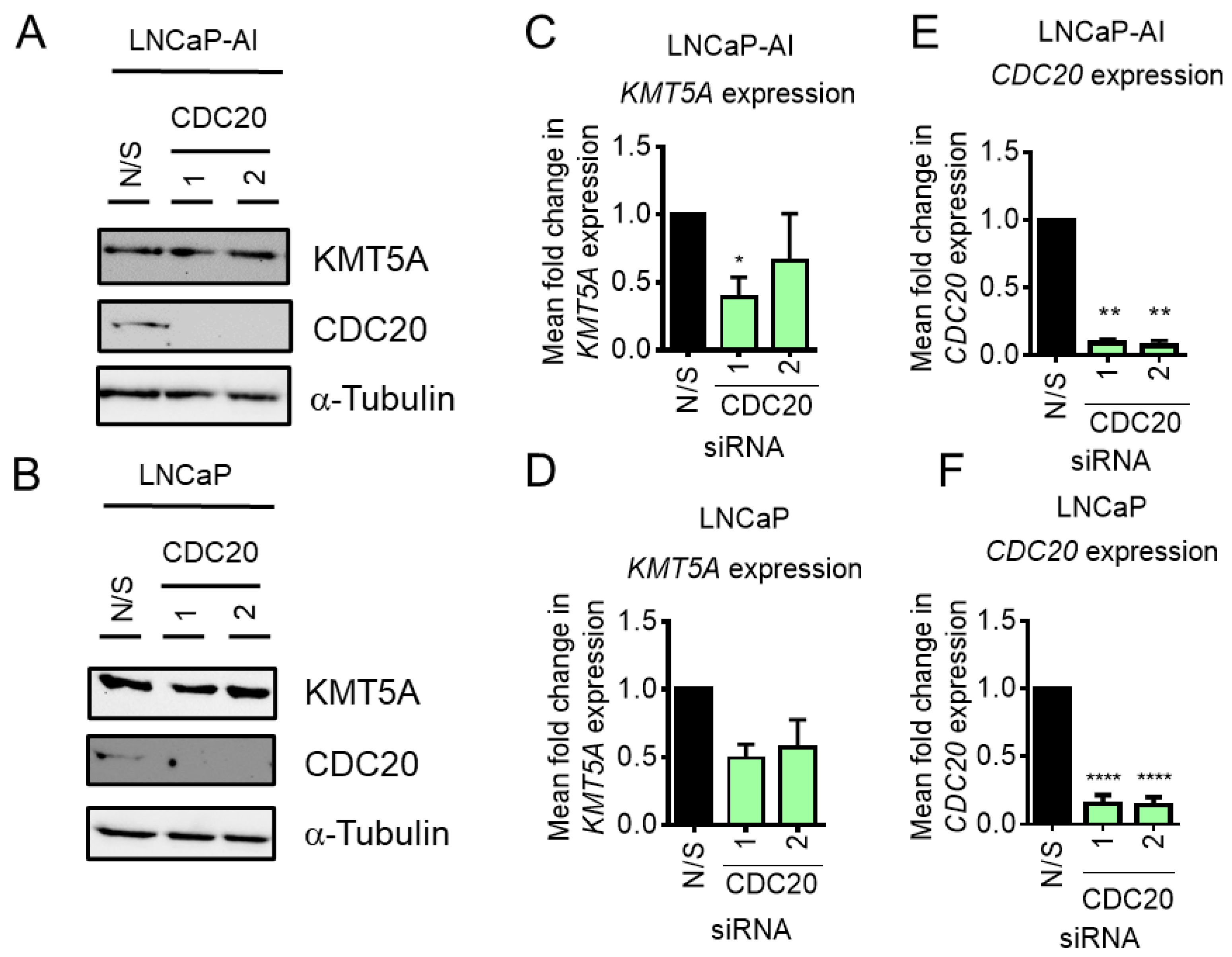
3.3. CDC20 Depletion Does Not Enhance KMT5A Protein Expression
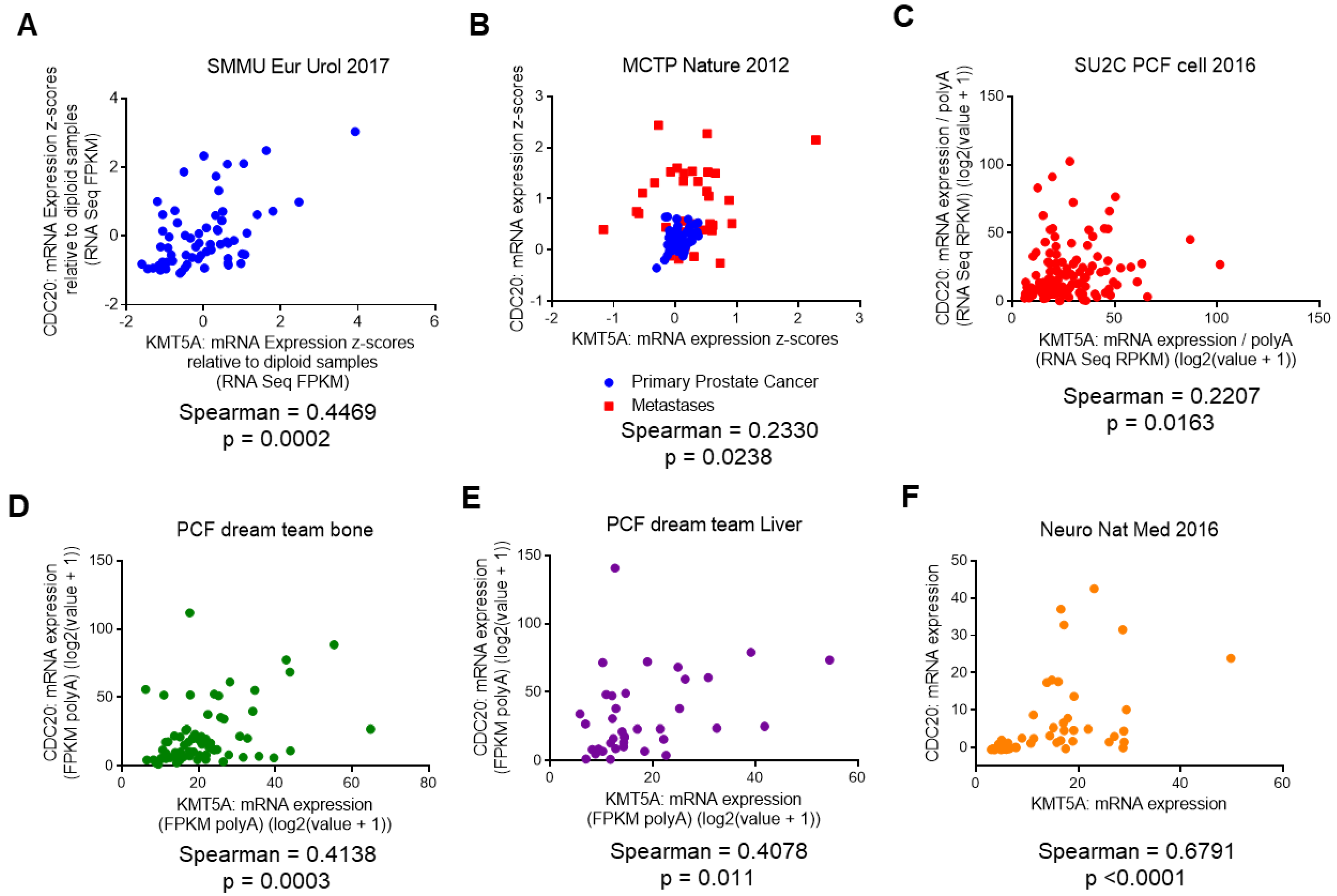
3.4. KMT5A Expression Correlates with CDC20 Expression in Prostate Cancer Patients
3.5. KMT5A Inhibition Reduces CDC20 Expression and Reduces Prostate Cancer Cell Proliferation
3.6. KMT5A Knockdown Reduces H4K20Me1 at the CDC20 Promoter
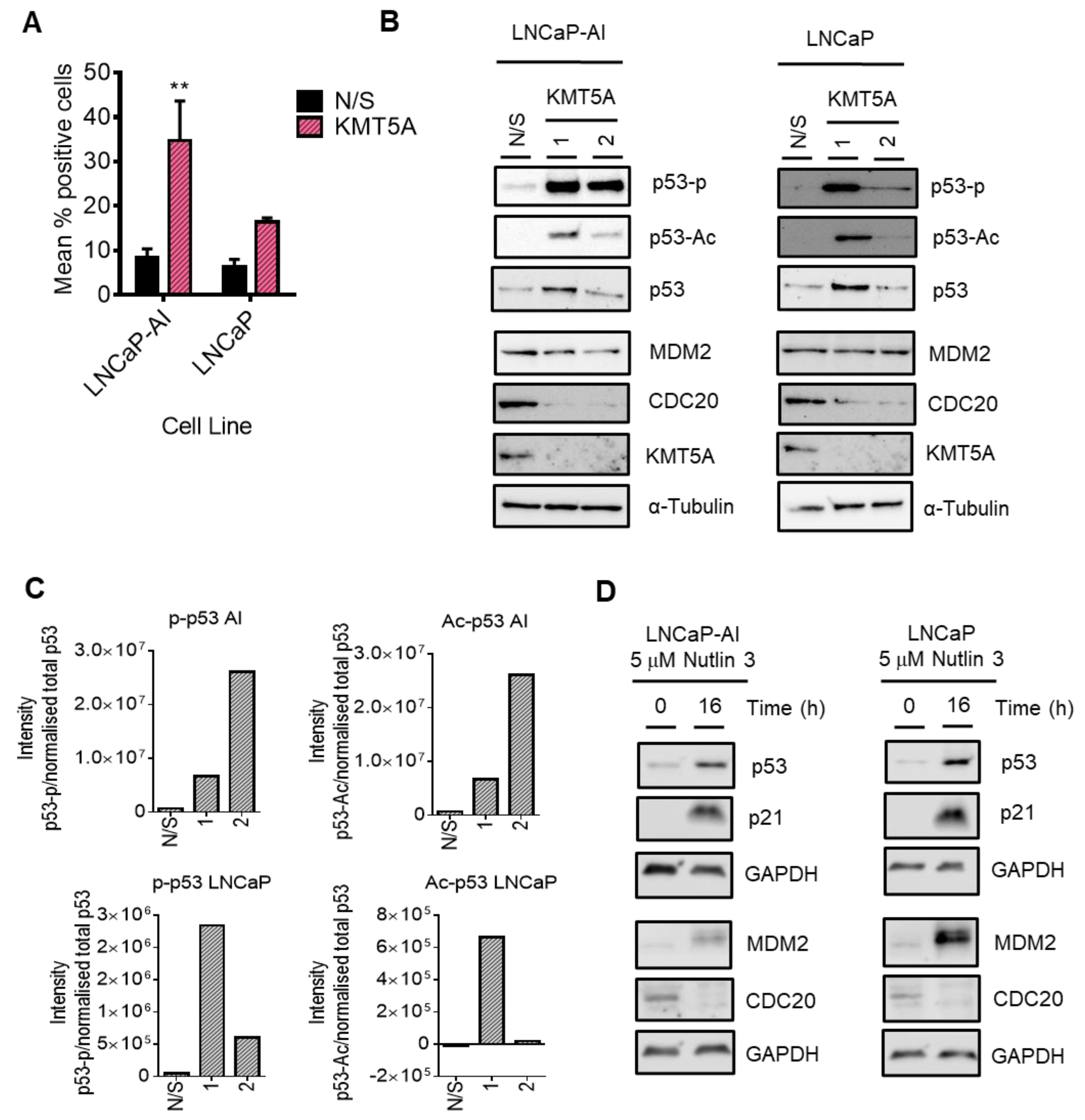
3.7. p53 Mediates KMT5A Regulation of CDC20 Expression
3.8. CDC20 Is Down-Regulated by Protein Turnover in the Absence of p53
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Veschi, V.; Liu, Z.; Voss, T.C.; Ozbun, L.; Gryder, B.; Yan, C.; Hu, Y.; Ma, A.; Jin, J.; Mazur, S.J.; et al. Epigenetic siRNA and Chemical Screens Identify SETD8 Inhibition as a Therapeutic Strategy for p53 Activation in High-Risk Neuroblastoma. Cancer Cell 2017, 31, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Qiao, K.; Du, Y.; Zhang, X.; Cheng, H.; Peng, L.; Guo, Z. Downregulation of histone methyltransferase SET8 inhibits progression of hepatocellular carcinoma. Sci. Rep. 2020, 10, 4490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hou, W.; Chai, M.; Zhao, H.; Jia, J.; Sun, X.; Zhao, B.; Wang, R. MicroRNA-127-3p inhibits proliferation and invasion by targeting SETD8 in human osteosarcoma cells. Biochem. Biophys. Res. Commun. 2016, 469, 1006–1011. [Google Scholar] [CrossRef]
- Yao, L.; Li, Y.; Du, F.; Han, X.; Li, X.; Niu, Y.; Ren, S.; Sun, Y. Histone H4 Lys 20 methyltransferase SET8 promotes androgen receptor-mediated transcription activation in prostate cancer. Biochem. Biophys. Res. Commun. 2014, 450, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Li, Q.; Yu, Y.; Li, M.; Zhang, D. SET8 induces epithelialmesenchymal transition and enhances prostate cancer cell metastasis by cooperating with ZEB1. Mol. Med. Rep. 2016, 13, 1681–1688. [Google Scholar] [CrossRef]
- Shi, X.; Kachirskaia, I.; Yamaguchi, H.; West, L.E.; Wen, H.; Wang, E.W.; Dutta, S.; Appella, E.; Gozani, O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 2007, 27, 636–646. [Google Scholar] [CrossRef]
- Wu, S.; Wang, W.; Kong, X.; Congdon, L.M.; Yokomori, K.; Kirschner, M.W.; Rice, J.C. Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 2010, 24, 2531–2542. [Google Scholar] [CrossRef]
- Wu, S.; Rice, J.C. A new regulator of the cell cycle: The PR-Set7 histone methyltransferase. Cell Cycle 2011, 10, 68–72. [Google Scholar] [CrossRef]
- Cho, H.J.; Lee, E.H.; Han, S.H.; Chung, H.J.; Jeong, J.H.; Kwon, J.; Kim, H. Degradation of human RAP80 is cell cycle regulated by Cdc20 and Cdh1 ubiquitin ligases. Mol. Cancer Res. 2012, 10, 615–625. [Google Scholar] [CrossRef]
- Chun, A.C.; Kok, K.H.; Jin, D.Y. REV7 is required for anaphase-promoting complex-dependent ubiquitination and degradation of translesion DNA polymerase REV1. Cell Cycle 2013, 12, 365–378. [Google Scholar] [CrossRef]
- Banerjee, T.; Nath, S.; Roychoudhury, S. DNA damage induced p53 downregulates Cdc20 by direct binding to its promoter causing chromatin remodeling. Nucleic Acids Res. 2009, 37, 2688–2698. [Google Scholar] [CrossRef] [PubMed]
- Kidokoro, T.; Tanikawa, C.; Furukawa, Y.; Katagiri, T.; Nakamura, Y.; Matsuda, K. CDC20, a potential cancer therapeutic target, is negatively regulated by p53. Oncogene 2008, 27, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.J.; Dimova, N.V.; Tan, M.K.; Sigoillot, F.D.; King, R.W.; Shi, Y. The G2/M regulator histone demethylase PHF8 is targeted for degradation by the anaphase-promoting complex containing CDC20. Mol. Cell Biol. 2013, 33, 4166–4180. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Wan, L.; Zhou, X.; Wang, Z.; Wei, W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol. Ther. 2015, 151, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Li, K.; Lu, L.; Si-Tu, J.; Lu, M.; Gao, X. Overexpression of Cdc20 in clinically localized prostate cancer: Relation to high Gleason score and biochemical recurrence after laparoscopic radical prostatectomy. Cancer Biomark. 2016, 16, 351–358. [Google Scholar] [CrossRef]
- Wu, F.; Dai, X.; Gan, W.; Wan, L.; Li, M.; Mitsiades, N.; Wei, W.; Ding, Q.; Zhang, J. Prostate cancer-associated mutation in SPOP impairs its ability to target Cdc20 for poly-ubiquitination and degradation. Cancer Lett. 2017, 385, 207–214. [Google Scholar] [CrossRef]
- Li, K.; Mao, Y.; Lu, L.; Hu, C.; Wang, D.; Si-Tu, J.; Lu, M.; Peng, S.; Qiu, J.; Gao, X. Silencing of CDC20 suppresses metastatic castration-resistant prostate cancer growth and enhances chemosensitivity to docetaxel. Int. J. Oncol. 2016, 49, 1679–1685. [Google Scholar] [CrossRef]
- Li, J.; Karki, A.; Hodges, K.B.; Ahmad, N.; Zoubeidi, A.; Strebhardt, K.; Ratliff, T.L.; Konieczny, S.F.; Liu, X. Cotargeting Polo-Like Kinase 1 and the Wnt/beta-Catenin Signaling Pathway in Castration-Resistant Prostate Cancer. Mol. Cell Biol. 2015, 35, 4185–4198. [Google Scholar] [CrossRef]
- Coffey, K.; Rogerson, L.; Ryan-Munden, C.; Alkharaif, D.; Stockley, J.; Heer, R.; Sahadevan, K.; O’Neill, D.; Jones, D.; Darby, S.; et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013, 41, 4433–4446. [Google Scholar] [CrossRef]
- Bainbridge, A.; Walker, S.; Smith, J.; Patterson, K.; Dutt, A.; Ng, Y.M.; Thomas, H.D.; Wilson, L.; McCullough, B.; Jones, D.; et al. IKBKE activity enhances AR levels in advanced prostate cancer via modulation of the Hippo pathway. Nucleic Acids Res. 2020, 48, 5366–5382. [Google Scholar] [CrossRef]
- Coffey, K.; Blackburn, T.J.; Cook, S.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Hewitt, L.; Huberman, K.; McNeill, H.V.; Newell, D.R.; et al. Characterisation of a Tip60 specific inhibitor, NU9056, in prostate cancer. PLoS ONE 2012, 7, e45539. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple hypothesis testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Labaf, M.; Li, M.; Ting, L.; Karno, B.; Zhang, S.; Gao, S.; Patalano, S.; Macoska, J.A.; Zarringhalam, K.; Han, D.; et al. Increased AR expression in castration-resistant prostate cancer rapidly induces AR signaling reprogramming with the collaboration of EZH2. Front. Oncol. 2022, 12, 1021845. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Schmidt, D.; Wilson, M.D.; Spyrou, C.; Brown, G.D.; Hadfield, J.; Odom, D.T. ChIP-seq: Using high-throughput sequencing to discover protein-DNA interactions. Methods 2009, 48, 240–248. [Google Scholar] [CrossRef]
- Xie, Q.; Wu, Q.; Mack, S.C.; Yang, K.; Kim, L.; Hubert, C.G.; Flavahan, W.A.; Chu, C.; Bao, S.; Rich, J.N. CDC20 maintains tumor initiating cells. Oncotarget 2015, 6, 13241–13254. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Wei, G.H.; Liu, D.; Wang, L.; Hou, Y.; Zhu, S.; Peng, L.; Zhang, Q.; Cheng, Y.; Su, H.; et al. Whole-genome and Transcriptome Sequencing of Prostate Cancer Identify New Genetic Alterations Driving Disease Progression. Eur. Urol. 2018, 73, 322–339. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Jorgensen, S.; Elvers, I.; Trelle, M.B.; Menzel, T.; Eskildsen, M.; Jensen, O.N.; Helleday, T.; Helin, K.; Sorensen, C.S. The histone methyltransferase SET8 is required for S-phase progression. J. Cell Biol. 2007, 179, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Tardat, M.; Murr, R.; Herceg, Z.; Sardet, C.; Julien, E. PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J. Cell Biol. 2007, 179, 1413–1426. [Google Scholar] [CrossRef]
- Blum, G.; Ibanez, G.; Rao, X.; Shum, D.; Radu, C.; Djaballah, H.; Rice, J.C.; Luo, M. Small-molecule inhibitors of SETD8 with cellular activity. ACS Chem. Biol. 2014, 9, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.; Yu, W.; Xiong, Y.; Butler, K.V.; Brown, P.J.; Jin, J. Structure-activity relationship studies of SETD8 inhibitors. Medchemcomm 2014, 5, 1892–1898. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Chowdhury, A.; Dey, S.; Roychoudhury, A.; Ganguly, A.; Bhattacharyya, D.; Roychoudhury, S. Deregulation of Rb-E2F1 axis causes chromosomal instability by engaging the transactivation function of Cdc20-anaphase-promoting complex/cyclosome. Mol. Cell Biol. 2015, 35, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Christensen, J.; Rappsilber, J.; Nielsen, A.L.; Johansen, J.V.; Helin, K. The histone lysine demethylase JMJD3/KDM6B is recruited to p53 bound promoters and enhancer elements in a p53 dependent manner. PLoS ONE 2014, 9, e96545. [Google Scholar] [CrossRef] [PubMed]
- Trojer, P.; Li, G.; Sims, R.J., 3rd; Vaquero, A.; Kalakonda, N.; Boccuni, P.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Nimer, S.D.; et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 2007, 129, 915–928. [Google Scholar] [CrossRef]
- Shoaib, M.; Walter, D.; Gillespie, P.J.; Izard, F.; Fahrenkrog, B.; Lleres, D.; Lerdrup, M.; Johansen, J.V.; Hansen, K.; Julien, E.; et al. Histone H4K20 methylation mediated chromatin compaction threshold ensures genome integrity by limiting DNA replication licensing. Nat. Commun. 2018, 9, 3704. [Google Scholar] [CrossRef]
- Oda, H.; Hubner, M.R.; Beck, D.B.; Vermeulen, M.; Hurwitz, J.; Spector, D.L.; Reinberg, D. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol. Cell 2010, 40, 364–376. [Google Scholar] [CrossRef]
- Nishioka, K.; Rice, J.C.; Sarma, K.; Erdjument-Bromage, H.; Werner, J.; Wang, Y.; Chuikov, S.; Valenzuela, P.; Tempst, P.; Steward, R.; et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 2002, 9, 1201–1213. [Google Scholar] [CrossRef]
- Yang, F.; Sun, L.; Li, Q.; Han, X.; Lei, L.; Zhang, H.; Shang, Y. SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 2012, 31, 110–123. [Google Scholar] [CrossRef]
- Li, Z.; Nie, F.; Wang, S.; Li, L. Histone H4 Lys 20 monomethylation by histone methylase SET8 mediates Wnt target gene activation. Proc. Natl. Acad. Sci. USA 2011, 108, 3116–3123. [Google Scholar] [CrossRef]
- Vakoc, C.R.; Sachdeva, M.M.; Wang, H.; Blobel, G.A. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol. Cell Biol. 2006, 26, 9185–9195. [Google Scholar] [CrossRef]
- Shoaib, M.; Chen, Q.; Shi, X.; Nair, N.; Prasanna, C.; Yang, R.; Walter, D.; Frederiksen, K.S.; Einarsson, H.; Svensson, J.P.; et al. Histone H4 lysine 20 mono-methylation directly facilitates chromatin openness and promotes transcription of housekeeping genes. Nat. Commun. 2021, 12, 4800. [Google Scholar] [CrossRef] [PubMed]
- Kapoor-Vazirani, P.; Vertino, P.M. A dual role for the histone methyltransferase PR-SET7/SETD8 and histone H4 lysine 20 monomethylation in the local regulation of RNA polymerase II pausing. J. Biol. Chem. 2014, 289, 7425–7437. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Shang, W.H.; Toyoda, A.; Misu, S.; Monma, N.; Ikeo, K.; Molina, O.; Vargiu, G.; Fujiyama, A.; Kimura, H.; et al. Histone H4 Lys 20 monomethylation of the CENP-A nucleosome is essential for kinetochore assembly. Dev. Cell 2014, 29, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Sun, Y.; Chen, J.; Li, H.; Yao, K.; Liu, Y.; Liu, Q.; Lu, J. The Oncogenic Role of APC/C Activator Protein Cdc20 by an Integrated Pan-Cancer Analysis in Human Tumors. Front. Oncol. 2021, 11, 721797. [Google Scholar] [CrossRef]
- Liang, X.; Hu, K.; Li, D.; Wang, Y.; Liu, M.; Wang, X.; Zhu, W.; Wang, X.; Yang, Z.; Lu, J. Identification of Core Genes and Potential Drugs for Castration-Resistant Prostate Cancer Based on Bioinformatics Analysis. DNA Cell Biol. 2020, 39, 836–847. [Google Scholar] [CrossRef]
- Dai, L.; Song, Z.X.; Wei, D.P.; Zhang, J.D.; Liang, J.Q.; Wang, B.B.; Ma, W.T.; Li, L.Y.; Dang, Y.L.; Zhao, L.; et al. CDC20 and PTTG1 are Important Biomarkers and Potential Therapeutic Targets for Metastatic Prostate Cancer. Adv. Ther. 2021, 38, 2973–2989. [Google Scholar] [CrossRef]
- Gu, P.; Yang, D.; Zhu, J.; Zhang, M.; He, X. Bioinformatics analysis identified hub genes in prostate cancer tumorigenesis and metastasis. Math. Biosci. Eng. 2021, 18, 3180–3196. [Google Scholar] [CrossRef]
- Wei, J.; Yin, Y.; Deng, Q.; Zhou, J.; Wang, Y.; Yin, G.; Yang, J.; Tang, Y. Integrative Analysis of MicroRNA and Gene Interactions for Revealing Candidate Signatures in Prostate Cancer. Front. Genet. 2020, 11, 176. [Google Scholar] [CrossRef]
- Luo, C.; Chen, J.; Chen, L. Exploration of gene expression profiles and immune microenvironment between high and low tumor mutation burden groups in prostate cancer. Int. Immunopharmacol. 2020, 86, 106709. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Shi, Q.; Zhu, Y.; Ma, J.; Chang, K.; Ding, D.; Bai, Y.; Gao, K.; Zhang, P.; Mo, R.; Feng, K.; et al. Prostate Cancer-associated SPOP mutations enhance cancer cell survival and docetaxel resistance by upregulating Caprin1-dependent stress granule assembly. Mol. Cancer 2019, 18, 170. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Lin, Y.; Cui, P.; Li, H.; Zhang, L.; Sun, Z.; Huang, S.; Li, S.; Huang, S.; Zhao, Q.; et al. Cdc20/p55 mediates the resistance to docetaxel in castration-resistant prostate cancer in a Bim-dependent manner. Cancer Chemother. Pharmacol. 2018, 81, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Tuzon, C.T.; Spektor, T.; Kong, X.; Congdon, L.M.; Wu, S.; Schotta, G.; Yokomori, K.; Rice, J.C. Concerted activities of distinct H4K20 methyltransferases at DNA double-strand breaks regulate 53BP1 nucleation and NHEJ-directed repair. Cell Rep. 2014, 8, 430–438. [Google Scholar] [CrossRef]
- Dulev, S.; Tkach, J.; Lin, S.; Batada, N.N. SET8 methyltransferase activity during the DNA double-strand break response is required for recruitment of 53BP1. EMBO Rep. 2014, 15, 1163–1174. [Google Scholar] [CrossRef]
- Lu, X.; Xu, M.; Zhu, Q.; Zhang, J.; Liu, G.; Bao, Y.; Gu, L.; Tian, Y.; Wen, H.; Zhu, W.G. RNF8-ubiquitinated KMT5A is required for RNF168-induced H2A ubiquitination in response to DNA damage. FASEB J. 2021, 35, e21326. [Google Scholar] [CrossRef]
- Jimenez-Vacas, J.M.; Herrero-Aguayo, V.; Montero-Hidalgo, A.J.; Gomez-Gomez, E.; Fuentes-Fayos, A.C.; Leon-Gonzalez, A.J.; Saez-Martinez, P.; Alors-Perez, E.; Pedraza-Arevalo, S.; Gonzalez-Serrano, T.; et al. Dysregulation of the splicing machinery is directly associated to aggressiveness of prostate cancer. EBioMedicine 2020, 51, 102547. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alebady, Z.A.H.; Azizyan, M.; Nakjang, S.; Lishman-Walker, E.; Al-Kharaif, D.; Walker, S.; Choo, H.X.; Garnham, R.; Scott, E.; Johnson, K.L.; et al. CDC20 Is Regulated by the Histone Methyltransferase, KMT5A, in Castration-Resistant Prostate Cancer. Cancers 2023, 15, 3597. https://doi.org/10.3390/cancers15143597
Alebady ZAH, Azizyan M, Nakjang S, Lishman-Walker E, Al-Kharaif D, Walker S, Choo HX, Garnham R, Scott E, Johnson KL, et al. CDC20 Is Regulated by the Histone Methyltransferase, KMT5A, in Castration-Resistant Prostate Cancer. Cancers. 2023; 15(14):3597. https://doi.org/10.3390/cancers15143597
Chicago/Turabian StyleAlebady, Zainab A. H., Mahsa Azizyan, Sirintra Nakjang, Emma Lishman-Walker, Dhuha Al-Kharaif, Scott Walker, Hui Xian Choo, Rebecca Garnham, Emma Scott, Katya L. Johnson, and et al. 2023. "CDC20 Is Regulated by the Histone Methyltransferase, KMT5A, in Castration-Resistant Prostate Cancer" Cancers 15, no. 14: 3597. https://doi.org/10.3390/cancers15143597
APA StyleAlebady, Z. A. H., Azizyan, M., Nakjang, S., Lishman-Walker, E., Al-Kharaif, D., Walker, S., Choo, H. X., Garnham, R., Scott, E., Johnson, K. L., Robson, C. N., & Coffey, K. (2023). CDC20 Is Regulated by the Histone Methyltransferase, KMT5A, in Castration-Resistant Prostate Cancer. Cancers, 15(14), 3597. https://doi.org/10.3390/cancers15143597