
The Emerging Role of Metformin in the Treatment of Hepatocellular Carcinoma: Is There Any Value in Repurposing Metformin for HCC Immunotherapy?

 , ,
, ,  ,
,  ,
, 

Abstract
Simple Summary
Abstract
1. Introduction
2. Mechanism of Action
3. Clinical Significance of Metformin
3.1. Meta-Analyses about Metformin in HCC
3.2. Clinical Studies for Metformin in HCC Risk in Diabetes, NAFLD and Viral Hepatitis
3.3. Clinical Studies about Metformin Use after Primary Treatment
4. Preclinical Evidence about Metformin Influence on HCC Hallmarks
4.1. The Effects of Metformin on HCC Proliferation and Apoptosis
4.2. Metformin in Potentiation of Sorafenib
4.3. The Effects of Metformin on HCC Autophagy
4.4. The Effects of Metformin in HCC Metastasis
4.5. Metformin and Epigenetic Regulation of HCC
5. Metformin as an Immunotherapy Enhancer
5.1. Enhancing the Efficacy of Anti-PD-1
5.2. Reversing of Effector T-Cell Exhaustion State
5.3. Metformin Effects on HCC Prevention/Reduction of Metastatic Potential
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Wege, H.; Li, J.; Ittrich, H. Treatment Lines in Hepatocellular Carcinoma. Visc. Med. 2019, 35, 266–272. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Machairas, N.; Tsilimigras, D.I.; Pawlik, T.M. State-of-the-art surgery for hepatocellular carcinoma. Langenbeck’s Arch. Surg. 2021, 406, 2151–2162. [Google Scholar] [CrossRef]
- Kotsifa, E.; Vergadis, C.; Vailas, M.; Machairas, N.; Kykalos, S.; Damaskos, C.; Garmpis, N.; Lianos, G.D.; Schizas, D. Transarterial Chemoembolization for Hepatocellular Carcinoma: Why, When, How? J. Pers. Med. 2022, 12, 436. [Google Scholar] [CrossRef]
- Machairas, N.; Tsilimigras, D.I.; Pawlik, T.M. Current Landscape of Immune Checkpoint Inhibitor Therapy for Hepatocellular Carcinoma. Cancers 2022, 14, 2018. [Google Scholar] [CrossRef]
- Machairas, N.; Papaconstantinou, D.; Dorovinis, P.; Tsilimigras, D.I.; Keramida, M.D.; Kykalos, S.; Schizas, D.; Pawlik, T.M. Meta-Analysis of Repeat Hepatectomy versus Radiofrequency Ablation for Recurrence of Hepatocellular Carcinoma. Cancers 2022, 14, 5398. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Hampel, H.; Javadi, F. The association between diabetes and hepatocellular carcinoma: A systematic review of epidemiologic evidence. Clin. Gastroenterol. Hepatol. 2006, 4, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Yao, S.K. Diabetes mellitus: A “true” independent risk factor for hepatocellular carcinoma? Hepatobiliary Pancreat. Dis. Int. 2009, 8, 465–473. [Google Scholar]
- Kawamura, Y.; Ikeda, K.; Arase, Y.; Yatsuji, H.; Sezaki, H.; Hosaka, T.; Akuta, N.; Kobayashi, M.; Saitoh, S.; Suzuki, F.; et al. Diabetes mellitus worsens the recurrence rate after potentially curative therapy in patients with hepatocellular carcinoma associated with nonviral hepatitis. J. Gastroenterol. Hepatol. 2008, 23, 1739–1746. [Google Scholar] [CrossRef] [PubMed]
- Kasmari, A.J.; Welch, A.; Liu, G.; Leslie, D.; McGarrity, T.; Riley, T. Independent of Cirrhosis, Hepatocellular Carcinoma Risk Is Increased with Diabetes and Metabolic Syndrome. Am. J. Med. 2017, 130, 746.e1–746.e7. [Google Scholar] [CrossRef]
- Fujita, K.; Iwama, H.; Miyoshi, H.; Tani, J.; Oura, K.; Tadokoro, T.; Sakamoto, T.; Nomura, T.; Morishita, A.; Yoneyama, H.; et al. Diabetes mellitus and metformin in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 6100–6113. [Google Scholar] [CrossRef]
- Mallik, R.; Chowdhury, T.A. Metformin in cancer. Diabetes Res. Clin. Pract. 2018, 143, 409–419. [Google Scholar] [CrossRef]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Mäkelä, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J.A. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006, 29, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Cunha, V.; Cotrim, H.P.; Rocha, R.; Carvalho, K.; Lins-Kusterer, L. Metformin in the prevention of hepatocellular carcinoma in diabetic patients: A systematic review. Ann. Hepatol. 2020, 19, 232–237. [Google Scholar] [CrossRef]
- Zhou, J.; Ke, Y.; Lei, X.; Wu, T.; Li, Y.; Bao, T.; Tang, H.; Zhang, C.; Wu, X.; Wang, G.; et al. Meta-analysis: The efficacy of metformin and other anti-hyperglycemic agents in prolonging the survival of hepatocellular carcinoma patients with type 2 diabetes. Ann. Hepatol. 2020, 19, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.S.; Jonker, J.W.; Kato, Y.; Kusuhara, H.; Schinkel, A.H.; Sugiyama, Y. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J. Pharmacol. Exp. Ther. 2002, 302, 510–515. [Google Scholar] [CrossRef]
- Tanihara, Y.; Masuda, S.; Sato, T.; Katsura, T.; Ogawa, O.; Inui, K.-I. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H+-organic cation antiporters. Biochem. Pharmacol. 2007, 74, 359–371. [Google Scholar] [CrossRef]
- Chen, Y.; Teranishi, K.; Li, S.; Yee, S.W.; Hesselson, S.; Stryke, D.; Johns, S.J.; Ferrin, T.E.; Kwok, P.; Giacomini, K.M. Genetic variants in multidrug and toxic compound extrusion-1, hMATE1, alter transport function. Pharm. J. 2009, 9, 127–136. [Google Scholar] [CrossRef]
- Tsuda, M.; Terada, T.; Ueba, M.; Sato, T.; Masuda, S.; Katsura, T.; Inui, K.-I. Involvement of Human Multidrug and Toxin Extrusion 1 in the Drug Interaction between Cimetidine and Metformin in Renal Epithelial Cells. J. Pharmacol. Exp. Ther. 2009, 329, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef]
- Baur, J.A.; Birnbaum, M.J. Control of gluconeogenesis by metformin: Does redox trump energy charge? Cell Metab. 2014, 20, 197–199. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.-Q.; Lin, S.-Y.; Lin, S.-C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Mynett, K.J.; Page, T. Importance of the intestine as a site of metformin-stimulated glucose utilization. Br. J. Pharmacol. 1994, 112, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Sundelin, E.I.O.; Gormsen, L.C.; Jensen, J.B.; Vendelbo, M.H.; Jakobsen, S.; Munk, O.L.; Christensen, M.M.H.; Brøsen, K.; Frøkiær, J.; Jessen, N. Genetic Polymorphisms in Organic Cation Transporter 1 Attenuates Hepatic Metformin Exposure in Humans. Clin. Pharmacol. Ther. 2017, 102, 841–848. [Google Scholar] [CrossRef]
- Buse, J.B.; DeFronzo, R.A.; Rosenstock, J.; Kim, T.; Burns, C.; Skare, S.; Baron, A.; Fineman, M. The primary glucose-lowering effect of metformin resides in the gut, not the circulation: Results from short-term pharmacokinetic and 12-week dose-ranging studies. Diabetes Care 2016, 39, 198–205. [Google Scholar] [CrossRef]
- Massollo, M.; Marini, C.; Brignone, M.; Emionite, L.; Salani, B.; Riondato, M.; Capitanio, S.; Fiz, F.; Democrito, A.; Amaro, A.; et al. Metformin temporal and localized effects on gut glucose metabolism assessed using 18F-FDG PET in mice. J. Nucl. Med. 2013, 54, 259–266. [Google Scholar] [CrossRef]
- Preiss, D.; Dawed, A.; Welsh, P.; Heggie, A.; Jones, A.G.; Dekker, J.; Koivula, R.; Hansen, T.H.; Stewart, C.; Holman, R.R.; et al. Sustained influence of metformin therapy on circulating glucagon-like peptide-1 levels in individuals with and without type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 356–363. [Google Scholar] [CrossRef]
- Duca, F.A.; Côté, C.D.; Rasmussen, B.A.; Zadeh-Tahmasebi, M.; Rutter, G.A.; Filippi, B.M.; Lam, T.K. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med. 2015, 21, 506–511. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Krogh Pedersen, H.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.; Savinko, T.; Wong, A.K.; et al. Anti-Inflammatory Effects of Metformin Irrespective of Diabetes Status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Metformin and Other Biguanides in Oncology: Advancing the Research Agenda. Cancer Prev. Res. 2010, 3, 1060–1065. [Google Scholar] [CrossRef]
- Ma, R.; Yi, B.; Riker, A.I.; Xi, Y. Metformin and cancer immunity. Acta Pharmacol. Sin. 2020, 41, 1403–1409. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin Selectively Targets Cancer Stem Cells, and Acts Together with Chemotherapy to Block Tumor Growth and Prolong Remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.-Z.; Qu, S.-B.; Wang, D.-S. Reprogramming of glucose metabolism in hepatocellular carcinoma: Progress and prospects. World J. Gastroenterol. 2016, 22, 9933. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, F.; Tian, Y.; Cao, L.; Gao, Q.; Zhang, C.; Zhang, K.; Shen, C.; Ping, Y.; Maimela, N.R.; et al. Metformin Enhances the Antitumor Activity of CD8+ T Lymphocytes via the AMPK–miR-107–Eomes–PD-1 Pathway. J. Immunol. 2020, 204, 2575–2588. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyàs, E.; Martin-Castillo, B.; Menendez, J.A. Metformin as an archetype immuno-metabolic adjuvant for cancer immunotherapy. Oncoimmunology 2019, 8, e1633235. [Google Scholar] [CrossRef]
- Facciorusso, A. The Influence of Diabetes in the Pathogenesis and the Clinical Course of Hepatocellular Carcinoma: Recent Findings and New Perspectives. Curr. Diabetes Rev. 2013, 9, 382–386. [Google Scholar] [CrossRef]
- Facciorusso, A.; Abd El Aziz, M.A.; Singh, S.; Pusceddu, S.; Milione, M.; Giacomelli, L.; Sacco, R. Statin Use Decreases the Incidence of Hepatocellular Carcinoma: An Updated Meta-Analysis. Cancers 2020, 12, 874. [Google Scholar] [CrossRef]
- Kramer, J.R.; Natarajan, Y.; Dai, J.; Yu, X.; Li, L.; El-Serag, H.B.; Kanwal, F. Effect of diabetes medications and glycemic control on risk of hepatocellular cancer in patients with nonalcoholic fatty liver disease. Hepatology 2022, 75, 1420–1428. [Google Scholar] [CrossRef]
- Cho, Y.Y.; Yu, S.J.; Lee, H.W.; Kim, D.Y.; Kang, W.; Paik, Y.-H.; Sung, P.S.; Bae, S.H.; Park, S.C.; Doh, Y.S.; et al. Clinical Characteristics of Long-Term Survivors After Sorafenib Treatment for Unresectable Hepatocellular Carcinoma: A Korean National Multicenter Retrospective Cohort Study. J. Hepatocell. Carcinoma 2021, 8, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Ostwal, V.; Ramaswamy, A.; Gota, V.; Bhargava, P.G.; Srinivas, S.; Shriyan, B.; Jadhav, S.; Goel, M.; Patkar, S.; Mandavkar, S.; et al. Phase I Study Evaluating Dose De-escalation of Sorafenib with Metformin and Atorvastatin in Hepatocellular Carcinoma (SMASH). Oncologist 2022, 27, 165–222. [Google Scholar] [CrossRef]
- Zeng, R.W.; Yong, J.N.; Tan, D.J.H.; Fu, C.E.; Lim, W.H.; Xiao, J.; Chan, K.E.; Tan, C.; Goh, X.L.; Chee, D.; et al. Meta-analysis: Chemoprevention of hepatocellular carcinoma with statins, aspirin and metformin. Aliment. Pharmacol. Ther. 2023, 57, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Ma, J.; Wang, J.; Hao, J. The effect of metformin usage on survival outcomes for hepatocellular carcinoma patients with type 2 diabetes mellitus after curative therapy. Front. Endocrinol. 2022, 13, 12374. [Google Scholar] [CrossRef] [PubMed]
- Memel, Z.N.; Arvind, A.; Moninuola, O.; Philpotts, L.; Chung, R.T.; Corey, K.E.; Simon, T.G. Aspirin Use Is Associated with a Reduced Incidence of Hepatocellular Carcinoma: A Systematic Review and Meta-analysis. Hepatol. Commun. 2021, 5, 133–143. [Google Scholar] [CrossRef]
- Campbell, C.; Wang, T.; McNaughton, A.L.; Barnes, E.; Matthews, P.C. Risk factors for the development of hepatocellular carcinoma (HCC) in chronic hepatitis B virus (HBV) infection: A systematic review and meta-analysis. J. Viral Hepat. 2021, 28, 493–507. [Google Scholar] [CrossRef]
- Li, Q.; Xu, H.; Sui, C.; Zhang, H. Impact of metformin use on risk and mortality of hepatocellular carcinoma in diabetes mellitus. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101781. [Google Scholar] [CrossRef]
- Valenti, L.; Pelusi, S.; Aghemo, A.; Gritti, S.; Pasulo, L.; Bianco, C.; Iegri, C.; Cologni, G.; Degasperi, E.; D’ambrosio, R.; et al. Dysmetabolism, Diabetes and Clinical Outcomes in Patients Cured of Chronic Hepatitis C: A Real-Life Cohort Study. Hepatol. Commun. 2022, 6, 867–877. [Google Scholar] [CrossRef]
- Tsai, P.-C.; Kuo, H.-T.; Hung, C.-H.; Tseng, K.-C.; Lai, H.-C.; Peng, C.-Y.; Wang, J.-H.; Chen, J.-J.; Lee, P.-L.; Chien, R.-N.; et al. Metformin reduces hepatocellular carcinoma incidence after successful antiviral therapy in patients with diabetes and chronic hepatitis C in Taiwan. J. Hepatol. 2022, 78, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wong, V.W.S.; Castellanos, M.; Aller-de la Fuente, R.; Eslam, M.; Wong, G.L.-H.; George, J.; Romero-Gomez, M.; Adams, L.A. Type 2 Diabetes and Metformin Use Associate With Outcomes of Patients With Nonalcoholic Steatohepatitis–Related, Child–Pugh A Cirrhosis. Clin. Gastroenterol. Hepatol. 2021, 19, 136–145.e6. [Google Scholar] [CrossRef]
- Antwi, S.O.; Li, Z.; Mody, K.; Roberts, L.R.; Patel, T. Independent and Joint Use of Statins and Metformin by Elderly Patients with Diabetes and Overall Survival following HCC Diagnosis. J. Clin. Gastroenterol. 2020, 54, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.H.; Kao, T.Y.; Hsieh, T.H.; Kao, C.C.; Peng, C.Y.; Lai, H.C.; Chuang, P.-H.; Kao, J.-T. Prognostic roles of diabetes mellitus and hypertension in advanced hepatocellular carcinoma treated with sorafenib. PLoS ONE 2020, 15, e0244293. [Google Scholar] [CrossRef]
- Azit, N.A.; Sahran, S.; Voon Meng, L.; Subramaniam, M.K.; Mokhtar, S.; Mohammed Nawi, A. The survival outcomes and prognostic factors of hepatocellular carcinoma among type 2 diabetes patients: A two-centre retrospective cohort study. Turkish J. Med. Sci. 2022, 52, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Hydes, T.J.; Cuthbertson, D.J.; Graef, S.; Berhane, S.; Teng, M.; Skowronska, A.; Singh, P.; Dhanaraj, S.; Tahrani, A.; Johnson, P.J. The Impact of Diabetes and Glucose-Lowering Therapies on Hepatocellular Carcinoma Incidence and Overall Survival. Clin. Ther. 2022, 44, 257–268. [Google Scholar] [CrossRef]
- Cao, J.Z.; Wang, Z.G.; Yu, J.; Tao, Y.P.; Yang, Y.; Liu, H.; Zhou, W.-P.; Lu, J.; Huang, Q. Antidiabetic treatment improves prognosis after radical resection in hepatocellular carcinoma patients with diabetes mellitus: A retrospective cohort study from 2000 to 2013. J. Gastrointest. Oncol. 2022, 13, 1330–1339. [Google Scholar] [CrossRef]
- Luo, C.S.; Lin, Y.; Zhou, W.P.; Shi, J. Survival advantage associated with metformin usage in hepatocellular carcinoma patients with diabetes mellitus receiving radical resection: A propensity score matching analysis. Eur. J. Gastroenterol. Hepatol. 2020, 32, 1030–1035. [Google Scholar] [CrossRef]
- Kang, W.; Tak, E.; Hwang, S.; Song, G.; Jwa, E.; Lee, Y.; Kim, K.; Ahn, C.; Moon, D.; Ha, T.; et al. Metformin-associated Chemopreventive Effects on Recurrence After Hepatic Resection of Hepatocellular Carcinoma: From In Vitro to a Clinical Study. Anticancer. Res. 2018, 38, 2399–2407. [Google Scholar] [CrossRef]
- Chan, K.M.; Kuo, C.F.; Hsu JTe Chiou, M.J.; Wang, Y.C.; Wu, T.H.; Lee, C.F.; Wu, T.J.; Chou, H.S.; Lee, W.C. Metformin confers risk reduction for developing hepatocellular carcinoma recurrence after liver resection. Liver Int. 2017, 37, 434–441. [Google Scholar] [CrossRef]
- Chen, M.L.; Wu, C.X.; Zhang, J.B.; Zhang, H.; Sun, Y.D.; Tian, S.L.; Han, J.J. Transarterial chemoembolization combined with metformin improves the prognosis of hepatocellular carcinoma patients with type 2 diabetes. Front. Endocrinol. 2022, 13, 996228. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.J.; Jang, S.; Choi, W.J.; Park, J.; Choi, G.H.; Jang, E.S.; Jeong, S.-H.; Lee, J.H.; Yoon, C.J.; Kim, J.-W. Metformin administration is associated with enhanced response to transarterial chemoembolization for hepatocellular carcinoma in type 2 diabetes patients. Sci. Rep. 2022, 12, 14482. [Google Scholar] [CrossRef]
- Elsayed, M.; Wagstaff, W.; Behbahani, K.; Villalobos, A.; Bercu, Z.; Majdalany, B.S.; Akce, M.; Schuster, D.M.; Mao, H.; Kokabi, N. Improved Tumor Response in Patients on Metformin Undergoing Yttrium-90 Radioembolization Segmentectomy for Hepatocellular Carcinoma. Cardiovasc. Interv. Radiol. 2021, 44, 1937–1944. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.M.; Lin, C.C.; Huang, P.T.; Wen, C.F. Metformin associated with lower mortality in diabetic patients with early stage hepatocellular carcinoma after radiofrequency ablation. J. Gastroenterol. Hepatol. 2011, 26, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Yang, W.; Wu, F.; Wang, C.; Yu, L.; Tang, L.; Qiu, B.; Li, Y.; Guo, L.; Wu, M.; et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 5372–5380. [Google Scholar] [CrossRef]
- Donadon, V.; Balbi, M.; Mas, M.D.; Casarin, P.; Zanette, G. Metformin and reduced risk of hepatocellular carcinoma in diabetic patients with chronic liver disease. Liver Int. 2010, 30, 750–758. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Li, C.Q.; Liu, Z.Q.; Liu, S.S.; Zhang, G.T.; Jiang, L.; Chen, C.; Luo, D.-Q. Transcriptome Analysis of Liver Cancer Cell Huh-7 Treated With Metformin. Front. Pharmacol. 2022, 13, 822023. [Google Scholar] [CrossRef]
- Cai, X.; Hu, X.; Cai, B.; Wang, Q.; Li, Y.; Tan, X.; Hu, H.; Chen, X.; Huang, J.; Cheng, J.; et al. Metformin suppresses hepatocellular carcinoma cell growth through induction of cell cycle G1/G0 phase arrest and p21CIP and p27KIP expression and downregulation of cyclin D1 in vitro and in vivo. Oncol. Rep. 2013, 30, 2449–2457. [Google Scholar] [CrossRef]
- Miyoshi, H.; Kato, K.; Iwama, H.; Maeda, E.; Sakamoto, T.; Fujita, K.; Toyota, Y.; Tani, J.; Nomura, T.; Mimura, S.; et al. Effect of the anti-diabetic drug metformin in hepatocellular carcinoma in vitro and in vivo. Int. J. Oncol. 2014, 45, 322–332. [Google Scholar] [CrossRef]
- Cai, H.; Li, H.; Li, J.; Li, X.; Li, Y.; Shi, Y.; Wang, D. Sonic hedgehog signaling pathway mediates development of hepatocellular carcinoma. Tumour Biol. 2016, 37, 16199–16205. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Hu, Z.; Ye, J.; Liu, Y.; Lai, Z.; Zhang, M.; Ji, W.; Huang, L.; Zou, H.; Chen, B.; et al. Metformin exerts anti-tumor effects via Sonic hedgehog signaling pathway by targeting AMPK in HepG2 cells. Biochem. Cell Biol. 2022, 100, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Xia, L.; Geng, W.; Xu, D.; Zhong, C.; Zhang, J.; Xia, Q. Metformin suppresses interleukin-22 induced hepatocellular carcinoma by upregulating Hippo signaling pathway. J. Gastroenterol. Hepatol. 2021, 36, 3469–3476. [Google Scholar] [CrossRef]
- Vacante, F.; Senesi, P.; Montesano, A.; Paini, S.; Luzi, L.; Terruzzi, I. Metformin Counteracts HCC Progression and Metastasis Enhancing KLF6/p21 Expression and Downregulating the IGF Axis. Int. J. Endocrinol. 2019, 2019, 7570146. [Google Scholar] [CrossRef]
- Zhuo, H.; Miao, S.; Jin, Z.; Zhu, D.; Xu, Z.; Sun, D.; Ji, J.; Tan, Z. Metformin Suppresses Hepatocellular Carcinoma through Regulating Alternative Splicing of LGR4. J. Oncol. 2022, 2022, 1774095. [Google Scholar] [CrossRef] [PubMed]
- Wang MDa Wang, N.Y.; Zhang, H.L.; Sun, L.Y.; Xu, Q.R.; Liang, L.; Li, C.; Huang, D.-S.; Zhu, H.; Yang, T. Fatty acid transport protein-5 (FATP5) deficiency enhances hepatocellular carcinoma progression and metastasis by reprogramming cellular energy metabolism and regulating the AMPK-mTOR signaling pathway. Oncogenesis 2021, 10, 74. [Google Scholar] [CrossRef]
- Cheng, L.; Deepak, R.N.V.K.; Wang, G.; Meng, Z.; Tao, L.; Xie, M.; Chi, W.; Zhang, Y.; Yang, M.; Liao, Y.; et al. Hepatic mitochondrial NAD+ transporter SLC25A47 activates AMPKα mediating lipid metabolism and tumorigenesis. Hepatology 2023, 10, 1097. [Google Scholar] [CrossRef]
- Abdelhamid, A.M.; Saber, S.; Youssef, M.E.; Gaafar, A.G.A.; Eissa, H.; Abd-Eldayem, M.A.; Alqarni, M.; Batiha, G.E.-S.; Obaidullah, A.J.; Shahien, M.A.; et al. Empagliflozin adjunct with metformin for the inhibition of hepatocellular carcinoma progression: Emerging approach for new application. Biomed. Pharmacother 2022, 145, 112455. [Google Scholar] [CrossRef]
- Sun, R.; Zhai, R.; Ma, C.; Miao, W. Combination of aloin and metformin enhances the antitumor effect by inhibiting the growth and invasion and inducing apoptosis and autophagy in hepatocellular carcinoma through PI3K/AKT/mTOR pathway. Cancer Med. 2020, 9, 1141–1151. [Google Scholar] [CrossRef]
- Tawfik, S.M.; Abdollah, M.R.A.; Elmazar, M.M.; El-Fawal, H.A.N.; Abdelnaser, A. Effects of Metformin Combined With Antifolates on HepG2 Cell Metabolism and Cellular Proliferation. Front. Oncol. 2022, 12, 164. [Google Scholar] [CrossRef]
- Kim, T.S.; Lee, M.; Park, M.; Kim, S.Y.; Shim, M.S.; Lee, C.Y.; Choi, D.H.; Cho, Y. Metformin and dichloroacetate suppress proliferation of liver cancer cells by inhibiting mTOR complex 1. Int. J. Mol. Sci. 2021, 22, 10027. [Google Scholar] [CrossRef] [PubMed]
- Saber, S.; Ghanim, A.M.H.; El-Ahwany, E.; El-Kader, E.M.A. Novel complementary antitumour effects of celastrol and metformin by targeting IκBκB, apoptosis and NLRP3 inflammasome activation in diethylnitrosamine-induced murine hepatocarcinogenesis. Cancer Chemother. Pharmacol. 2020, 85, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhou, W.; Huang, Y.; Ren, M.; Xu, F.; Wang, H. Systemic hypoxia potentiates anti-tumor effects of metformin in hepatocellular carcinoma in mice. Acta Biochim. Biophys. Sin. 2020, 52, 421–429. [Google Scholar] [CrossRef]
- Yang, Q.; Guo, X.; Yang, L.L. Metformin enhances the effect of regorafenib and inhibits recurrence and metastasis of hepatic carcinoma after liver resection via regulating expression of hypoxia inducible factors 2α (Hif-2α) and 30 kDa HIV tat-interacting protein (TIP30). Med. Sci. Monit. 2018, 24, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Su, T.; Han, Y.; Yang, Z.; Wei, J.; Jin, L.; Fan, H. A convergent synthetic platform for dual anticancer drugs functionalized by reduced graphene nanocomposite delivery for hepatocellular cancer. Drug Deliv. 2021, 28, 1982–1994. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Harati, R.; Vandamme, M.; Blanchet, B.; Bardin, C.; Praz, F.; Hamoudi, R.A.; Desbois-Mouthon, C. Drug-drug interaction between metformin and sorafenib alters antitumor effect in hepatocellular carcinoma cells. Mol. Pharmacol. 2021, 100, 32–45. [Google Scholar] [CrossRef]
- Huang, L.; Xiao, D.; Wu, T.; Hu, X.; Deng, J.; Yan, X.; Wu, J.; Xu, S.; Yang, X.; Li, G. Phenformin synergistically sensitizes liver cancer cells to sorafenib by downregulating CRAF/ERK and PI3K/AKT/mTOR pathways. Am. J. Transl. Res. 2021, 13, 7508–7523. [Google Scholar]
- Siddharth, S.; Kuppusamy, P.; Wu, Q.; Nagalingam, A.; Saxena, N.K.; Sharma, D. Metformin Enhances the Anti-Cancer Efficacy of Sorafenib via Suppressing MAPK/ERK/Stat3 Axis in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 8083. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ling, S.; Song, L.; Fan, N.; Feng, T.; Liu, L.; Yang, X.; Wang, M.; Li, Y.; Tian, Y.; et al. Combination of metformin and sorafenib suppresses proliferation and induces autophagy of hepatocellular carcinoma via targeting the mTOR pathway. Int. J. Oncol. 2017, 50, 297–309. [Google Scholar] [CrossRef]
- Tang, K.; Chen, Q.; Liu, Y.; Wang, L.; Lu, W. Combination of Metformin and Sorafenib Induces Ferroptosis of Hepatocellular Carcinoma Through p62-Keap1-Nrf2 Pathway. J. Cancer 2022, 13, 3234–3243. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Gu, Y.K.; Li, Z.; Xu, G.Z.; Zhang, Y.M.; Dong, M.X.; Wang, Y.; Zhou, X.B. CXCR3 confers sorafenib resistance of HCC cells through regulating metabolic alteration and AMPK pathway. Am. J. Transl. Res. 2020, 12, 825–836. [Google Scholar] [PubMed]
- Cui, J.; Shen, H.M.; Lim, L.H.K. The role of autophagy in liver cancer: Crosstalk in signaling pathways and potential therapeutic targets. Pharmaceuticals 2020, 13, 432. [Google Scholar] [CrossRef]
- Song, Z.B.; Zhang, G.P.; Yu, Y.; Li, S.Q. A Prognostic Autophagy-Related Gene Pair Signature and Small-Molecule Drugs for Hepatocellular Carcinoma. Front. Genet. 2021, 12, 689801. [Google Scholar] [CrossRef]
- Lai, H.Y.; Tsai, H.H.; Yen, C.J.; Hung, L.Y.; Yang, C.C.; Ho, C.H.; Liang, H.-Y.; Chen, F.-W.; Li, C.-F.; Wang, J.-M. Metformin Resensitizes Sorafenib-Resistant HCC Cells Through AMPK-Dependent Autophagy Activation. Front. Cell Dev. Biol. 2021, 8, 596655. [Google Scholar] [CrossRef]
- Gao, C.; Fang, L.; Zhang, H.; Zhang, W.S.; Li, X.O.; Du, S.Y. Metformin induces autophagy via the ampk-mtor signaling pathway in human hepatocellular carcinoma cells. Cancer Manag. Res. 2020, 12, 5803–5811. [Google Scholar] [CrossRef]
- Papadakos, S.P.; Tsagkaris, C.; Papadakis, M.; Papazoglou, A.S.; Moysidis, D.V.; Zografos, C.G.; Theocharis, S. Angiogenesis in gastrointestinal stromal tumors: From bench to bedside. World J. Gastrointest. Oncol. 2022, 14, 1469–1477. [Google Scholar] [CrossRef]
- Niu, Z.S.; Wang, W.H.; Niu, X.J. Recent progress in molecular mechanisms of postoperative recurrence and metastasis of hepatocellular carcinoma. World J. Gastroenterol. 2022, 28, 6433–6477. [Google Scholar] [CrossRef]
- Xiong, C.; Wang, G.; Bai, D. A novel prognostic models for identifying the risk of hepatocellular carcinoma based on epithelial-mesenchymal transition-associated genes. Bioengineered 2020, 11, 1034–1146. [Google Scholar] [CrossRef]
- Zhu, G.; Xia, H.; Tang, Q.; Bi, F. An epithelial-mesenchymal transition-related 5-gene signature predicting the prognosis of hepatocellular carcinoma patients. Cancer Cell Int. 2021, 21, 166. [Google Scholar] [CrossRef]
- Gao, X.; Qiao, X.; Xing, X.; Huang, J.; Qian, J.; Wang, Y.; Zhang, Y.; Zhang, X.; Li, M.; Cui, J.; et al. Matrix Stiffness-Upregulated MicroRNA-17-5p Attenuates the Intervention Effects of Metformin on HCC Invasion and Metastasis by Targeting the PTEN/PI3K/Akt Pathway. Front. Oncol. 2020, 10, 1563. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, G.P.; Dariya, B.; Kasa, P.; Peela, S.; El-Rayes, B.F. Epigenetics in hepatocellular carcinoma. Semin. Cancer Biol. 2022, 86, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Men, Y.; Lu, L.; Geng, T.; Zhou, J.; Mitsuhashi, A.; Shozu, M.; Maihle, N.J.; Carmichael, G.G.; Taylor, H.S.; et al. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene 2017, 36, 2345–2354. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Hou, M.; Wu, Z.; Wang, J.; Zhou, M.; Zhuang, X.; Xing, J.; Tao, Q.; Huang, L.; Zhou, F.; et al. Plasma exosomal miR-122 regulates the efficacy of metformin via AMPK in type 2 diabetes and hepatocellular carcinoma. Heliyon 2022, 8, e11503. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, K.; Mitroulis, I.; Chatzigeorgiou, A.; Elefsiniotis, I.; Germanidis, G. The Liver Cancer Immune Microenvironment: Emerging Concepts for Myeloid Cell Profiling with Diagnostic and Therapeutic Implications. Cancers 2023, 15, 1522. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Fulgenzi, C.A.M.; D’Alessio, A.; Cheon, J.; Nishida, N.; Saeed, A.; Wietharn, B.; Cammarota, A.; Pressiani, T.; Personeni, N.; et al. Neutrophil-to-Lymphocyte and Platelet-to-Lymphocyte Ratios as Prognostic Biomarkers in Unresectable Hepatocellular Carcinoma Treated with Atezolizumab plus Bevacizumab. Cancers 2022, 14, 5834. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Heinrich, B.; Brown, Z.J.; Diggs, L.P.; Vormehr, M.; Ma, C.; Subramanyam, V.; Rosato, U.; Ruf, B.; Walz, J.S.; McVey, J.C.; et al. Steatohepatitis Impairs T-cell-Directed Immunotherapies Against Liver Tumors in Mice. Gastroenterology 2021, 160, 331–345.e6. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Koletsa, T.; Mitroulis, I.; Germanidis, G. Tumor-Associated Macrophages in Hepatocellular Carcinoma Pathogenesis, Prognosis and Therapy. Cancers 2022, 14, 226. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, K.; Mitroulis, I.; Germanidis, G. Tumor-associated neutrophils in hepatocellular carcinoma pathogenesis, prognosis, and therapy. Cancers 2021, 13, 2899. [Google Scholar] [CrossRef] [PubMed]
- Papadakos, S.P.; Dedes, N.; Kouroumalis, E.; Theocharis, S. The Role of the NLRP3 Inflammasome in HCC Carcinogenesis and Treatment: Harnessing Innate Immunity. Cancers 2022, 14, 3150. [Google Scholar] [CrossRef]
- Lujambio, A.; Sarobe, P. Metformin keeps CD8+ T cells active and moving in NASH-HCC immunotherapy. J. Hepatol. 2022, 77, 593–595. [Google Scholar] [CrossRef]
- Shen, Z.; Zhou, H.; Li, A.; Wu, T.; Ji, X.; Guo, L.; Zhu, X.; Zhang, D.; He, X. Metformin inhibits hepatocellular carcinoma development by inducing apoptosis and pyroptosis through regulating FOXO3. Aging 2021, 13, 22120–22133. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef]
- Montironi, C.; Castet, F.; Haber, P.K.; Pinyol, R.; Torres-Martin, M.; Torrens Fontanals, L.; Mesropian, A.; Wang, H.; Puigvehi, M.; Maeda, M.; et al. Inflamed and non-inflamed classes of HCC: A revised immunogenomic classification. Gut 2022, 72, 129–140. [Google Scholar] [CrossRef]
- Wabitsch, S.; McCallen, J.D.; Kamenyeva, O.; Ruf, B.; McVey, J.C.; Kabat, J.; Walz, J.S.; Rotman, Y.; Bauer, K.C.; Craig, A.J.; et al. Metformin treatment rescues CD8+ T-cell response to immune checkpoint inhibitor therapy in mice with NAFLD. J. Hepatol. 2022, 77, 748–760. [Google Scholar] [CrossRef]
- Kanda, Y.; Okazaki, T.; Katakai, T. Motility dynamics of T cells in tumor-draining lymph nodes: A rational indicator of antitumor response and immune checkpoint blockade. Cancers 2021, 13, 4616. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef]
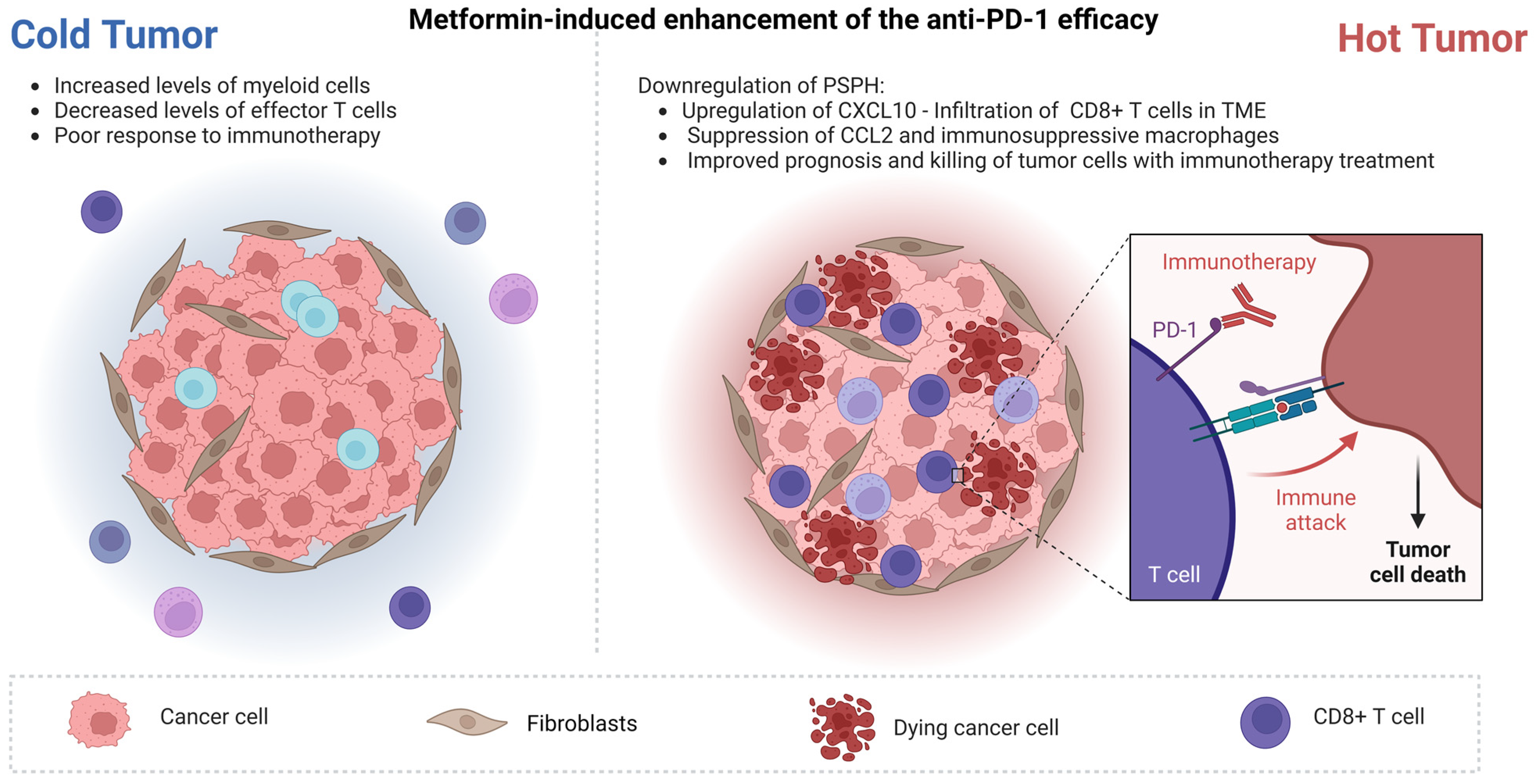
- Peng, Z.P.; Liu, X.C.; Ruan, Y.H.; Jiang, D.; Huang, A.Q.; Ning, W.R.; Jiang, Z.Z.; Zheng, L.; Wu, Y. Downregulation of phosphoserine phosphatase potentiates tumor immune environments to enhance immune checkpoint blockade therapy. J. Immunother. Cancer 2023, 11, e005986. [Google Scholar] [CrossRef]
- Gao, S.; Li, A.; Liu, F.; Chen, F.; Williams, M.; Zhang, C.; Kelley, Z.; Wu, C.-L.; Luo, R.; Xiao, H. NCOA5 Haploinsufficiency Results in Glucose Intolerance and Subsequent Hepatocellular Carcinoma. Cancer Cell 2013, 24, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Liu, X.; Zhang, Y.; Reske, J.; Bahal, D.; Gohl, T.G.; Hollern, D.; Ensink, E.; Kiupel, M.; Luo, R.; et al. NCOA5 deficiency promotes a unique liver protumorigenic microenvironment through p21WAF1/CIP1 overexpression, which is reversed by metformin. Oncogene 2020, 39, 3821–3836. [Google Scholar] [CrossRef] [PubMed]
- Rohr-Udilova, N.; Klinglmüller, F.; Schulte-Hermann, R.; Stift, J.; Herac, M.; Salzmann, M.; Finotello, F.; Timelthaler, G.; Oberhuber, G.; Pinter, M.; et al. Deviations of the immune cell landscape between healthy liver and hepatocellular carcinoma. Sci. Rep. 2018, 8, 6220. [Google Scholar] [CrossRef]
- Arelaki, S.; Koletsa, T.; Sinakos, E.; Papadopoulos, V.; Arvanitakis, K.; Skendros, P.; Akriviadis, E.; Ritis, K.; Germanidis, G.; Hytiroglou, P. Neutrophil extracellular traps enriched with IL-1β and IL-17A participate in the hepatic inflammatory process of patients with non-alcoholic steatohepatitis. Virchows Arch 2022, 481, 455–465. [Google Scholar] [CrossRef]
- Velliou, R.-I.; Mitroulis, I.; Chatzigeorgiou, A. Neutrophil extracellular traps contribute to the development of hepatocellular carcinoma in NASH by promoting Treg differentiation. Hepatobiliary Surg. Nutr. 2022, 11, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H.; Wang, Y.; Brown, Z.J.; Xia, Y.; Huang, Z.; Shen, C.; Hu, Z.; Beane, J.; Ansa-Addo, E.A.; et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 1271–1283. [Google Scholar] [CrossRef]
- Jiang, Z.-Z.; Peng, Z.-P.; Liu, X.-C.; Guo, H.-F.; Zhou, M.-M.; Jiang, D.; Ning, W.-R.; Huang, Y.-F.; Zheng, L.; Wu, Y. Neutrophil extracellular traps induce tumor metastasis through dual effects on cancer and endothelial cells. Oncoimmunology 2022, 11, 2052418. [Google Scholar] [CrossRef]
- Yang, L.Y.; Shen, X.T.; Sun, H.T.; Zhu, W.W.; Zhang, J.B.; Lu, L. Neutrophil extracellular traps in hepatocellular carcinoma are enriched in oxidized mitochondrial DNA which is highly pro-inflammatory and pro-metastatic. J. Cancer 2022, 13, 1261–1271. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Meserve, J.; Facciorusso, A.; Holmer, A.K.; Annese, V.; Sandborn, W.J.; Singh, S. Systematic review with meta-analysis: Safety and tolerability of immune checkpoint inhibitors in patients with pre-existing inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2021, 53, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Rimassa, L.; Finn, R.S.; Sangro, B. Combination immunotherapy for hepatocellular carcinoma. J. Hepatol. 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Melero, I.; Yau, T.C.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Choo, S.; Trojan, J.; Welling, T.; Meyer, T.; et al. Impact of antitumor activity on survival outcomes, and nonconventional benefit, with nivolumab (NIVO) in patients with advanced hepatocellular carcinoma (aHCC): Subanalyses of CheckMate-040. J. Clin. Oncol. 2018, 36, 475. [Google Scholar] [CrossRef]
- Kim, C.G.; Kim, C.; Yoon, S.E.; Kim, K.H.; Choi, S.J.; Kang, B.; Kim, H.R.; Park, S.-H.; Shin, E.-C.; Kim, Y.-Y.; et al. Hyperprogressive disease during PD-1 blockade in patients with advanced hepatocellular carcinoma. J. Hepatol. 2021, 74, 350–359. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Van Dao, T.; De Toni, E.N.; et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef]
- Lee, S.W.; Yang, S.S.; Lien, H.C.; Peng, Y.C.; Tung, C.F.; Lee, T.Y. The Combining of Tyrosine Kinase Inhibitors and Immune Checkpoint Inhibitors as First-Line Treatment for Advanced Stage Hepatocellular Carcinoma. J. Clin. Med. 2022, 11, 4874. [Google Scholar] [CrossRef]
- Lord, S.R.; Harris, A.L. Is it still worth pursuing the repurposing of metformin as a cancer therapeutic? Br. J. Cancer 2023, 128, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Yang, W.H.; Xia, W.; Wei, Y.; Chan, L.C.; Lim, S.O.; Li, C.W.; Kim, T.; Chang, S.S.; Lee, H.H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef]
- Chiang, C.F.; Chao, T.T.; Su, Y.F.; Hsu, C.C.; Chien, C.Y.; Chiu, K.C.; Shiah, S.G.; Lee, C.H.; Liu, S.Y.; Shieh, Y.S. Metformin-treated cancer cells modulate macrophage polarization through AMPK-NF-κB signaling. Oncotarget 2017, 8, 20706–20718. [Google Scholar] [CrossRef] [PubMed]
- Orel, V.B.; Papazoglou, A.S.; Tsagkaris, C.; Moysidis, D.V.; Papadakos, S.; Galkin, O.Y.; Orel, V.E.; Syvak, L.A. Nanotherapy based on magneto-mechanochemical modulation of tumor redox state. WIREs Nanomed. Nanobiotechnology 2022, 15, e1868. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Author; Year | Methods | Outcomes | Ref |
|---|---|---|---|
| Zhou; 2020 | Six retrospective cohort studies: four studies involving curative treatments for HCC and two studies involving noncurative treatments. The curative treatment studies: 618 patients on metformin and 532 patients on other antihyperglycemic agents, while the noncurative treatment studies: 92 patients on metformin and 57 patients on other antihyperglycemic agents. | Patients treated with metformin had significantly longer OS rates compared to those treated with other antihyperglycemic agents after curative therapies. The odds ratios (ORs) for one-year, three-year and five-year OS were 2.62 (95% confidence interval (CI): 1.76–3.90), 3.14 (95% CI: 2.33–4.24) and 3.31 (95% CI: 2.39–4.59). Similarly, metformin treatment was associated with improved recurrence-free survival (RFS) rates after curative therapies. The OR for one-year and three-year RFS were 2.52 (95% CI: 1.84–3.44) and 2.87 (95% CI: 2.15–3.84). | [20] |
| Li; 2022 | Nine case-control studies (248,433 participants) and fifteen cohort studies (1,203,832 participants) to investigate the influence of metformin usage on the risk of HCC in DM patients. Nine studies (11,375 participants) examined the impact of metformin usage on the mortality of HCC in DM patients. | Metformin was associated with a reduced risk of HCC in DM patients OR/RR 0.59 (95% CI: 0.51–0.68) with a high degree of heterogeneity (I2 = 96.5%). Metformin usage was linked to a decreased risk of all-cause mortality in individuals with DM who had been diagnosed with HCC HR 0.74 (95% CI: 0.66–0.83) with moderate heterogeneity (I2 = 49.6%). | [59] |
| Memel; 2021 | The analysis encompassed a total of 2,389,019 participants (20,479 incident HCC cases). They assessed the combined RRs and corresponding 95% CIs to assess the association between aspirin usage and the risk of incident HCC. | The beneficial effect of aspirin was notably stronger in studies that accounted for the concurrent use of statins and/or metformin (RR = 0.45, 95% CI: 0.28–0.64) compared to studies that did not consider these factors. | [57] |
| Yuan; 2022 | The effectiveness of metformin in improving the OS and RFS among HCC patients diagnosed with T2DM following curative treatment: six studies—5936 patients. | The utilization of metformin was associated with significant improvements in the 3-year (OR = 1.50, 95% CI: 1.22–1.83) and 5-year (OR = 1.88, 95% CI: 1.47–2.41) OS rates. Metformin usage was linked to reduced rates of recurrence at 1-year (OR = 1.31, 95% CI: 1.08–1.59,), 3-year (OR = 1.88, 95% CI: 1.48–2.37) and 5-year (OR = 1.83, 95% CI: 1.40–2.40) intervals. | [56] |
| Campbell; 2021 | The aim was to evaluate the risk factors contributing to the progression of HCC in individuals with chronic hepatitis B virus (HBV) infection: 68 studies, 25,447 cases of HCC and 576,792 patients with chronic HBV. | In studies that accounted for the use of metformin, the relationship between DM and an increased risk of HCC became less pronounced. In the analysis that was limited to adjusted studies for metformin, DM participants had a 16% higher risk of HCC compared to non-DM individuals (HR 1.16, 95% CI: 1.04–1.29). | [58] |
| Zeng; 2022 | The association between metformin and the risk of HCC: 3 studies, 125,458 patients. | The utilization of metformin did not demonstrate an association with a reduced overall risk of HCC (HR: 0.57, 95% CI: 0.31–1.06). | [55] |
| Author; Year | Country | Type of Study | Groups/N of Patients | Outcome | Ref |
|---|---|---|---|---|---|
| Antwi; 2019 | USA | Retrospective study | Nonusers (N = 1193) Statins Only (N = 582) Metformin Only (N = 295) Both (N = 429) | A 28% lower risk of death after HCC diagnosis (HR, 0.72; 95% CI: 0.58–0.91) associated with an average daily metformin dose of ≤ 1500 mg before diagnosis compared to nonusers. | [63] |
| HsiehI; 2020 | Taiwan | Retrospective study | Control (N = 353), DM (N = 91), HTN (N = 184) and DM + HTN (N = 105)/metformin (N = 63), OHA (n = 104) and RI/NPH (n = 29) | Control group vs. metformin group (7.70 vs. 12.60 months, p = 0.011) control group vs. non-metformin oral hypoglycemic agents group (7.70 vs. 10.80 months, p = 0.016) control group vs. insulin glargine/NPH group (7.70 vs. 15.20 months, p = 0.026). | [64] |
| Azit; 2022 | Malaysia | Retrospective study | Metformin (Ν = 130), insulin (Ν = 71), sulphonylureas (Ν = 90) | Patients on metformin had a 1.44-times greater risk of mortality (AHR = 1.44, 95% CI: 1.03–2.00). | [65] |
| Hydes; 2022 | UK | Retrospective study | Diet (Ν = 91), metformin (Ν = 171) thiazolidinedione (Ν= 1), DPP4 inhibitor (Ν= 7), sulfonylureas (Ν = 117), insulin (N = 126) | In diabetic patients with HCC, metformin was linked to a better OS, as evidenced by a mean survival of 31 months vs. 24 months (p = 0.016) and a hazard ratio (HR) for death of 0.75 (p = 0.032). | [66] |
| Author; Year | Country | Type of Study | Groups | N of Patients | Overall Survival (%) | Ref | ||
|---|---|---|---|---|---|---|---|---|
| 1-Year | 3-Year | 5-Year | ||||||
| Curative intent surgery | ||||||||
| Cao; 2022 | China | RSC | Antidiabetic treatment (metformin or insulin or both) vs. no treatment | 292 vs. 106 | 82.7 vs. 80.1 | 65.6 vs. 58.7 | 46.4 vs. 29.2 | [67] |
| Luo; 2019 | China | RSC | Metformin vs. other | 63 vs. 113 | 94 vs. 77 | 76 vs. 49 | 53 vs. 29 | [68] |
| Kang; 2018 | South Korea | RSC | Metformin vs. other | 45 vs. 225 | NA | 97.8 vs. 89.5 | 83.2 vs. 67.8 | [69] |
| Chan; 2017 | Taiwan | ND | Metformin vs. other | 1632 vs. 2978 | Among DM, those on metformin had significantly improved RFS and OS. | [70] | ||
| A reduction in the risk of HCC recurrence after liver resection was positively correlated with the amount and duration of metformin use. | ||||||||
| RFA/SBRT/ HFRT/ Radioembolization/ TACE | ||||||||
| Chen; 2022 | China | RSC | Metformin vs. other | 39 matched | 43 vs. 35 months | [71] | ||
| Jung; 2022 | South Korea | RSC | Metformin vs. other | 47 vs. 47 | No difference. | [72] | ||
| Elsayed; 2021 | USA | RSC | Metformin vs. other | 19 vs. 93 | NA | 38.2 months vs. 40.3 months | NA | [73] |
| Chen; 2011 | Taiwan | RSC | Metformin vs. other | 21 vs. 32 | 95 vs. 74.5 | 69.2 vs. 44.8 | 60.5 vs. 26.2 | [74] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadakos, S.P.; Ferraro, D.; Carbone, G.; Frampton, A.E.; Vennarecci, G.; Kykalos, S.; Schizas, D.; Theocharis, S.; Machairas, N. The Emerging Role of Metformin in the Treatment of Hepatocellular Carcinoma: Is There Any Value in Repurposing Metformin for HCC Immunotherapy? Cancers 2023, 15, 3161. https://doi.org/10.3390/cancers15123161
Papadakos SP, Ferraro D, Carbone G, Frampton AE, Vennarecci G, Kykalos S, Schizas D, Theocharis S, Machairas N. The Emerging Role of Metformin in the Treatment of Hepatocellular Carcinoma: Is There Any Value in Repurposing Metformin for HCC Immunotherapy? Cancers. 2023; 15(12):3161. https://doi.org/10.3390/cancers15123161
Chicago/Turabian StylePapadakos, Stavros P., Daniele Ferraro, Gabriele Carbone, Adam Enver Frampton, Giovanni Vennarecci, Stylianos Kykalos, Dimitrios Schizas, Stamatios Theocharis, and Nikolaos Machairas. 2023. "The Emerging Role of Metformin in the Treatment of Hepatocellular Carcinoma: Is There Any Value in Repurposing Metformin for HCC Immunotherapy?" Cancers 15, no. 12: 3161. https://doi.org/10.3390/cancers15123161
APA StylePapadakos, S. P., Ferraro, D., Carbone, G., Frampton, A. E., Vennarecci, G., Kykalos, S., Schizas, D., Theocharis, S., & Machairas, N. (2023). The Emerging Role of Metformin in the Treatment of Hepatocellular Carcinoma: Is There Any Value in Repurposing Metformin for HCC Immunotherapy? Cancers, 15(12), 3161. https://doi.org/10.3390/cancers15123161