
The Development of p53-Targeted Therapies for Human Cancers

Abstract
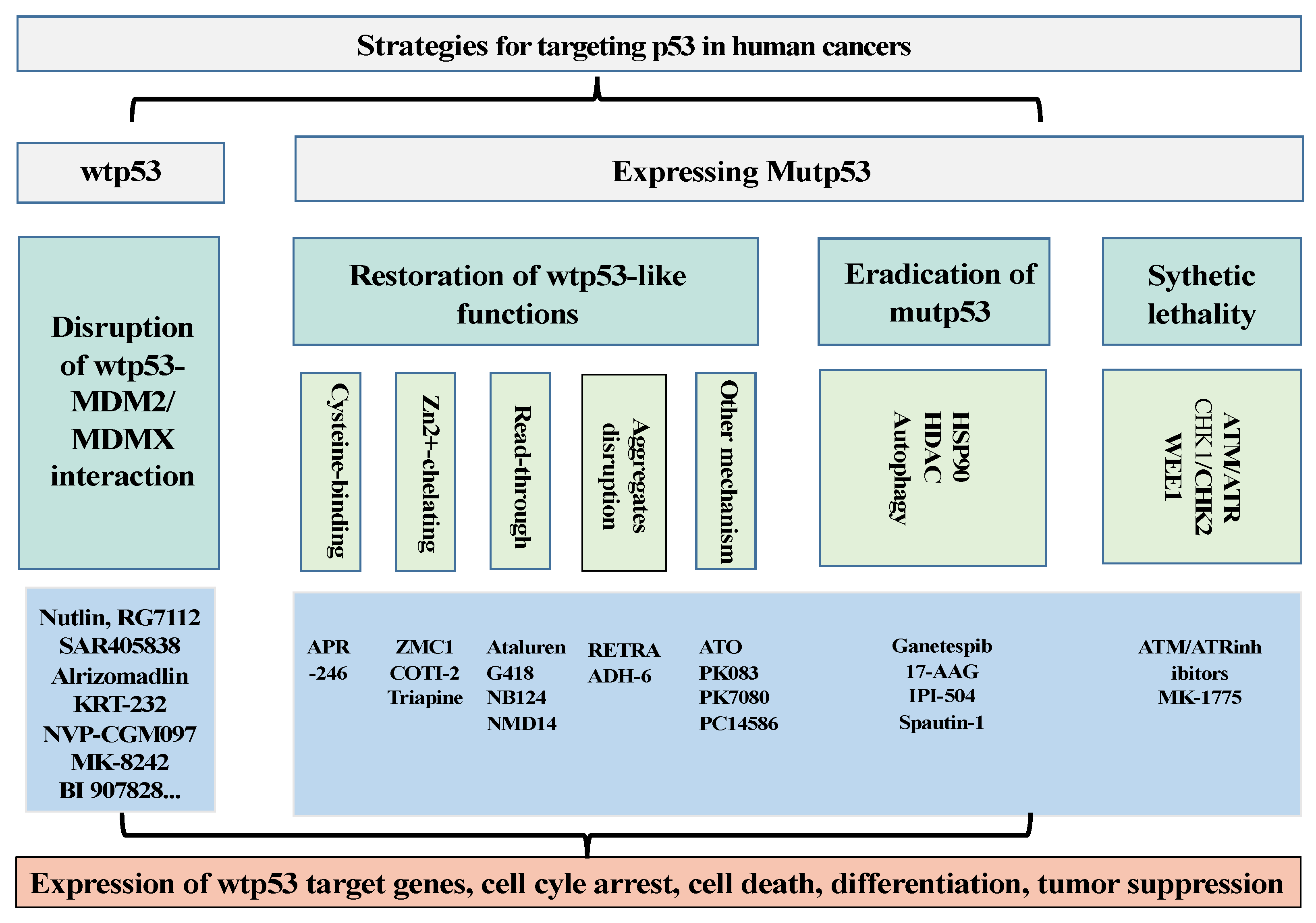
Simple Summary
Abstract
1. Introduction
2. p53 Targeting Therapies
2.1. Development of Drugs to Restore the Activities of wtp53 by Targeting p53–MDM2/MDMX Interaction
2.2. Development of Drugs to Target p53 Mutants (Mutp53)
2.2.1. Development of Drugs to Restore wtp53-Dependent Activities
Cysteine-Binding Compounds
Zn2+-Chelating Compounds
p53 Nonsense Mutation Read-Through Drugs
Dispersion of p53 Aggregates
Other Compounds
2.2.2. To Develop Strategies to Eradicate Mutp53
2.3. Drugs Induce Synthetic Lethality with mutp53
2.4. Cancer Immunotherapy for Mutp53
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Chene, P. The role of tetramerization in p53 function. Oncogene 2001, 20, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef]
- Vikhanskaya, F.; Lee, M.K.; Mazzoletti, M.; Broggini, M.; Sabapathy, K. Cancer-derived p53 mutants suppress p53-target gene expression—Potential mechanism for gain of function of mutant p53. Nucleic Acids Res. 2007, 35, 2093–2104. [Google Scholar] [CrossRef]
- Espadinha, M.; Barcherini, V.; Lopes, E.A.; Santos, M.M.M. An Update on MDMX and Dual MDM2/X Inhibitors. Curr. Top. Med. Chem. 2018, 18, 647–660. [Google Scholar] [CrossRef]
- Marine, J.C.; Lozano, G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010, 17, 93–102. [Google Scholar] [CrossRef]
- Chen, J.D.; Marechal, V.; Levine, A.J. Mapping of the p53 and mdm-2 interaction domains. Mol. Cell. Biol. 1993, 13, 4107–4114. [Google Scholar]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gvuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumor suppressor-p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef]
- Lu, W.G.; Pochampally, R.; Chen, L.H.; Traidej, M.; Wang, Y.L.; Chen, J.D. Nuclear exclusion of p53 in a subset of tumors requires MDM2 function. Oncogene 2000, 19, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Chen, J.D. MDM2 promotes ubiquitination and degradation of MDMX. Mol. Cell. Biol. 2003, 23, 5113–5121. [Google Scholar] [CrossRef]
- Sharp, D.A.; Kratowicz, S.A.; Sank, M.J.; George, D.L. Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J. Biol. Chem. 1999, 274, 38189–38196. [Google Scholar] [CrossRef] [PubMed]
- Stad, R.; Little, N.A.; Xirodimas, D.P.; Frenk, R.; van der Eb, A.J.; Lane, D.P.; Saville, M.K.; Jochemsen, A.G. Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep. 2001, 2, 1029–1034. [Google Scholar] [CrossRef]
- Shinohara, T.; Uesugi, M. In-vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Tanpakushitsu Kakusan Koso. Protein Nucleic Acid Enzym. 2007, 52 (Suppl. S13), 1816–1817. [Google Scholar]
- Van Maerken, T.; Speleman, F.; Vermeulen, J.; Lambertz, I.; De Clercq, S.; De Smet, E.; Yigit, N.; Coppens, V.; Philippe, J.; De Paepe, A.; et al. Small-molecule MDM2 antagonists as a new therapy concept for neuroblastoma. Cancer Res. 2006, 66, 9646–9655. [Google Scholar] [CrossRef]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef]
- Montesinos, P.; Beckermann, B.M.; Catalani, O.; Esteve, J.; Gamel, K.; Konopleva, M.Y.; Martinelli, G.; Monnet, A.; Papayannidis, C.; Park, A.; et al. MIRROS: A randomized, placebo-controlled, Phase III trial of cytarabine ± idasanutlin in relapsed or refractory acute myeloid leukemia. Future Oncol. 2020, 16, 807–815. [Google Scholar] [CrossRef]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barriere, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef]
- Arnhold, V.; Schmelz, K.; Proba, J.; Winkler, A.; Wunschel, J.; Toedling, J.; Deubzer, H.E.; Kunkele, A.; Eggert, A.; Schulte, J.H.; et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget 2018, 9, 2304–2319. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.M.; Bauer, T.M.; Schwartz, G.K.; LoRusso, P.; Kumar, P.; Kato, K.; Tao, B.; Hong, Y.; Patel, P.; Hong, D. Milademetan, an oral MDM2 inhibitor, in well-differentiated/dedifferentiated liposarcoma: Results from a phase 1 study in patients with solid tumors or lymphomas. Eur. J. Cancer 2020, 138, S3–S4. [Google Scholar] [CrossRef]
- Dumbrava, E.E.; Hanna, G.J.; Cote, G.M.; Stinchcombe, T.; Johnson, M.L.; Chen, C.; Devarakonda, S.; Shah, N.; Xu, F.; Doebele, R.C.; et al. A phase 2 study of the MDM2 inhibitor milademetan in patients with TP53-wild type and MDM2-amplified advanced or metastatic solid tumors (MANTRA-2). J. Clin. Oncol. 2022, 40, 16. [Google Scholar] [CrossRef]
- McKean, M.; Tolcher, A.W.; Reeves, J.A.; Chmielowski, B.; Shaheen, M.F.; Beck, J.T.; Orloff, M.M.; Somaiah, N.; Van Tine, B.A.; Drabick, J.J.; et al. Newly updated activity results of alrizomadlin (APG-115), a novel MDM2/p53 inhibitor, plus pembrolizumab: Phase 2 study in adults and children with various solid tumors. J. Clin. Oncol. 2022, 40, 9517. [Google Scholar] [CrossRef]
- Wong, M.K.K.; Burgess, M.A.; Chandra, S.; Schadendorf, D.; Silk, A.W.; Olszanski, A.J.; Grob, J.-J.; Jang, S.; Grewal, J.S.; Lewis, K.D.; et al. Navtemadlin (KRT-232) activity after failure of anti-PD-1/L1 therapy in patients (pts) with TP53WT Merkel cell carcinoma (MCC). J. Clin. Oncol. 2022, 40, 9506. [Google Scholar] [CrossRef]
- Bauer, S.; Demetri, G.D.; Halilovic, E.; Dummer, R.; Meille, C.; Tan, D.S.W.; Guerreiro, N.; Jullion, A.; Ferretti, S.; Jeay, S.; et al. Pharmacokinetic-pharmacodynamic guided optimisation of dose and schedule of CGM097, an HDM2 inhibitor, in preclinical and clinical studies. Br. J. Cancer 2021, 125, 687–698. [Google Scholar] [CrossRef]
- Kang, M.H.; Reynolds, C.P.; Kolb, E.A.; Gorlick, R.; Carol, H.; Lock, R.; Keir, S.T.; Maris, J.M.; Wu, J.R.; Lyalin, D.; et al. Initial Testing (Stage 1) of MK-8242A Novel MDM2 Inhibitorby the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2016, 63, 1744–1752. [Google Scholar] [CrossRef]
- Ravandi, F.; Gojo, I.; Patnaik, M.M.; Minden, M.D.; Kantarjian, H.; Johnson-Levonas, A.O.; Fancourt, C.; Lam, R.; Jones, M.B.; Knox, C.D.; et al. A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk. Res. 2016, 48, 92–100. [Google Scholar] [CrossRef]
- Wagner, A.J.; Banerji, U.; Mahipal, A.; Somaiah, N.; Hirsch, H.; Fancourt, C.; Johnson-Levonas, A.O.; Lam, R.; Meister, A.K.; Russo, G.; et al. Phase I Trial of the Human Double Minute 2 Inhibitor MK-8242 in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2017, 35, 1304–1311. [Google Scholar] [CrossRef]
- Gounder, M.M.; Yamamoto, N.; Patel, M.R.; Bauer, T.M.; Schoffski, P.; Grempler, R.; Durland-Busbice, S.; Geng, J.; Maerten, A.; LoRusso, P. A phase Ia/Ib, dose-escalation/expansion study of the MDM2-p53 antagonist BI 907828 in patients with solid tumors, including advanced/metastatic liposarcoma (LPS). J. Clin. Oncol. 2022, 40, 3004. [Google Scholar] [CrossRef]
- Chapeau, E.A.; Gembarska, A.; Durand, E.Y.; Mandon, E.; Estadieu, C.; Romanet, V.; Wiesmann, M.; Tiedt, R.; Lehar, J.; de Weck, A.; et al. Resistance mechanisms to TP53-MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an Arf−/− mouse model. Proc. Natl. Acad. Sci. USA 2017, 114, 3151–3156. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DeAngelo, D.J.; Chromik, J.; Chatterjee, M.; Bauer, S.; Lin, C.C.; Suarez, C.; de Vos, F.; Steeghs, N.; Cassier, P.A.; et al. Results from a First-in-Human Phase I Study of Siremadlin (HDM201) in Patients with Advanced Wild-Type TP53 Solid Tumors and Acute Leukemia. Clin. Cancer Res. 2022, 28, 870–881. [Google Scholar] [CrossRef]
- Abdul Razak, A.R.; Bauer, S.; Suarez, C.; Lin, C.C.; Quek, R.; Hutter-Kronke, M.L.; Cubedo, R.; Ferretti, S.; Guerreiro, N.; Jullion, A.; et al. Co-Targeting of MDM2 and CDK4/6 with Siremadlin and Ribociclib for the Treatment of Patients with Well-Differentiated or Dedifferentiated Liposarcoma: Results from a Proof-of-Concept, Phase Ib Study. Clin. Cancer Res. 2022, 28, 1087–1097. [Google Scholar] [CrossRef]
- Marine, J.C. MDM2 and MDMX in cancer and development. Curr. Top. Dev. Biol. 2011, 94, 45–75. [Google Scholar] [PubMed]
- Pairawan, S.; Zhao, M.; Yuca, E.; Annis, A.; Evans, K.; Sutton, D.; Carvajal, L.; Ren, J.G.; Santiago, S.; Guerlavais, V.; et al. First in class dual MDM2/MDMX inhibitor ALRN-6924 enhances antitumor efficacy of chemotherapy in TP53 wild-type hormone receptor-positive breast cancer models. Breast Cancer Res. 2021, 23, 29. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Quah, S.T.; Jong, J.; Goh, A.M.; Chiam, P.C.; Khoo, K.H.; Choong, M.L.; Lee, M.A.; Yurlova, L.; Zolghadr, K.; et al. Stapled Peptides with Improved Potency and Specificity That Activate p53. ACS Chem. Biol. 2013, 8, 506–512. [Google Scholar] [CrossRef]
- Yuen, T.Y.; Brown, C.J.; Xue, Y.Z.; Tan, Y.S.; Gago, F.J.F.; Lee, X.E.; Neo, J.Y.; Thean, D.; Kaan, H.Y.K.; Partridge, A.W.; et al. Stereoisomerism of stapled peptide inhibitors of the p53-Mdm2 interaction: An assessment of synthetic strategies and activity profiles. Chem. Sci. 2019, 10, 6457–6466. [Google Scholar] [CrossRef]
- Spiegelberg, D.; Mortensen, A.C.; Lundsten, S.; Brown, C.J.; Lane, D.P.; Nestor, M. The MDM2/MDMX-p53 Antagonist PM2 Radiosensitizes Wild-Type p53 Tumors. Cancer Res. 2018, 78, 5084–5093. [Google Scholar] [CrossRef]
- Wei, S.J.; Chee, S.; Yurlova, L.; Lane, D.; Verma, C.; Brown, C.; Ghadessy, F. Avoiding drug resistance through extended drug target interfaces: A case for stapled peptides. Oncotarget 2016, 7, 32232–32246. [Google Scholar] [CrossRef]
- Lundsten, S.; Hernandez, V.A.; Gedda, L.; Saren, T.; Brown, C.J.; Lane, D.P.; Edwards, K.; Nestor, M. Tumor-Targeted Delivery of the p53-Activating Peptide VIP116 with PEG-Stabilized Lipodisks. Nanomaterials 2020, 10, 783. [Google Scholar] [CrossRef]
- Graves, B.; Thompson, T.; Xia, M.; Janson, C.; Lukacs, C.; Deo, D.; Di Lello, P.; Fry, D.; Garvie, C.; Huang, K.S.; et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc. Natl. Acad. Sci. USA 2012, 109, 11788–11793. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, X.; Yuan, W.; Zou, Y.; Guo, Z.; Chai, Y.; Lu, W. Rapid identification of dual p53-MDM2/MDMX interaction inhibitors through virtual screening and hit-based substructure search. RSC Adv. 2017, 7, 9989–9997. [Google Scholar] [CrossRef]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Read, M.; Cullinane, C.; Azar, W.J.; Fennell, C.M.; Montgomery, K.G.; Haupt, S.; Haupt, Y.; Wiman, K.G.; Duong, C.P.; et al. APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut 2015, 64, 1506–1516. [Google Scholar] [CrossRef]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef]
- Synnott, N.C.; Murray, A.; McGowan, P.M.; Kiely, M.; Kiely, P.A.; O’Donovan, N.; O’Connor, D.P.; Gallagher, W.M.; Crown, J.; Duffy, M.J. Mutant p53: A novel target for the treatment of patients with triple-negative breast cancer? Int. J. Cancer 2017, 140, 234–246. [Google Scholar] [CrossRef]
- Zache, N.; Lambert, J.M.R.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1(MET) inhibits growth of mouse tumors carrying mutant p53. Anal. Cell. Oncol. 2008, 30, 411–418. [Google Scholar]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Rangel, L.P.; Ferretti, G.D.S.; Costa, C.L.; Andrade, S.; Carvalho, R.S.; Costa, D.C.F.; Silva, J.L. p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J. Biol. Chem. 2019, 294, 3670–3682. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.Y.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhang, M.Q.Z.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.N.; Arner, E.S.J.; Wiman, K.G. APR-246/PRIMA-1(MET) inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef] [PubMed]
- Rokaeus, N.; Shen, J.; Eckhardt, I.; Bykov, V.J.; Wiman, K.G.; Wilhelm, M.T. PRIMA-1(MET)/APR-246 targets mutant forms of p53 family members p63 and p73. Oncogene 2010, 29, 6442–6451. [Google Scholar] [CrossRef] [PubMed]
- Shalom-Feuerstein, R.; Serror, L.; Aberdam, E.; Muller, F.J.; van Bokhoven, H.; Wiman, K.G.; Zhou, H.; Aberdam, D.; Petit, I. Impaired epithelial differentiation of induced pluripotent stem cells from ectodermal dysplasia-related patients is rescued by the small compound APR-246/PRIMA-1MET. Proc. Natl. Acad. Sci. USA 2013, 110, 2152–2156. [Google Scholar] [CrossRef]
- Shen, J.; van den Bogaard, E.H.; Kouwenhoven, E.N.; Bykov, V.J.; Rinne, T.; Zhang, Q.; Tjabringa, G.S.; Gilissen, C.; van Heeringen, S.J.; Schalkwijk, J.; et al. APR-246/PRIMA-1(MET) rescues epidermal differentiation in skin keratinocytes derived from EEC syndrome patients with p63 mutations. Proc. Natl. Acad. Sci. USA 2013, 110, 2157–2162. [Google Scholar] [CrossRef]
- Gourley, C.; Green, J.; Gabra, H.; Vergote, I.; Basu, B.; Brenton, J.D.; Bjorklund, U.; Smith, A.M.; Von Euler, M. PISARRO: A EUTROC phase Ib study of APR-246 in combination with carboplatin (C) and pegylated liposomal doxorubicin (PLD) in platinum sensitive relapsed high grade serous ovarian cancer (HGSOC). J. Clin. Oncol. 2016, 34, 5571. [Google Scholar] [CrossRef]
- Sallman, D.A.; Komrokji, R.S.; De Zern, A.E.; Sebert, M.; Garcia-Manero, G.; Rahme, R.; Steensma, D.P.; Che, J.L.; Roboz, G.J.; Madelaine, I.; et al. Long Term Follow-up and Combined Phase 2 Results of Eprenetapopt (APR-246) and Azacitidine (AZA) in Patients with TP53 mutant Myelodysplastic Syndromes (MDS) and Oligoblastic Acute Myeloid Leukemia (AML). Blood 2021, 138, 246. [Google Scholar] [CrossRef]
- Loh, S.N. The missing zinc: p53 misfolding and cancer. Metallomics 2010, 2, 442–449. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Yu, X.; Blanden, A.R.; Narayanan, S.; Jayakumar, L.; Lubin, D.; Augeri, D.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Small molecule restoration of wildtype structure and function of mutant p53 using a novel zinc-metallochaperone based mechanism. Oncotarget 2014, 5, 8879–8892. [Google Scholar] [CrossRef]
- Blanden, A.R.; Yu, X.; Wolfe, A.J.; Gilleran, J.A.; Augeri, D.J.; O’Dell, R.S.; Olson, E.C.; Kimball, S.D.; Emge, T.J.; Movileanu, L.; et al. Synthetic metallochaperone ZMC1 rescues mutant p53 conformation by transporting zinc into cells as an ionophore. Mol. Pharmacol. 2015, 87, 825–831. [Google Scholar] [CrossRef]
- Blanden, A.R.; Yu, X.; Loh, S.N.; Levine, A.J.; Carpizo, D.R. Reactivating mutant p53 using small molecules as zinc metallochaperones: Awakening a sleeping giant in cancer. Drug Discov. Today 2015, 20, 1391–1397. [Google Scholar] [CrossRef]
- Yu, X.; Blanden, A.; Tsang, A.T.; Zaman, S.; Liu, Y.; Gilleran, J.; Bencivenga, A.F.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Thiosemicarbazones Functioning as Zinc Metallochaperones to Reactivate Mutant p53. Mol. Pharmacol. 2017, 91, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef]
- Maleki Vareki, S.; Salim, K.Y.; Danter, W.R.; Koropatnick, J. Novel anti-cancer drug COTI-2 synergizes with therapeutic agents and does not induce resistance or exhibit cross-resistance in human cancer cell lines. PLoS ONE 2018, 13, e0191766. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef]
- Bordeira-Carrico, R.; Pego, A.P.; Santos, M.; Oliveira, C. Cancer syndromes and therapy by stop-codon readthrough. Trends Mol. Med. 2012, 18, 667–678. [Google Scholar] [CrossRef]
- Martin, L.; Grigoryan, A.; Wang, D.; Wang, J.; Breda, L.; Rivella, S.; Cardozo, T.; Gardner, L.B. Identification and characterization of small molecules that inhibit nonsense-mediated RNA decay and suppress nonsense p53 mutations. Cancer Res. 2014, 74, 3104–3113. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018, 24, 25. [Google Scholar] [CrossRef]
- Das, A.; Makarov, D.E. Effect of Mutation on an Aggregation-Prone Segment of p53: From Monomer to Dimer to Multimer. J. Phys. Chem. B 2016, 120, 11665–11673. [Google Scholar] [CrossRef]
- Ghosh, S.; Ghosh, D.; Ranganathan, S.; Anoop, A.; Kumar, P.S.; Jha, N.N.; Padinhateeri, R.; Maji, S.K. Investigating the intrinsic aggregation potential of evolutionarily conserved segments in p53. Biochemistry 2014, 53, 5995–6010. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef]
- Palanikumar, L.; Karpauskaite, L.; Al-Sayegh, M.; Chehade, I.; Alam, M.; Hassan, S.; Maity, D.; Ali, L.; Kalmouni, M.; Hunashal, Y.; et al. Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function. Nat. Commun. 2021, 12, 3962. [Google Scholar] [CrossRef]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.V.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Prabhu, V.V.; Zhang, S.; van den Heuvel, A.P.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Prodigiosin rescues deficient p53 signaling and antitumor effects via upregulating p73 and disrupting its interaction with mutant p53. Cancer Res. 2014, 74, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, L.; El-Deiry, W.S. Small-Molecule NSC59984 Induces Mutant p53 Degradation through a ROS-ERK2-MDM2 Axis in Cancer Cells. Mol. Cancer Res. 2022, 20, 622–636. [Google Scholar] [CrossRef]
- Song, H.; Wu, J.; Tang, Y.; Dai, Y.; Xiang, X.; Li, Y.; Wu, L.; Wu, J.; Liang, Y.; Xing, Y.; et al. Diverse rescue potencies of p53 mutations to ATO are predetermined by intrinsic mutational properties. Sci. Transl. Med. 2023, 15, eabn9155. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.L.; Liang, Y.; Tang, Y.G.; Song, H.X.; Wu, L.L.; Xing, Y.F.; Yan, N.; Li, Y.T.; Wang, Z.Y.; et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2021, 39, 225–239.e8. [Google Scholar] [CrossRef]
- Tang, Y.; Song, H.; Wang, Z.; Xiao, S.; Xiang, X.; Zhan, H.; Wu, L.; Wu, J.; Xing, Y.; Tan, Y.; et al. Repurposing antiparasitic antimonials to noncovalently rescue temperature-sensitive p53 mutations. Cell Rep. 2022, 39, 110622. [Google Scholar] [CrossRef]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Dumbrava, E.E.; Johnson, M.L.; Tolcher, A.W.; Shapiro, G.; Thompson, J.A.; El-Khoueiry, A.B.; Vandross, A.L.; Kummar, S.; Parikh, A.R.; Munster, P.N.; et al. First-in-human study of PC14586, a small molecule structural corrector of Y220C mutant p53, in patients with advanced solid tumors harboring a TP53 Y220C mutation. J. Clin. Oncol. 2022, 40, 3003. [Google Scholar] [CrossRef]
- Guiley, K.Z.; Shokat, K.M. A Small Molecule Reacts with the p53 Somatic Mutant Y220C to Rescue Wild-type Thermal Stability. Cancer Discov. 2023, 13, 56–69. [Google Scholar] [CrossRef]
- Zhao, C.Y.; Szekely, L.; Bao, W.; Selivanova, G. Rescue of p53 function by small-molecule RITA in cervical carcinoma by blocking E6-mediated degradation. Cancer Res. 2010, 70, 3372–3381. [Google Scholar] [CrossRef]
- Weilbacher, A.; Gutekunst, M.; Oren, M.; Aulitzky, W.E.; van der Kuip, H. RITA can induce cell death in p53-defective cells independently of p53 function via activation of JNK/SAPK and p38. Cell Death Dis. 2014, 5, e1318. [Google Scholar] [CrossRef] [PubMed]
- Burmakin, M.; Shi, Y.; Hedstrom, E.; Kogner, P.; Selivanova, G. Dual targeting of wild-type and mutant p53 by small molecule RITA results in the inhibition of N-Myc and key survival oncogenes and kills neuroblastoma cells in vivo and in vitro. Clin. Cancer Res. 2013, 19, 5092–5103. [Google Scholar] [CrossRef] [PubMed]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef]
- Hiraki, M.; Hwang, S.Y.; Cao, S.; Ramadhar, T.R.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.U.; Kolev, V.; et al. Small-Molecule Reactivation of Mutant p53 to Wild-Type-like p53 through the p53-Hsp40 Regulatory Axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef]
- Punganuru, S.R.; Madala, H.R.; Venugopal, S.N.; Samala, R.; Mikelis, C.; Srivenugopal, K.S. Design and synthesis of a C7-aryl piperlongumine derivative with potent antimicrotubule and mutant p53-reactivating properties. Eur. J. Med. Chem. 2016, 107, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, S.; Archer, T.K. Modulating molecular chaperone Hsp90 functions through reversible acetylation. Trends Cell Biol. 2005, 15, 565–567. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Saini, N.; Parris, A.B.; Zhao, M.; Yang, X. Ganetespib induces G2/M cell cycle arrest and apoptosis in gastric cancer cells through targeting of receptor tyrosine kinase signaling. Int. J. Oncol. 2017, 51, 967–974. [Google Scholar] [CrossRef]
- Richardson, P.G.; Badros, A.Z.; Jagannath, S.; Tarantolo, S.; Wolf, J.L.; Albitar, M.; Berman, D.; Messina, M.; Anderson, K.C. Tanespimycin with bortezomib: Activity in relapsed/refractory patients with multiple myeloma. Br. J. Haematol. 2010, 150, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Qian, X.G.; Sha, B.D. Heat Shock Protein 40: Structural Studies and Their Functional Implications. Protein Pept. Lett. 2009, 16, 606–612. [Google Scholar] [CrossRef]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef]
- Parrales, A.; Thoenen, E.; Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Ingallina, E.; Sorrentino, G.; Bertolio, R.; Lisek, K.; Zannini, A.; Azzolin, L.; Severino, L.U.; Scaini, D.; Mano, M.; Mantovani, F.; et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell Biol. 2018, 20, 28–35. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lin, J.H.; Chou, C.W.; Chang, Y.F.; Yeh, S.H.; Chen, C.C. Statins increase p21 through inhibition of histone deacetylase activity and release of promoter-associated HDAC1/2. Cancer Res. 2008, 68, 2375–2383. [Google Scholar] [CrossRef]
- Alalem, M.; Bhosale, M.; Ranjan, A.; Yamamoto, S.; Kaida, A.; Nishikawa, S.; Parrales, A.; Farooki, S.; Anant, S.; Padhye, S.; et al. Mutant p53 Depletion by Novel Inhibitors for HSP40/J-Domain Proteins Derived from the Natural Compound Plumbagin. Cancers 2022, 14, 4187. [Google Scholar] [CrossRef]
- Nishikawa, S.; Kaida, A.; Parrales, A.; Ranjan, A.; Alalem, M.; Ren, H.Y.; Schoenen, F.J.; Johnson, D.K.; Iwakuma, T. DNAJA1-and conformational mutant p53-dependent inhibition of cancer cell migration by a novel compound identified through a virtual screen. Cell Death Discov. 2022, 8, 437. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Tahaney, W.M.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecularly targeted therapies for p53-mutant cancers. Cell Mol. Life Sci. 2017, 74, 4171–4187. [Google Scholar] [CrossRef]
- Osman, A.A.; Monroe, M.M.; Ortega Alves, M.V.; Patel, A.A.; Katsonis, P.; Fitzgerald, A.L.; Neskey, D.M.; Frederick, M.J.; Woo, S.H.; Caulin, C.; et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol. Cancer Ther. 2015, 14, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Weberpals, J.I.; Provencher, D.M.; Grischke, E.-M.; Hall, M.; Uyar, D.; Estevez-Diz, M.D.P.; Marme, F.; Kuzmin, A.; Rosenberg, P.; et al. An international, biomarker-directed, randomized, phase II trial of AZD1775 plus paclitaxel and carboplatin (P/C) for the treatment of women with platinum-sensitive, TP53-mutant ovarian cancer. J. Clin. Oncol. 2015, 33, 5506. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients with TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef]
- Moore, K.N.; Chambers, S.K.; Hamilton, E.P.; Chen, L.-M.; Oza, A.M.; Ghamande, S.A.; Konecny, G.E.; Plaxe, S.C.; Spitz, D.L.; Geenen, J.J.J.; et al. Adavosertib with chemotherapy (CT) in patients (pts) with platinum-resistant ovarian cancer (PPROC): An open label, four-arm, phase II study. J. Clin. Oncol. 2019, 37, 5513. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.Y.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug Name | Combination with Other Therapy | Type of Malignancies | Clinical Trials | Clinical Trial ID |
|---|---|---|---|---|
| RG7112 | Hematologic | 1 | NCT00623870 | |
| Idasanutlin | Cytarabine | AML | 3 | NCT02545283 |
| Chemotherapy/ Venetoclax | AML Solid tumors | 2 | NCT04029688 | |
| Solid tumors | 2 | NCT04589845 | ||
| Glioblastoma | 2 | NCT03158389 | ||
| SAR405838 | Malignant neoplasm | 1 | NCT01636479 | |
| Malignant neoplasm | 1 | NCT01985191 | ||
| Milademetan | Advanced solid tumor, Lymphoma | 1 | NCT01877382 | |
| Solid tumor | 2 | NCT05012397 | ||
| Alrizomadlin | Pembrolizumab | Metastatic melanomas Advanced solid tumors | 2 | NCT03611868 |
| KRT-232 | MCC | 2 | NCT03787602 | |
| NVP-CGM097 | Solid tumors | 1 | NCT01760525 | |
| MK-8242 | Cytarabine | AML | 1 | NCT01451437 |
| Solid tumors | 1 | NCT01463696 | ||
| BI 907828 | Solid tumors | 1 | NCT03449381 | |
| Siremadlin | Advanced solid tumors Hematological neoplasm | 1 | NCT02143635 | |
| LEE011 | Solid tumors | 1 | NCT02343172 | |
| ALRN-6924 | Advanced solid tumors | 1 | NCT05622058 | |
| APR-246 | Refractory hematologic tumor Prostate cancer | 1 | NCT00900614 | |
| Carboplatin+ Pegylated Liposomal Doxorubicin Hydrochloride (PLD) | Platinum sensitive recurrent high-grade serous ovarian Cancer with p53 Mutations | 2 | NCT02098343 | |
| Myeloid Neoplasms with p53 mutations | 2 | NCT03072043 | ||
| Azacitidine | MDS with p53 mutations | 3 | NCT03745716 | |
| COTI-2 | Cisplatin | Advanced solid tumors with p53 mutations | 1 | NCT02433626 |
| Tanespimycin | Bortezomib | Multiple myeloma with p53 mutations | 3 | NCT00514371 |
| Atorvastatin Calcium | Colorectal Carcinoma Ulcerative Colitis with p53 mutations | 2 | NCT04767984 | |
| Atorvastatin | Advanced solid tumors with p53 mutations | 1 | NCT03560882 | |
| MK-1775 | Paclitaxel+ Carboplatin | Platinum-sensitive Ovarian Tumors with p53 Mutations | 2 | NCT01357161 |
| Carboplatin | Epithelial ovarian cancer with p53 Mutation | 2 | NCT01164995 | |
| Paclitaxel/Carboplatin/Gemcitabine/PLD | Ovarian, Fallopian Tube, Peritoneal cancer with p53 mutations | 2 | NCT02272790 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Wu, M.; Xu, Y.; Yu, L. The Development of p53-Targeted Therapies for Human Cancers. Cancers 2023, 15, 3560. https://doi.org/10.3390/cancers15143560
Lu Y, Wu M, Xu Y, Yu L. The Development of p53-Targeted Therapies for Human Cancers. Cancers. 2023; 15(14):3560. https://doi.org/10.3390/cancers15143560
Chicago/Turabian StyleLu, Yier, Meng Wu, Yang Xu, and Lili Yu. 2023. "The Development of p53-Targeted Therapies for Human Cancers" Cancers 15, no. 14: 3560. https://doi.org/10.3390/cancers15143560
APA StyleLu, Y., Wu, M., Xu, Y., & Yu, L. (2023). The Development of p53-Targeted Therapies for Human Cancers. Cancers, 15(14), 3560. https://doi.org/10.3390/cancers15143560