
p90RSK Regulates p53 Pathway by MDM2 Phosphorylation in Thyroid Tumors

 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Cell Lines and Transfection
2.3. Cell Proliferation Assay
2.4. Plasmids
2.5. Protein Extraction, Western Blotting, and Antibodies
2.6. In Vitro Kinase Assay
2.7. RNA Extraction and Real-Time PCR
- p53 Forward: TTCTTGCATTCTGGGACAGCC;
- Reverse: GCTTCTGACGCACACCTATTG;
- forward: AAGATGCGCGGGAAGTAGC;
- Reverse: GGTTTTGGTCTAACCTGGAGGC;
- GAPDH forward: CCATCACCATCTTCCAGGAGC;
- Reverse: AGAGATGATGACCCTTTTGGC.
2.8. TUNEL Assay
2.9. BrdU Assay and Immunofluorescence
2.10. RNA Silencing
2.11. Histological Examination and Immunohistochemistry
2.12. Statistical Analysis
3. Results
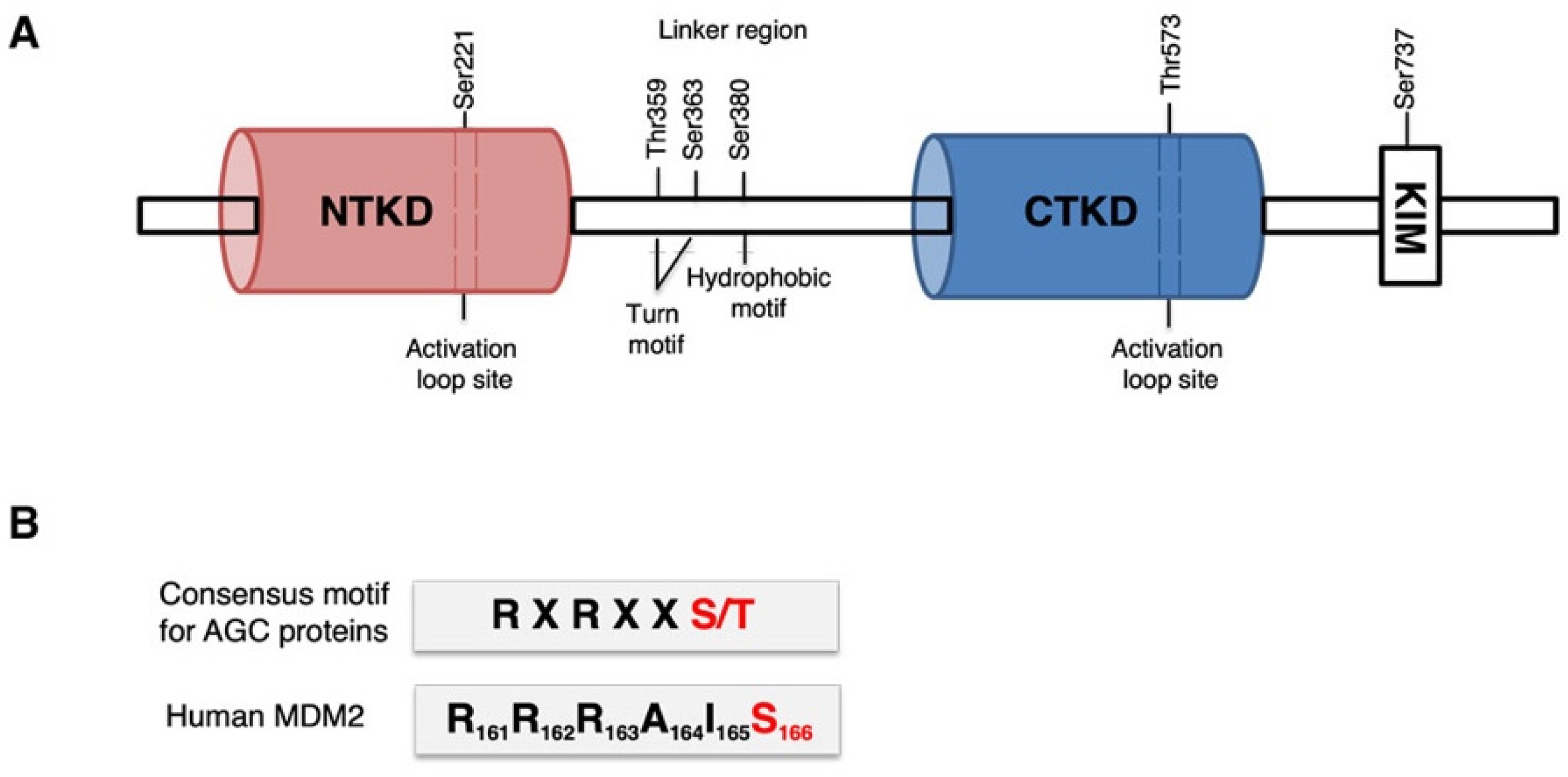
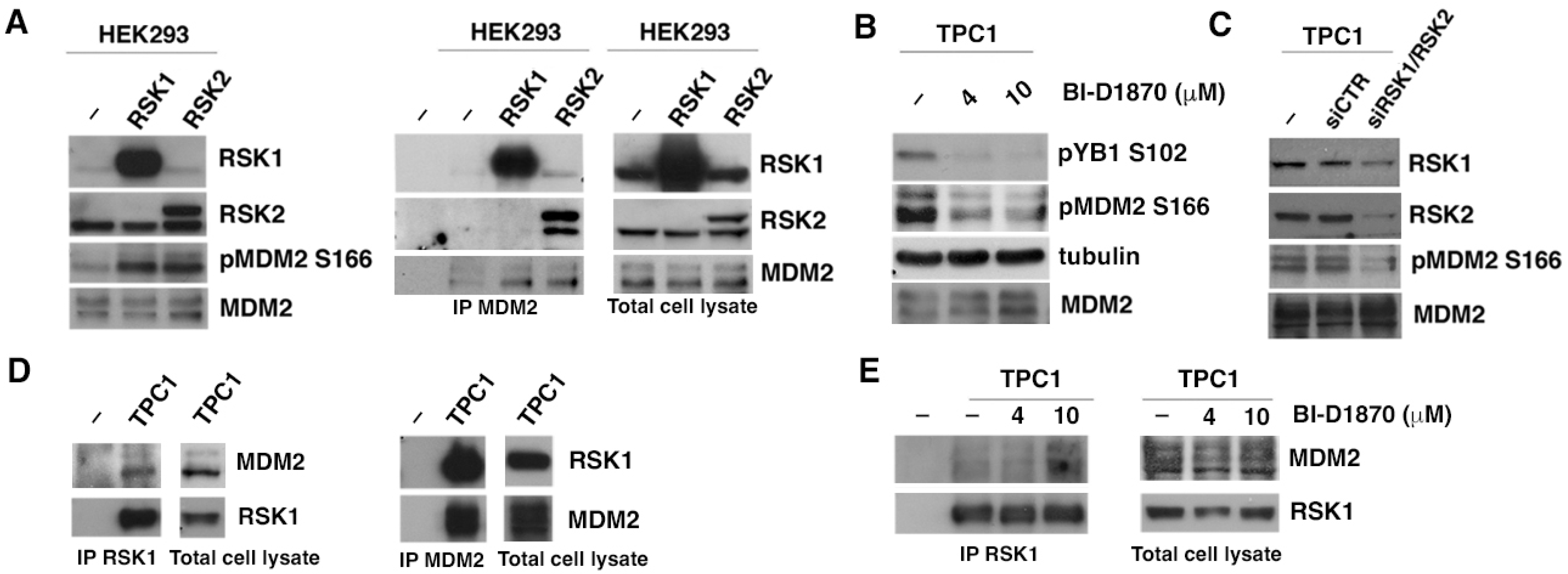
3.1. p90RSK Directly Binds and Phosphorylates MDM2 on S166 In Vitro
3.2. p90RSK Binds and Phosphorylates MDM2 on S166 In Vivo
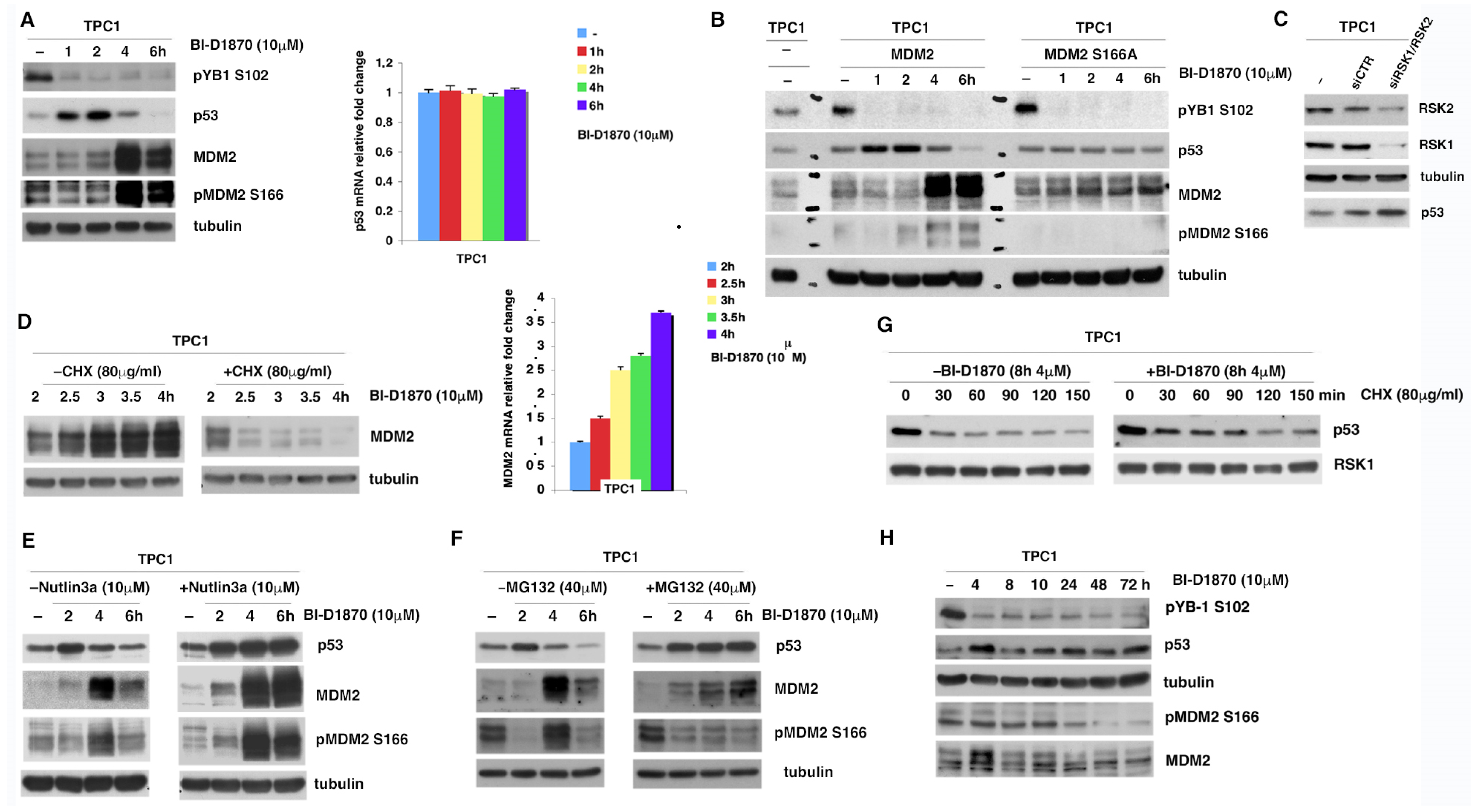
3.3. p90RSK Promotes Degradation of p53 via MDM2 Phosphorylation
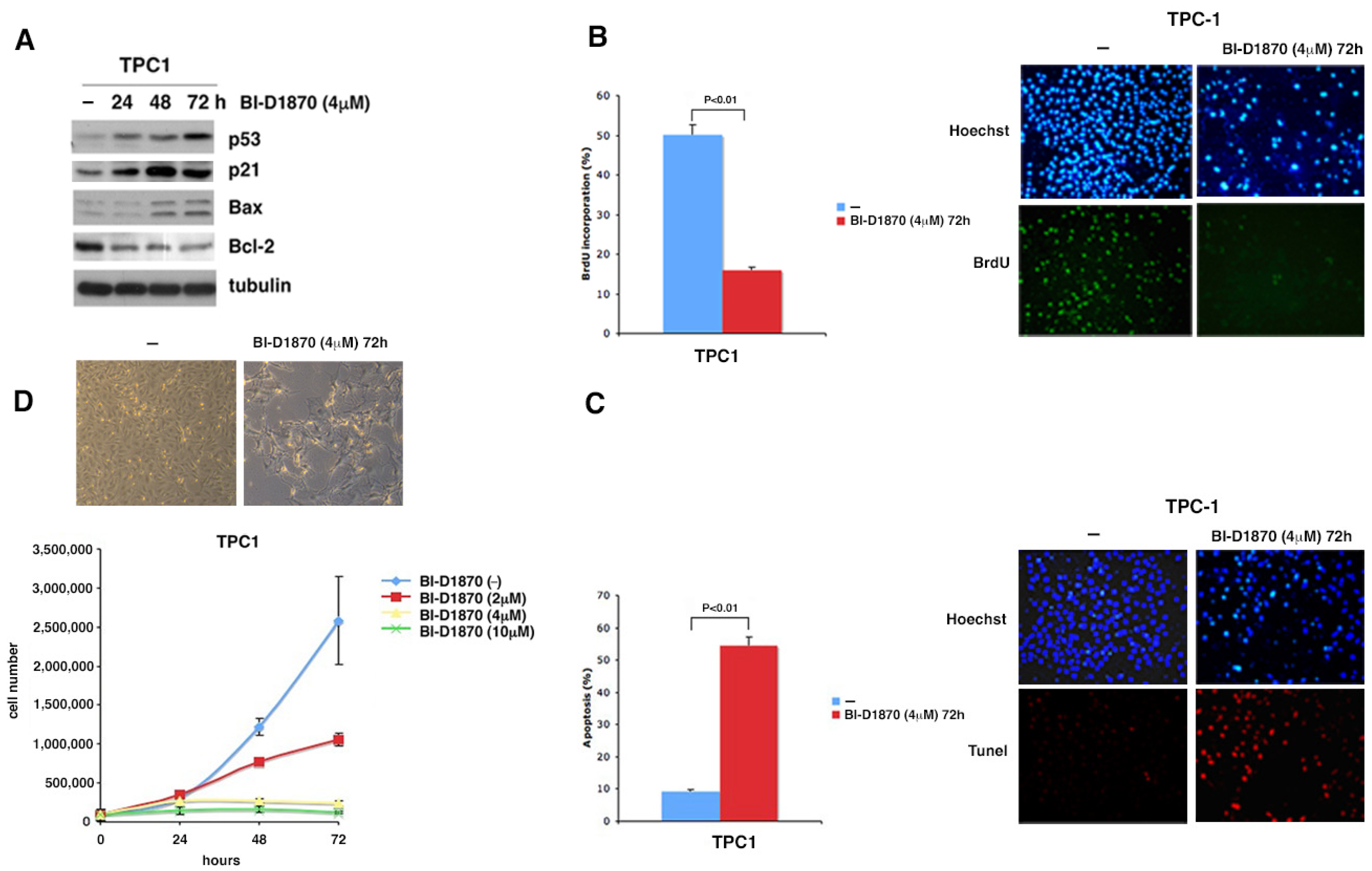
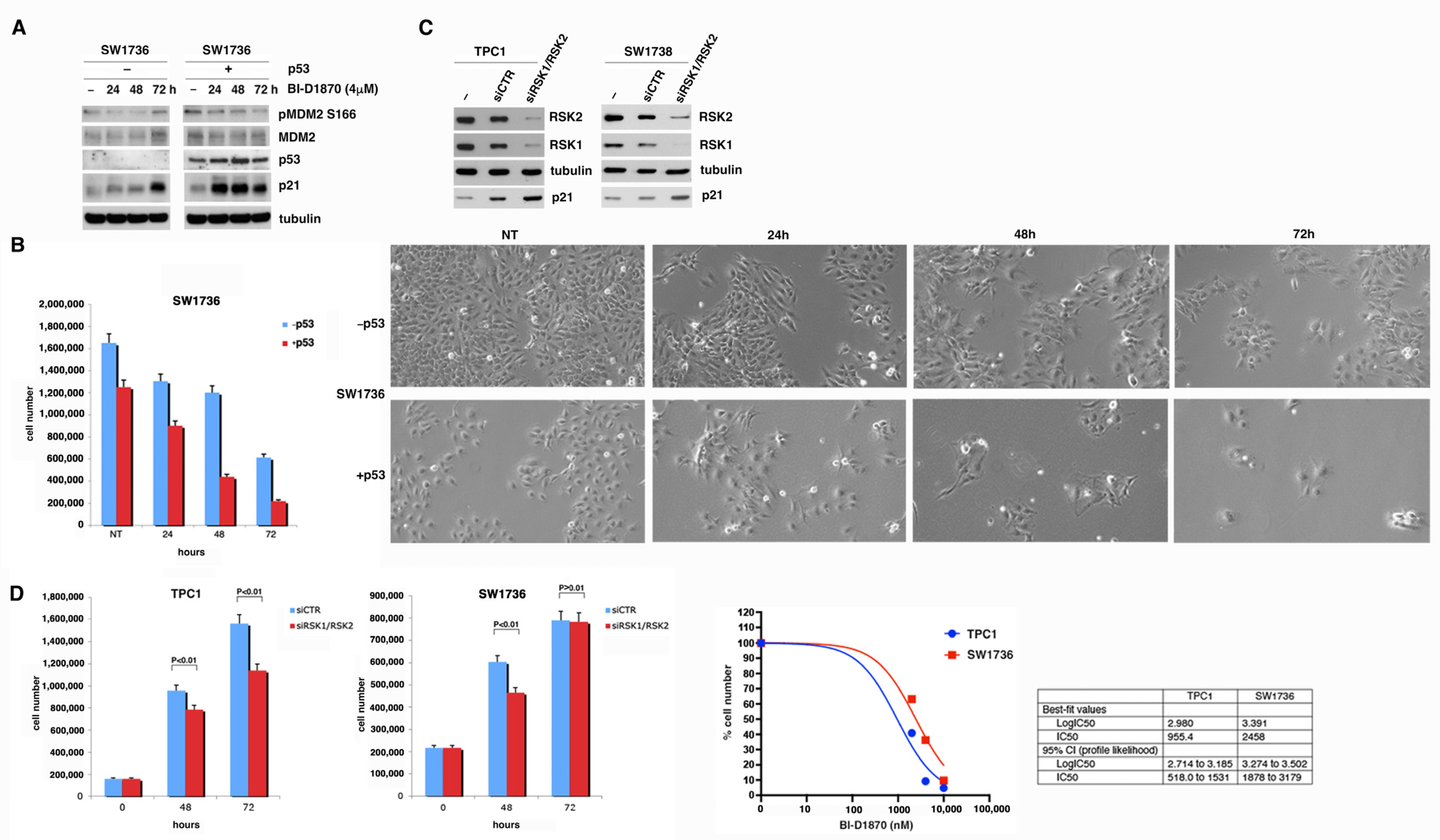
3.4. p90RSK Is Able to Regulate Proliferation and Apoptosis by Controlling p53 Levels
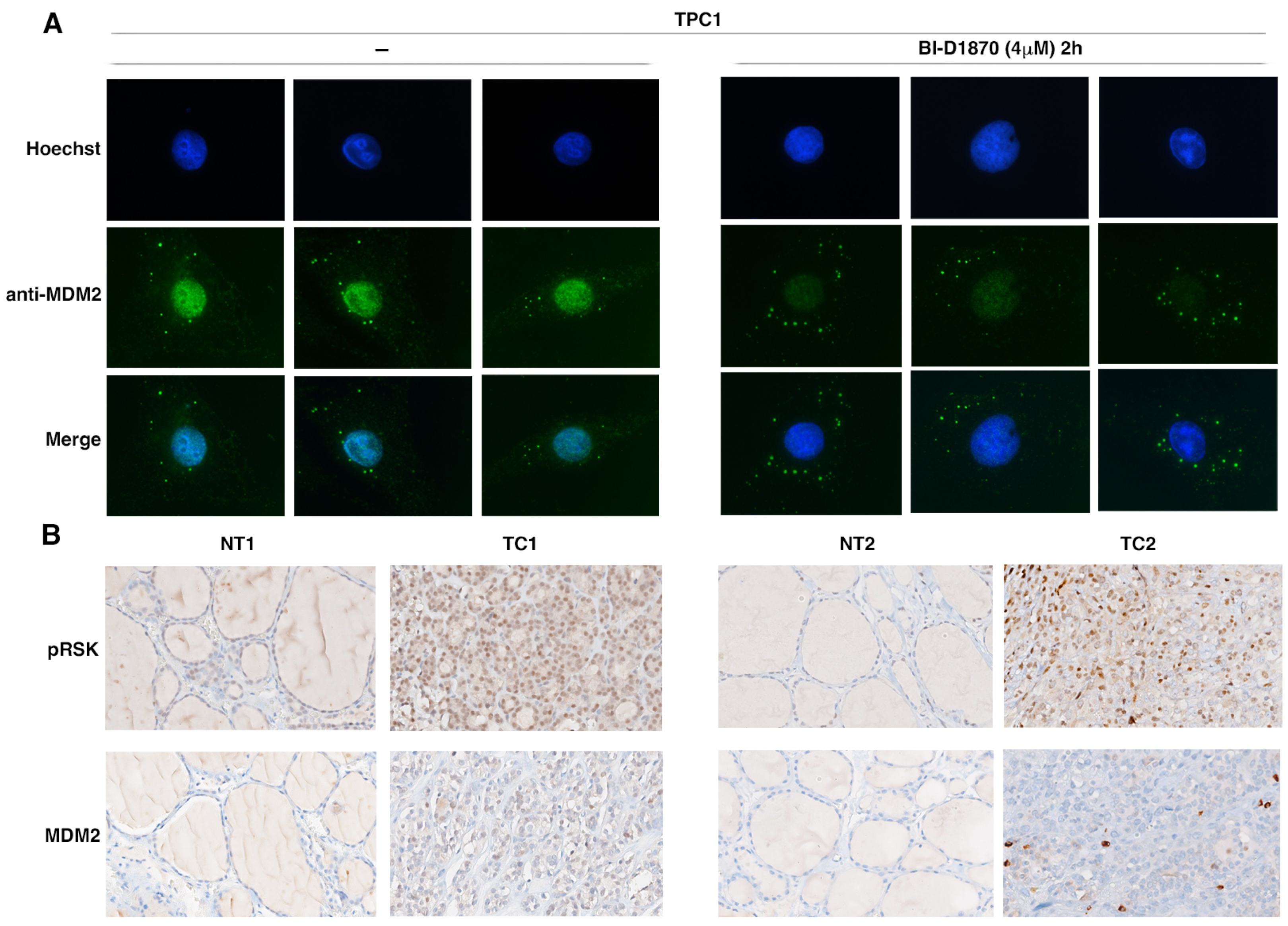
3.5. p90RSK Activation Is Associated to MDM2 Stabilization in TPC1 Cells and in Primary Thyroid Tumors
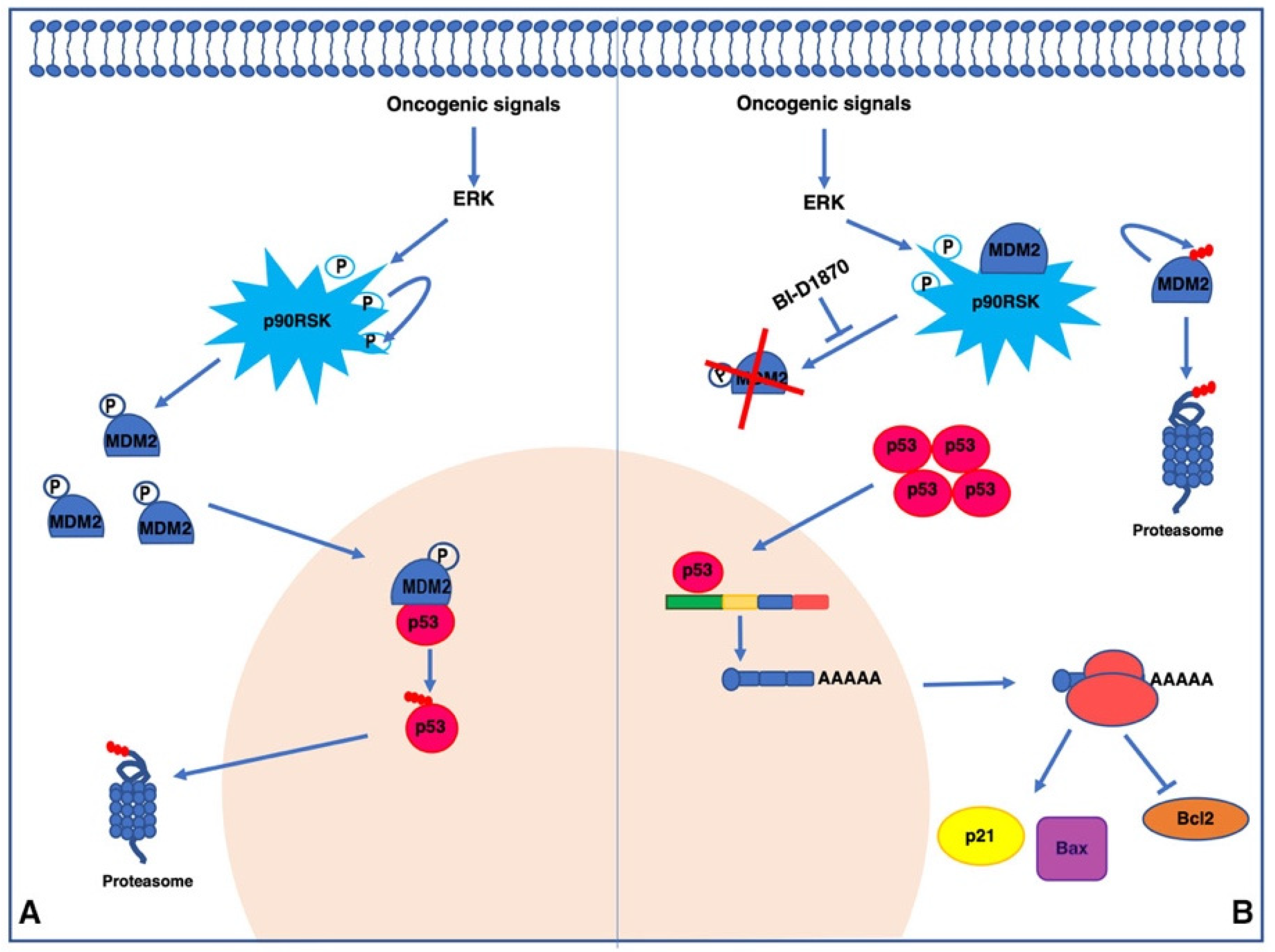
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frödin, M.; Gammeltoft, S. Role and Regulation of 90 KDa Ribosomal S6 Kinase (RSK) in Signal Transduction. Mol. Cell. Endocrinol. 1999, 151, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Leighton, I.A.; Dalby, K.N.; Barry Caudwell, F.; Cohen, P.T.W.; Cohen, P. Comparison of the Specificities of P70 S6 Kinase and MAPKAP Kinase-1 Identifies a Relatively Specific Substrate for P70 S6 Kinase: The N-Terminal Kinase Domain of MAPKAP Kinase-1 Is Essential for Peptide Phosphorylation. FEBS Lett. 1995, 375, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Poteet-Smith, C.E.; Malarkey, K.; Sturgill, T.W. Identification of an Extracellular Signal-Regulated Kinase (ERK) Docking Site in Ribosomal S6 Kinase, a Sequence Critical for Activation by ERK in Vivo. J. Biol. Chem. 1999, 274, 2893–2898. [Google Scholar] [CrossRef] [PubMed]
- Fisher, T.L.; Blenis, J. Evidence for Two Catalytically Active Kinase Domains in Pp90rsk. Mol. Cell. Biol. 1996, 16, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Dalby, K.N.; Morrice, N.; Caudwell, F.B.; Avruch, J.; Cohen, P. Identification of Regulatory Phosphorylation Sites in Mitogen-Activated Protein Kinase (MAPK)-Activated Protein Kinase-1a/P90rsk That Are Inducible by MAPK. J. Biol. Chem. 1998, 273, 1496–1505. [Google Scholar] [CrossRef]
- Jensen, C.J.; Buch, M.B.; Krag, T.O.; Hemmings, B.A.; Gammeltoft, S.; Fr√∂din, M. 90-KDa Ribosomal S6 Kinase Is Phosphorylated and Activated by 3-Phosphoinositide-Dependent Protein Kinase-1. J. Biol. Chem. 1999, 274, 27168–27176. [Google Scholar] [CrossRef]
- Lara, R.; Seckl, M.J.; Pardo, O.E. The P90 RSK Family Members: Common Functions and Isoform Specificity. Cancer Res. 2013, 73, 5301–5308. [Google Scholar] [CrossRef]
- Romeo, Y.; Zhang, X.; Roux, P.P. Regulation and Function of the RSK Family of Protein Kinases. Biochem. J. 2012, 441, 553–569. [Google Scholar] [CrossRef]
- Schouten, G.J.; Vertegaal, A.C.O.; Whiteside, S.T.; Israël, A.; Toebes, M.; Dorsman, J.C.; van der Eb, A.J.; Zantema, A. IκBα is a target for the mitogen-activated 90 kDa ribosomal S6 kinase. EMBO J. 1997, 16, 3133–3144. [Google Scholar] [CrossRef]
- Servillo, G.; Della Fazia, M.A.; Sassone-Corsi, P. Coupling cAMP signaling to transcription in the liver: Pivotal role of CREB and CREM. Exp. Cell. Res. 2002, 275, 143–154. [Google Scholar] [CrossRef]
- Joel, P.B.; Smith, J.; Sturgill, T.W.; Fisher, T.L.; Blenis, J.; Lannigan, D.A. Pp90rsk1 Regulates Estrogen Receptor-Mediated Transcription through Phosphorylation of Ser-167. Mol. Cell. Biol. 1998, 18, 1978–1984. [Google Scholar] [CrossRef] [PubMed]
- Yamnik, R.L.; Holz, M.K. MTOR/S6K1 and MAPK/RSK Signaling Pathways Coordinately Regulate Estrogen Receptor Alpha Serine 167 Phosphorylation. FEBS Lett. 2010, 584, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Blenis, J. ERK and P38 MAPK-Activated Protein Kinases: A Family of Protein Kinases with Diverse Biological Functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Gibson, T.B.; Robinson, F.; Silvestro, L.; Pearson, G.; Xu, B.E.; Wright, A.; Vanderbilt, C.; Cobb, M.H. MAP Kinases. Chem. Rev. 2001, 101, 2449–2476. [Google Scholar] [CrossRef] [PubMed]
- Romeo, Y.; Roux, P.P. Paving the way for targeting RSK in cancer. Expert Opin. Ther. Targets 2011, 15, 5–9. [Google Scholar] [CrossRef]
- David, J.P.; Mehic, D.; Bakiri, L.; Schilling, A.F.; Mandic, V.; Priemel, M.; Idarraga, M.H.; Reschke, M.O.; Hoffmann, O.; Amling, M.; et al. Essential role of RSK2 in c-Fos-dependent osteosarcoma development. J. Clin. Investig. 2005, 115, 664–672. [Google Scholar] [CrossRef]
- Kang, S.; Elf, S.; Lythgoe, K.; Hitosugi, T.; Taunton, J.; Zhou, W.; Xiong, L.; Wang, D.; Muller, S.; Fan, S.; et al. p90 ribosomal S6 kinase 2 promotes invasion and metastasis of human head and neck squamous cell carcinoma cells. J. Clin. Investig. 2010, 120, 1165–1177. [Google Scholar] [CrossRef]
- Serra, V.; Eichhorn, P.J.A.; Garcia-Garcia, C.; Ibrahim, Y.H.; Prudkin, L.; Sanchez, G.; Rodriguez, O.; Anton, P.; Parra, J.L.; Marlow, S.; et al. RSK3/4 Mediate Resistance to PI3K Pathway Inhibitors in Breast Cancer. J. Clin. Investig. 2013, 123, 2551–2563. [Google Scholar] [CrossRef]
- Bender, C.; Ullrich, A. PRKX, TTBK2 and RSK4 Expression Causes Sunitinib Resistance in Kidney Carcinoma- and Melanoma-Cell Lines. Int. J. Cancer 2012, 131, E45–E55. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Ramos, J.W. RSK Isoforms in Cancer Cell Invasion and Metastasis. Cancer Res. 2013, 73, 6099–6105. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Robertson, M. Ubiquitin Ligases and Beyond. BMC Biol. 2012, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Iwakuma, T.; Lozano, G. Subject Review MDM2, An Introduction. Mol. Cancer Res. 2003, 1, 993–1000. [Google Scholar] [PubMed]
- Grossman, S.R.; Perez, M.; Kung, A.L.; Joseph, M.; Mansur, C.; Xiao, Z.X.; Kumar, S.; Howley, P.M.; Livingston, D.M. P300/MDM2 Complexes Participate in MDM2-Mediated P53 Degradation. Mol. Cell 1998, 2, 405–415. [Google Scholar] [CrossRef]
- Toth, A.; Nickson, P.; Liu, L.Q.; Erhardt, P. Differential Regulation of Cardiomyocyte Survival and Hypertrophy by MDM2, an E3 Ubiquitin Ligase. J. Biol. Chem. 2006, 281, 3679–3689. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a Potential Mediator of P53 Tumor Suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Brugarolas, J.; Chandrasekaran, C.; Gordon, J.I.; Beach, D.; Jacks, T.; Hannon, G.J. Radiation-Induced Cell Cycle Arrest Compromised by P21 Deficiency. Nature 1995, 377, 552–557. [Google Scholar] [CrossRef]
- Toshiyuki, M.; Reed, J.C. Tumor Suppressor P53 Is a Direct Transcriptional Activator of the Human Bax Gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef]
- Hemann, M.T.; Lowe, S.W. The P53-Bcl-2 Connection. Cell Death Differ. 2006, 13, 1256. [Google Scholar] [CrossRef]
- David, W.; Meek, D.W.; Knippschild, U. Posttranslational Modification of MDM2. Mol. Cancer Res. 2003, 14, 1017–1026. [Google Scholar]
- De Falco, V.; Buonocore, P.; Muthu, M.; Torregrossa, L.; Basolo, F.; Billaud, M.; Gozgit, J.M.; Carlomagno, F.; Santoro, M. Ponatinib (AP24534) Is a Novel Potent Inhibitor of Oncogenic RET Mutants Associated with Thyroid Cancer. J. Clin. Endocrinol. Metab. 2013, 98, E811–E819. [Google Scholar] [CrossRef]
- Maric, I.; Viaggi, S.; Caria, P.; Frau, D.V.; Degan, P.; Vanni, R. Centrosomal and mitotic abnormalities in cell lines derived from papillary thyroid cancer harboring specific gene alterations. Mol. Cytogenet. 2011, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Schweppe, R.E.; Klopper, J.P.; Korch, C.; Pugazhenthi, U.; Benezra, M.; Knauf, J.A.; Fagin, J.A.; Marlow, L.; Copland, J.A.; Smallridge, R.C.; et al. Deoxyribonucleic Acid Profiling Analysis of 40 Human Thyroid Cancer Cell Lines Reveals Cross-Contamination Resulting in Cell Line Redundancy and Misidentification. J. Clin. Endocrinol. Metab. 2008, 93, 4331–4341. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.A.; Dreisbach, V.C.; Murphy, L.O.; Blenis, J. Characterization of Regulatory Events Associated with Membrane Targeting of p90 Ribosomal S6 Kinase 1. Mol. Cell. Biol. 2001, 21, 7470–7480. [Google Scholar] [CrossRef] [PubMed]
- De Falco, V.; Castellone, M.D.; De Vita, G.; Cirafici, A.M.; Hershman, J.M.; Guerrero, C.; Fusco, A.; Melillo, R.M.; Santoro, M. RET/Papillary Thyroid Carcinoma Oncogenic Signaling through the Rap1 Small GTPase. Cancer Res. 2007, 67, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Löber, C.; Lenz-Stöppler, C.; Dobbelstein, M. Adenovirus E1-transformed cells grow despite the continuous presence of transcriptionally active p53. J. Gen. Virol. 2002, 83, 2047–2057. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gnanasundram, S.V.; Malbert-Colas, L.; Chen, S.; Fusée, L.; Daskalogianni, C.; Muller, P.; Salomao, N.; Fåhraeus, R. MDM2’s dual mRNA binding domains co-ordinate its oncogenic and tumour suppressor activities. Nucleic Acids Res. 2020, 48, 6775–6787. [Google Scholar] [CrossRef]
- Feng, J.; Tamaskovic, R.; Yang, Z.; Brazil, D.P.; Merlo, A.; Hess, D.; Hemmings, B.A. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J. Biol. Chem. 2004, 279, 35510–35517. [Google Scholar] [CrossRef]
- Bourgarel-Rey, V.; Savry, A.; Hua, G.; Carré, M.; Bressin, C.; Chacon, C.; Imbert, J.; Braguer, D.; Barra, Y. Transcriptional Down-Regulation of Bcl-2 by Vinorelbine: Identification of a Novel Binding Site of P53 on Bcl-2 Promoter. Biochem. Pharmacol. 2009, 78, 1148–1156. [Google Scholar] [CrossRef]
- Lavra, L.; Ulivieri, A.; Rinaldo, C.; Dominici, R.; Volante, M.; Luciani, E.; Bartolazzi, A.; Frasca, F.; Soddu, S.; Sciacchitano, S. Gal-3 is stimulated by gain-of-function p53 mutations and modulates chemoresistance in anaplastic thyroid carcinomas. J. Pathol. 2009, 218, 66–75. [Google Scholar] [CrossRef]
- Zhang, Y.; Young, A.; Zhang, J.; Chen, X. P73 tumor suppressor and its targets, p21 and PUMA, are required for madin-darby canine kidney cell morphogenesis by maintaining an appropriate level of epithelial to mesenchymal transition. Oncotarget 2015, 6, 13994–14004. [Google Scholar] [CrossRef] [PubMed]
- Messina, R.L.; Sanfilippo, M.; Vella, V.; Pandini, G.; Vigneri, P.; Nicolosi, M.L.; Gianì, F.; Vigneri, R.; Frasca, F. Reactivation of p53 mutants by p53 reactivation and induction of massive apoptosis in thyroid cancer cells. Int. J. Cancer 2012, 130, 2259–2270. [Google Scholar] [CrossRef] [PubMed]
- Frasca, F.; Vella, V.; Aloisi, A.; Mandarino, A.; Mazzon, E.; Vigneri, R.; Vigneri, P. p73 tumor-suppressor activity is impaired in human thyroid cancer. Cancer Res. 2003, 63, 5829–5837. [Google Scholar] [PubMed]
- Rada, M.; Barlev, N.; Macip, S. BTK modulates p73 activity to induce apoptosis independently of p53. Cell Death Discov. 2018, 4, 95. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular Targeted Therapy: Treating Cancer with Specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Melillo, R.M.; Castellone, M.D.; Guarino, V.; De Falco, V.; Cirafici, A.M.; Salvatore, G.; Caiazzo, F.; Basolo, F.; Giannini, R.; Kruhoffer, M.; et al. The RET/PTC-RAS-BRAF Linear Signaling Cascade Mediates the Motile and Mitogenic Phenotype of Thyroid Cancer Cells. J. Clin. Investig. 2005, 115, 1068–1081. [Google Scholar] [CrossRef]
- Adeyinka, A.; Nui, Y.; Cherlet, T.; Snell, L.; Watson, P.H.; Murphy, L.C. Activated Mitogen-Activated Protein Kinase Expression during Human Breast Tumorigenesis and Breast Cancer Progression. Clin. Cancer Res. 2002, 8, 1747–1753. [Google Scholar]
- Cohen, C.; Zavala-Pompa, A.; Sequeira, J.H.; Shoji, M.; Sexton, D.G.; Cotsonis, G.; Cerimele, F.; Govindarajan, B.; Macaron, N.; Arbiser, J.L. Mitogen-Actived Protein Kinase Activation Is an Early Event in Melanoma Progression. Clin. Cancer Res. 2002, 8, 3728–3733. [Google Scholar]
- Sivaraman, V.S.; Wang, H.Y.; Nuovo, G.J.; Malbon, C.C. Hyperexpression of Mitogen-Activated Protein Kinase in Human Breast Cancer. J. Clin. Investig. 1997, 99, 1478–1483. [Google Scholar] [CrossRef]
- Brose, M.S.; Volpe, P.; Feldman, M.; Kumar, M.; Rishi, I.; Gerrero, R.; Einhorn, E.; Herlyn, M.; Minna, J.; Nicholson, A.; et al. BRAF and RAS Mutations in Human Lung Cancer and Melanoma. Cancer Res. 2002, 62, 6997–7000. [Google Scholar] [PubMed]
- Rajagopalan, H.; Bardelli, A.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E. Tumorigenesis: RAF/RAS Oncogenes and Mismatch-Repair Status. Nature 2002, 418, 934. [Google Scholar] [CrossRef] [PubMed]
- Tebbutt, N.; Pedersen, M.W.; Johns, T.G. Targeting the ERBB Family in Cancer: Couples Therapy. Nat. Rev. Cancer 2013, 13, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Farooq, A.; Zhou, M.M. Structure and Regulation of MAPK Phosphatases. Cell. Signal. 2004, 16, 769–779. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. RAS Oncogenes: The First 30 Years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK Mitogen-Activated Protein Kinase Cascade for the Treatment of Cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT Signalling Pathways: Role in Cancer Pathogenesis and Implications for Therapeutic Approaches. Expert Opin. Ther. Targets 2012, 16 (Suppl. 2), S17–S27. [Google Scholar] [CrossRef]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, H. Current Development Status of MEK Inhibitors. Molecules 2017, 22, 1551. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP Kinase Pathway Alterations in BRAF-Mutant Melanoma Patients with Acquired Resistance to Combined RAF/MEK Inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK Reactivation in Early Resistance to Dabrafenib/Trametinib Combination Therapy of BRAF-Mutant Metastatic Melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Wellbrock, C. Phenotype Plasticity as Enabler of Melanoma Progression and Therapy Resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Motti, M.L.; Minopoli, M.; Di Carluccio, G.; Ascierto, P.A.; Carriero, M.V. MicroRNAs as Key Players in Melanoma Cell Resistance to MAPK and Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 4544. [Google Scholar] [CrossRef]
- Crispo, F.; Notarangelo, T.; Pietrafesa, M.; Lettini, G.; Storto, G.; Sgambato, A.; Maddalena, F.; Landriscina, M. BRAF Inhibitors in Thyroid Cancer: Clinical Impact, Mechanisms of Resistance and Future Perspectives. Cancers 2019, 11, 1388. [Google Scholar] [CrossRef]
- Fratangelo, F.; Camerlingo, R.; Carriero, M.V.; Pirozzi, G.; Palmieri, G.; Gentilcore, G.; Ragone, C.; Minopoli, M.; Ascierto, P.A.; Motti, M.L. Effect of ABT-888 on the Apoptosis, Motility and Invasiveness of BRAFi-Resistant Melanoma Cells. Int. J. Oncol. 2018, 53, 1149–1159. [Google Scholar] [CrossRef]
- Poomakkoth, N.; Issa, A.; Abdulrahman, N.; Abdelaziz, S.G.; Mraiche, F. p90 ribosomal S6 kinase: A potential therapeutic target in lung cancer. J. Transl. Med. 2016, 14, 14. [Google Scholar] [CrossRef]
- Doehn, U.; Hauge, C.; Frank, S.R.; Jensen, C.J.; Duda, K.; Nielsen, J.V.; Cohen, M.S.; Johansen, J.V.; Winther, B.R.; Lund, L.R.; et al. RSK is a major effector of the RAS-ERK pathway to elicit a coordinated promotile/invasive gene program and phenotype in epithelial cells. Mol. Cell 2009, 35, 511–522. [Google Scholar] [CrossRef]
- Casalvieri, K.A.; Matheson, C.J.; Backos, D.S.; Reigan, P. Selective Targeting of RSK Isoforms in Cancer. Trends Cancer 2017, 3, 302–312. [Google Scholar] [CrossRef]
- Malmlöf, M.; Roudier, E.; Högberg, J.; Stenius, U. MEK-ERK-mediated phosphorylation of Mdm2 at Ser-166 in hepatocytes. Mdm2 is activated in response to inhibited Akt signaling. J. Biol. Chem. 2007, 282, 2288–2296. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maietta, I.; Del Peschio, F.; Buonocore, P.; Viscusi, E.; Laudati, S.; Iannaci, G.; Minopoli, M.; Motti, M.L.; De Falco, V. p90RSK Regulates p53 Pathway by MDM2 Phosphorylation in Thyroid Tumors. Cancers 2023, 15, 121. https://doi.org/10.3390/cancers15010121
Maietta I, Del Peschio F, Buonocore P, Viscusi E, Laudati S, Iannaci G, Minopoli M, Motti ML, De Falco V. p90RSK Regulates p53 Pathway by MDM2 Phosphorylation in Thyroid Tumors. Cancers. 2023; 15(1):121. https://doi.org/10.3390/cancers15010121
Chicago/Turabian StyleMaietta, Immacolata, Francesca Del Peschio, Preziosa Buonocore, Eleonora Viscusi, Stefano Laudati, Giuseppe Iannaci, Michele Minopoli, Maria Letizia Motti, and Valentina De Falco. 2023. "p90RSK Regulates p53 Pathway by MDM2 Phosphorylation in Thyroid Tumors" Cancers 15, no. 1: 121. https://doi.org/10.3390/cancers15010121
APA StyleMaietta, I., Del Peschio, F., Buonocore, P., Viscusi, E., Laudati, S., Iannaci, G., Minopoli, M., Motti, M. L., & De Falco, V. (2023). p90RSK Regulates p53 Pathway by MDM2 Phosphorylation in Thyroid Tumors. Cancers, 15(1), 121. https://doi.org/10.3390/cancers15010121