

Polygenic Risk Score Predicts Modified Risk in BRCA1 Pathogenic Variant c.4035del and c.5266dup Carriers in Breast Cancer Patients
 , ,
, ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Genotyping with OncoArray
2.3. Genotype Calling and Quality Control (QC)
2.4. Genotype Imputation
2.5. Polygenic Risk Score (PRS) Calculations
2.6. Statistical Analysis
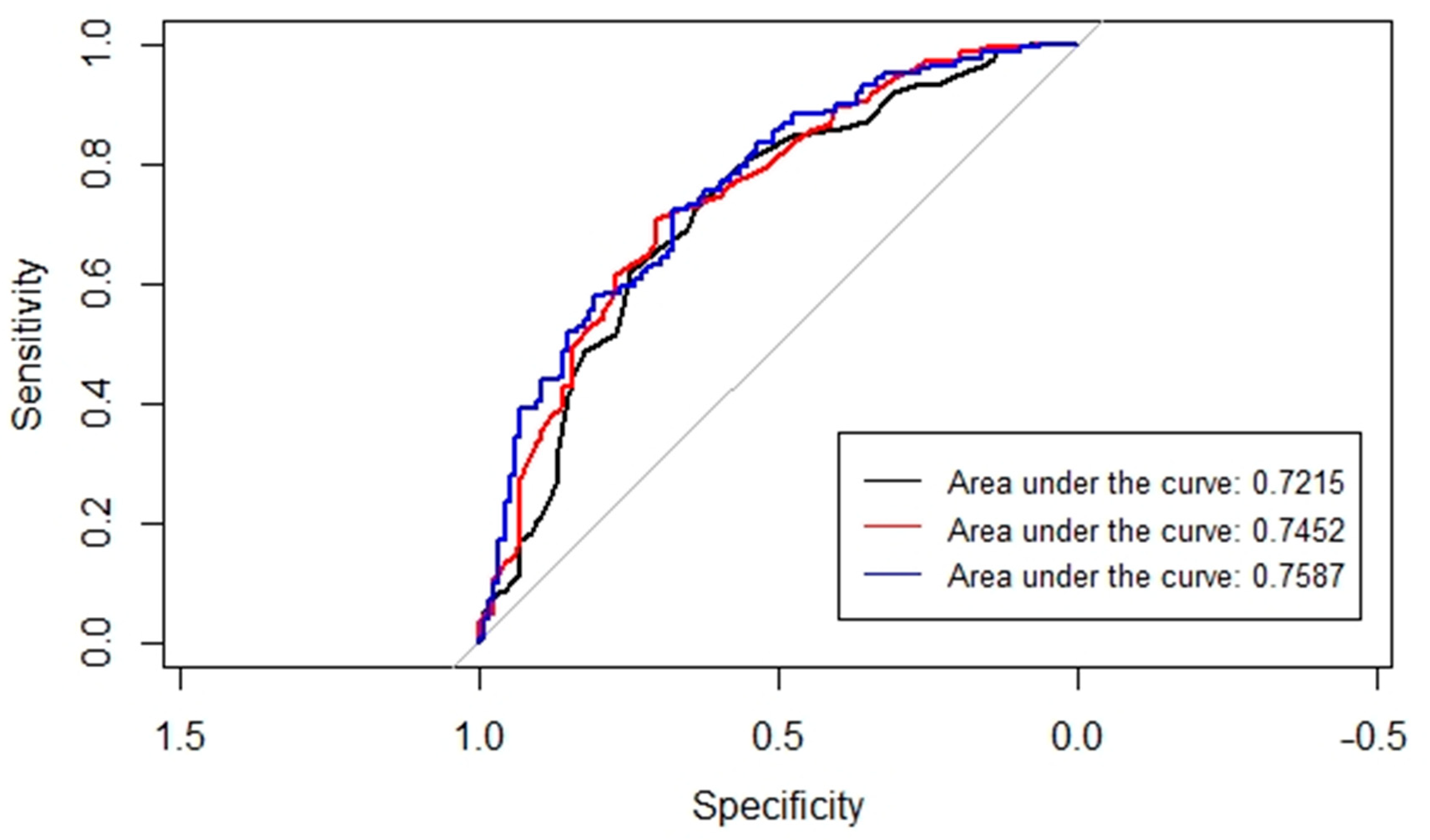
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mavaddat, N.; Michailidou, K.; Dennis, J.; Lush, M.; Fachal, L.; Lee, A.; Tyrer, J.P.; Chen, T.H.; Wang, Q.; Bolla, M.K.; et al. Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am. J. Hum. Genet. 2019, 104, 21–34. [Google Scholar] [CrossRef] [PubMed]
- SPKC. Statistikas Dati. Slimību Profilakses un Kontroles Centrs. Available online: https://www.spkc.gov.lv/lv/statistikas-dati (accessed on 3 March 2023).
- Dareng, E.O.; Tyrer, J.P.; Barnes, D.R.; Jones, M.R.; Yang, X.; Aben, K.K.H.; Adank, M.A.; Agata, S.; Andrulis, I.L.; Anton-Culver, H.; et al. Polygenic risk modeling for prediction of epithelial ovarian cancer risk. Eur. J. Hum. Genet. 2022, 30, 349–362. [Google Scholar] [CrossRef]
- Jürgens, H.; Roht, L.; Leitsalu, L.; Nõukas, M.; Palover, M.; Nikopensius, T.; Reigo, A.; Kals, M.; Kallak, K.; Kütner, R.; et al. Precise, Genotype-First Breast Cancer Prevention: Experience with Transferring Monogenic Findings from a Population Biobank to the Clinical Setting. Front. Genet. 2022, 13, 881100. [Google Scholar] [CrossRef] [PubMed]
- Borde, J.; Laitman, Y.; Blümcke, B.; Niederacher, D.; Weber-Lassalle, K.; Sutter, C.; Rump, A.; Arnold, N.; Wang-Gohrke, S.; Horváth, J.; et al. Polygenic risk scores indicate extreme ages at onset of breast cancer in female BRCA1/2 pathogenic variant carriers. BMC Cancer 2022, 22, 706. [Google Scholar] [CrossRef]
- Barnes, D.R.; Rookus, M.A.; McGuffog, L.; Leslie, G.; Mooij, T.M.; Dennis, J.; Mavaddat, N.; Adlard, J.; Ahmed, M.; Aittomäki, K.; et al. Polygenic risk scores and breast and epithelial ovarian cancer risks for carriers of BRCA1 and BRCA2 pathogenic variants. Genet. Med. 2020, 22, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef]
- Gardovskis, A.; Irmejs, A.; Miklasevics, E.; Borosenko, V.; Bitina, M.; Melbarde-Gorkusa, I.; Vanags, A.; Kurzawski, G.; Suchy, J.; Górski, B.; et al. Clinical, molecular and geographical features of hereditary breast/ovarian cancer in latvia. Hered. Cancer Clin. Pract. 2005, 3, 71–76. [Google Scholar] [CrossRef]
- Tikhomirova, L.; Sinicka, O.; Smite, D.; Eglitis, J.; Hodgson, S.V.; Stengrevics, A. High prevalence of two BRCA1 mutations, 4154delA and 5382insC, in Latvia. Fam. Cancer 2005, 4, 77–84. [Google Scholar] [CrossRef]
- Tamboom, K.; Kaasik, K.; Aršavskaja, J.; Tekkel, M.; Lilleorg, A.; Padrik, P.; Metspalu, A.; Veidebaum, T. BRCA1 mutations in women with familial or early-onset breast cancer and BRCA2 mutations in familial cancer in Estonia. Hered. Cancer Clin. Pract. 2010, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Janavičius, R.; Rudaitis, V.; Mickys, U.; Elsakov, P.; Griškevičius, L. Comprehensive BRCA1 and BRCA2 mutational profile in Lithuania. Cancer Genet. 2014, 207, 195–205. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; McGuffog, L.; Barrowdale, D.; Lee, A.; Soucy, P.; Dennis, J.; Domchek, S.M.; Robson, M.; Spurdle, A.B.; Ramus, S.J.; et al. Evaluation of Polygenic Risk Scores for Breast and Ovarian Cancer Risk Prediction in BRCA1 and BRCA2 Mutation Carriers. J. Natl. Cancer Inst. 2017, 109, djw302. [Google Scholar] [CrossRef]
- Chen, J.; Bae, E.; Zhang, L.; Hughes, K.; Parmigiani, G.; Braun, D.; Rebbeck, T.R. Penetrance of Breast and Ovarian Cancer in Women Who Carry a. JNCI Cancer Spectr. 2020, 4, pkaa029. [Google Scholar] [CrossRef]
- Lavoro, A.; Scalisi, A.; Candido, S.; Zanghì, G.N.; Rizzo, R.; Gattuso, G.; Caruso, G.; Libra, M.; Falzone, L. Identification of the most common BRCA alterations through analysis of germline mutation databases: Is droplet digital PCR an additional strategy for the assessment of such alterations in breast and ovarian cancer families? Int. J. Oncol. 2022, 60, 58. [Google Scholar] [CrossRef]
- Lee, A.; Moon, B.I.; Kim, T.H. BRCA1/BRCA2 Pathogenic Variant Breast Cancer: Treatment and Prevention Strategies. Ann. Lab. Med. 2020, 40, 114–121. [Google Scholar] [CrossRef]
- Doraczynska-Kowalik, A.; Michalowska, D.; Matkowski, R.; Czykalko, E.; Blomka, D.; Semeniuk, M.; Abrahamowska, M.; Janus-Szymanska, G.; Mlynarczykowska, P.; Szynglarewicz, B.; et al. Detection of BRCA1/2 pathogenic variants in patients with breast and/or ovarian cancer and their families. Analysis of 3458 cases from Lower Silesia (Poland) according to the diagnostic algorithm of the National Cancer Control Programme. Front. Genet. 2022, 13, 941375. [Google Scholar] [CrossRef]
- Mars, N.; Widén, E.; Kerminen, S.; Meretoja, T.; Pirinen, M.; Della Briotta Parolo, P.; Palta, P.; Palotie, A.; Kaprio, J.; Joensuu, H.; et al. The role of polygenic risk and susceptibility genes in breast cancer over the course of life. Nat. Commun. 2020, 11, 6383. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; Vries, J.D.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-wide association studies. Nat. Rev. Methods Prim. 2021, 1, 59. [Google Scholar] [CrossRef]
- Wang, Y.; Namba, S.; Lopera, E.; Kerminen, S.; Tsuo, K.; Läll, K.; Kanai, M.; Zhou, W.; Wu, K.-H.; Favé, M.-J.; et al. Global Biobank analyses provide lessons for developing polygenic risk scores across diverse cohorts. Cell Genom. 2023, 3, 100241. [Google Scholar] [CrossRef]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef]
- Szabo, C.; Masiello, A.; Ryan, J.F.; Brody, L.C. The breast cancer information core: Database design, structure, and scope. Hum. Mutat. 2000, 16, 123–131. [Google Scholar] [CrossRef]
- Orliac, E.J.; Trejo Banos, D.; Ojavee, S.E.; Läll, K.; Mägi, R.; Visscher, P.M.; Robinson, M.R. Improving GWAS discovery and genomic prediction accuracy in biobank data. Proc. Natl. Acad. Sci. USA 2022, 119, e2121279119. [Google Scholar] [CrossRef]
- Ojavee, S.E.; Kousathanas, A.; Trejo Banos, D.; Orliac, E.J.; Patxot, M.; Läll, K.; Mägi, R.; Fischer, K.; Kutalik, Z.; Robinson, M.R. Genomic architecture and prediction of censored time-to-event phenotypes with a Bayesian genome-wide analysis. Nat. Commun. 2021, 12, 2337. [Google Scholar] [CrossRef]
- Patxot, M.; Banos, D.T.; Kousathanas, A.; Orliac, E.J.; Ojavee, S.E.; Moser, G.; Holloway, A.; Sidorenko, J.; Kutalik, Z.; Mägi, R.; et al. Probabilistic inference of the genetic architecture underlying functional enrichment of complex traits. Nat. Commun. 2021, 12, 6972. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Guo, Y.; He, J.; Zhao, S.; Wu, H.; Zhong, X.; Sheng, Q.; Samuels, D.C.; Shyr, Y.; Long, J. Illumina human exome genotyping array clustering and quality control. Nat. Protoc. 2014, 9, 2643–2662. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Mitt, M.; Kals, M.; Pärn, K.; Gabriel, S.B.; Lander, E.S.; Palotie, A.; Ripatti, S.; Morris, A.P.; Metspalu, A.; Esko, T.; et al. Improved imputation accuracy of rare and low-frequency variants using population-specific high-coverage WGS-based imputation reference panel. Eur. J. Hum. Genet. 2017, 25, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Loh, P.R.; Palamara, P.F.; Price, A.L. Fast and accurate long-range phasing in a UK Biobank cohort. Nat. Genet. 2016, 48, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 27 May 2023).
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio, PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com/ (accessed on 27 May 2023).
- Carstensen, B.; Plummer, M.; Laara, E.; Hills, M. R Package, Version 2.46; Epi: A Package for Statistical Analysis in Epidemiology; Oxford University Press: Oxford, UK, 2022; Available online: https://CRAN.R-project.org/package=Epi (accessed on 22 May 2023).
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Metcalfe, K.; Sun, P.; Hanna, W.M.; Lynch, H.T.; Ghadirian, P.; Tung, N.; Olopade, O.I.; Weber, B.L.; McLennan, J.; et al. Estrogen receptor status in BRCA1- and BRCA2-related breast cancer: The influence of age, grade, and histological type. Clin. Cancer Res. 2004, 10, 2029–2034. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Au, W.W.; Henderson, B.R. Cytoplasmic mislocalization of BRCA1 caused by cancer-associated mutations in the BRCT domain. Exp. Cell. Res. 2004, 293, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Perrin-Vidoz, L.; Sinilnikova, O.M.; Stoppa-Lyonnet, D.; Lenoir, G.M.; Mazoyer, S. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum. Mol. Genet. 2002, 11, 2805–2814. [Google Scholar] [CrossRef] [PubMed]
- Plakhins, G.; Irmejs, A.; Gardovskis, A.; Subatniece, S.; Rozite, S.; Bitina, M.; Keire, G.; Purkalne, G.; Teibe, U.; Trofimovics, G.; et al. Genotype-phenotype correlations among BRCA1 4153delA and 5382insC mutation carriers from Latvia. BMC Med. Genet. 2011, 12, 147. [Google Scholar] [CrossRef]
- Rovite, V.; Wolff-Sagi, Y.; Zaharenko, L.; Nikitina-Zake, L.; Grens, E.; Klovins, J. Genome Database of the Latvian Population (LGDB): Design, Goals, and Primary Results. J. Epidemiol. 2018, 28, 353–360. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Total | BRCA1:c.4035del | BRCA1:c.5266dup | |
|---|---|---|---|
| Study sample | 406 | 161 (39.66%) | 245 (60.34%) |
| Breast cancer | 171 (42.12%) | 49 (12.07%) | 122 (30.05%) |
| Ovarian cancer | 121 (29.80%) | 64 (15.76%) | 57 (14.04%) |
| Controls | 114 (28.08%) | 48 (11.82%) | 66 (16.26%) |
| Mean age | 45.38 | 47.55 | 43.52 |
| Breast cancer | 46.67 | 49.53 | 45.52 |
| Ovarian cancer | 50.55 | 51.53 | 49.44 |
| Controls | 37.98 | 40.61 | 36.28 |
| Score | Description |
|---|---|
| score1 | The weighted effect size calculated in BC patients with the BayesW model |
| score2 | The weighted effect size calculated in BC patients with the BayesRR-RC model |
| score3 | The weighted effect size calculated in OC patients with the BayesW model |
| score4 | The weighted effect size calculated in OC patients with the BayesRR-RC model |
| OR | 95% CI | p Value | |
|---|---|---|---|
| BC + OC vs. Controls | |||
| score1 | 1.14 | 0.89–1.46 | 0.3119 |
| score2 | 1.11 | 0.86–1.42 | 0.4205 |
| score3 | 1.00 | 0.78–1.28 | 0.9781 |
| score4 | 0.89 | 0.69–1.14 | 0.3514 |
| BRCA1:c.5266dup | 1.73 | 1.03–2.91 | 0.0375 * |
| BC vs. Controls | |||
| score1 | 1.37 | 1.03–1.81 | 0.0291 * |
| score2 | 1.33 | 1.01–1.76 | 0.0423 * |
| score3 | 1.00 | 0.76–1.31 | 0.9825 |
| score4 | 0.95 | 0.72–1.25 | 0.7109 |
| BRCA1:c.5266dup | 2.55 | 1.44–4.53 | 0.0013 ** |
| OC vs. Controls | |||
| score1 | 0.94 | 0.68–1.31 | 0.7180 |
| score2 | 0.91 | 0.65–1.27 | 0.5800 |
| score3 | 0.99 | 0.71–1.38 | 0.9530 |
| score4 | 0.81 | 0.57–1.14 | 0.2250 |
| BRCA1:c.5266dup | 0.93 | 0.48–1.79 | 0.8170 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berga-Švītiņa, E.; Maksimenko, J.; Miklaševičs, E.; Fischer, K.; Vilne, B.; Mägi, R. Polygenic Risk Score Predicts Modified Risk in BRCA1 Pathogenic Variant c.4035del and c.5266dup Carriers in Breast Cancer Patients. Cancers 2023, 15, 2957. https://doi.org/10.3390/cancers15112957
Berga-Švītiņa E, Maksimenko J, Miklaševičs E, Fischer K, Vilne B, Mägi R. Polygenic Risk Score Predicts Modified Risk in BRCA1 Pathogenic Variant c.4035del and c.5266dup Carriers in Breast Cancer Patients. Cancers. 2023; 15(11):2957. https://doi.org/10.3390/cancers15112957
Chicago/Turabian StyleBerga-Švītiņa, Egija, Jeļena Maksimenko, Edvīns Miklaševičs, Krista Fischer, Baiba Vilne, and Reedik Mägi. 2023. "Polygenic Risk Score Predicts Modified Risk in BRCA1 Pathogenic Variant c.4035del and c.5266dup Carriers in Breast Cancer Patients" Cancers 15, no. 11: 2957. https://doi.org/10.3390/cancers15112957
APA StyleBerga-Švītiņa, E., Maksimenko, J., Miklaševičs, E., Fischer, K., Vilne, B., & Mägi, R. (2023). Polygenic Risk Score Predicts Modified Risk in BRCA1 Pathogenic Variant c.4035del and c.5266dup Carriers in Breast Cancer Patients. Cancers, 15(11), 2957. https://doi.org/10.3390/cancers15112957