

Dermatologic Manifestations of Neurofibromatosis Type 1 and Emerging Treatments
 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
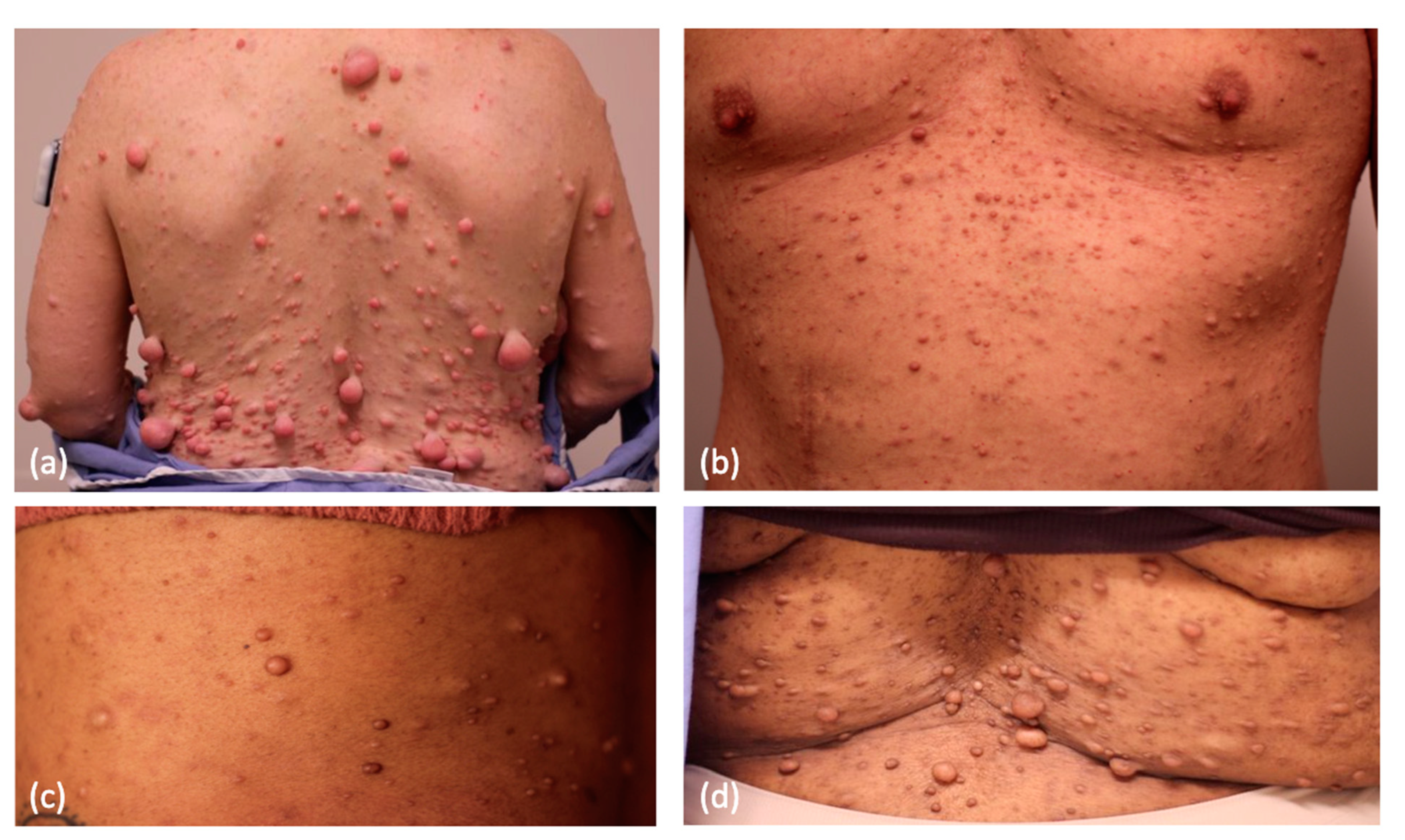
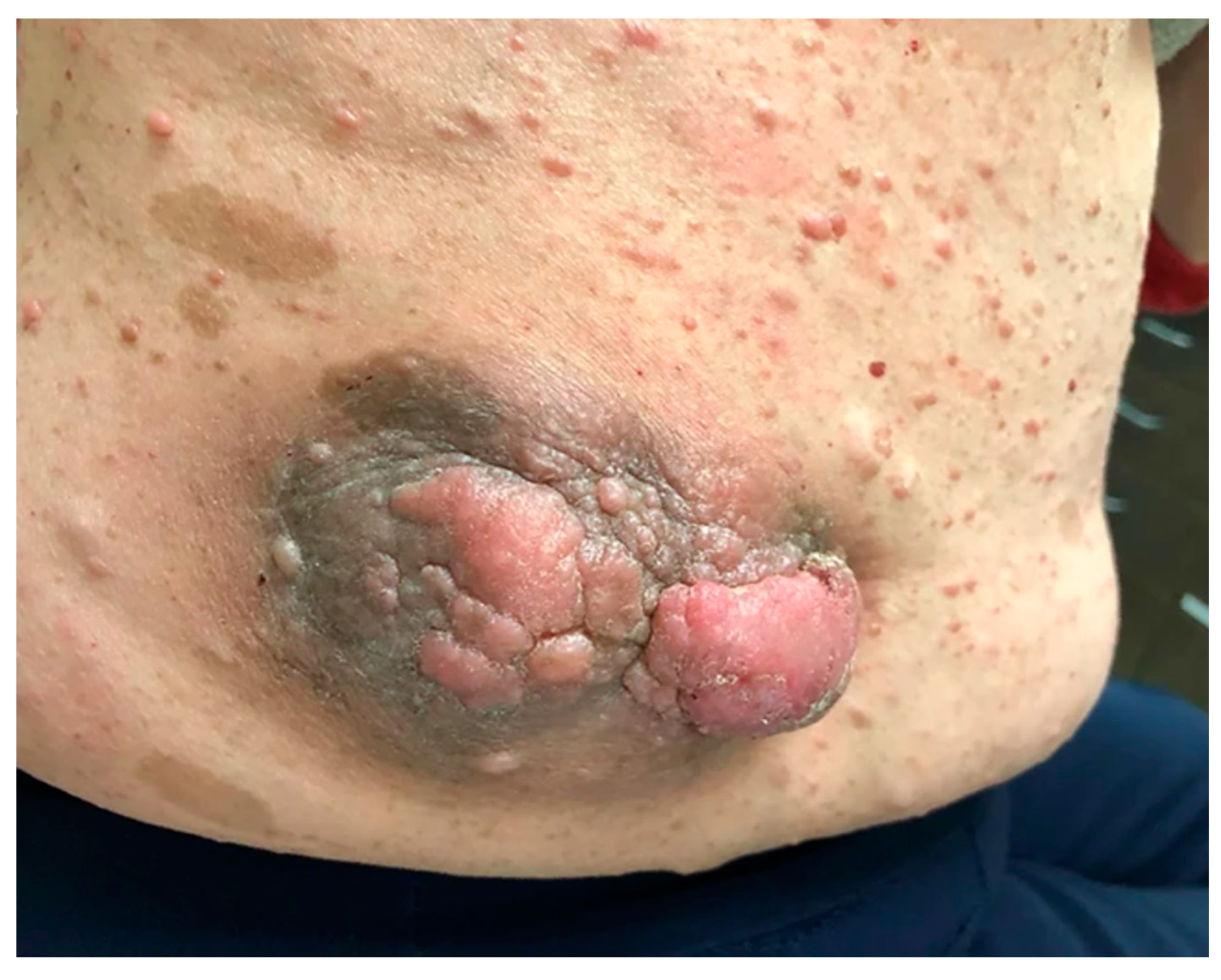
2. Cutaneous Neurofibroma
3. Systemic Therapies
4. Topical Treatments
5. Surgical Intervention
6. Electrodessication
7. Laser-Based Interventions
8. Light-Based Interventions
9. Plexiform, Diffuse, and Distinct Nodular Neurofibromas
10. Malignant Peripheral Nerve Sheath Tumor
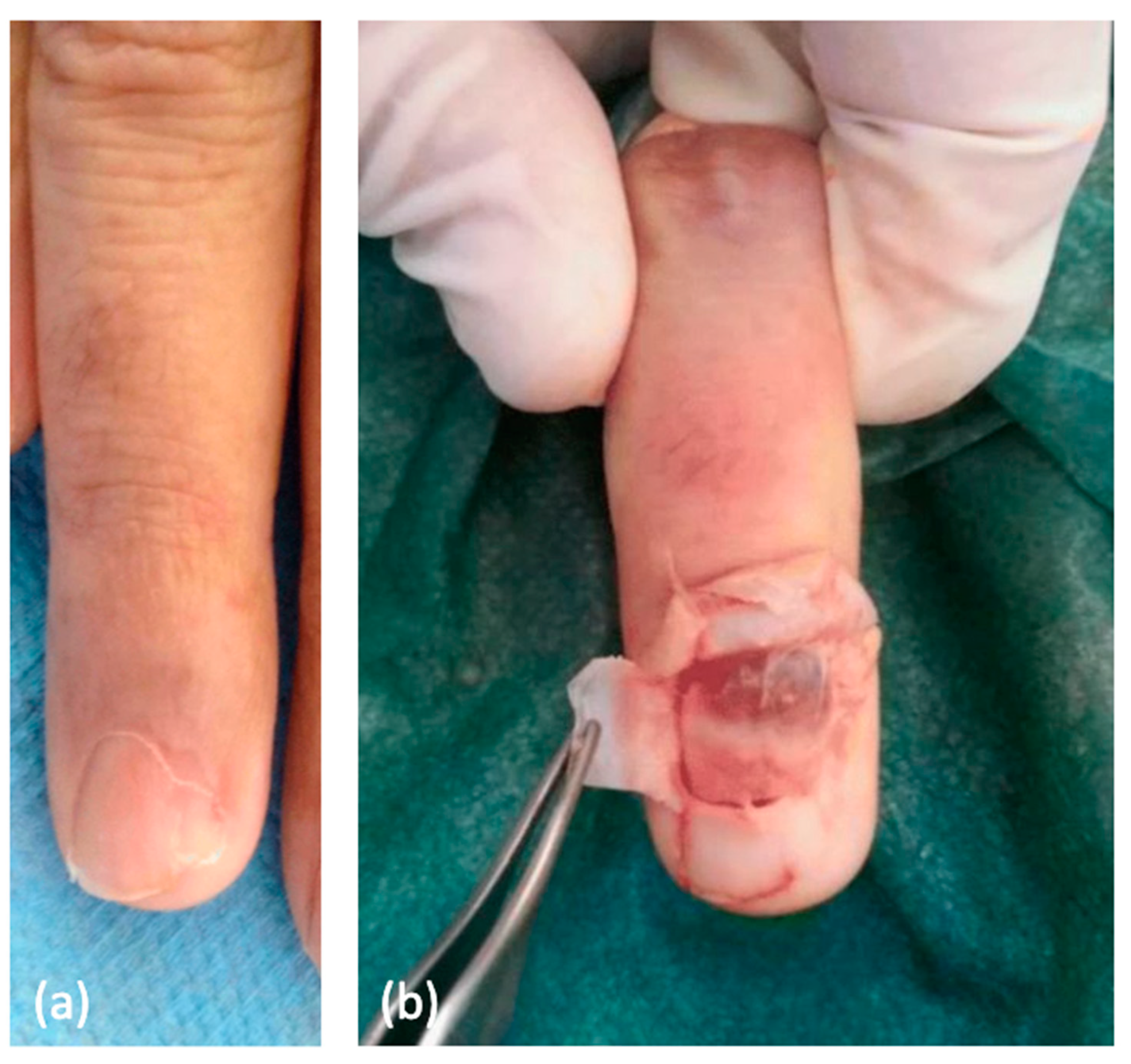
11. Glomus Tumor
12. Juvenile Xanthogranuloma
13. Skin Cancer
14. Cutaneous T Cell Lymphoma
15. Wound Healing and Scarring
16. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kresak, J.L.; Walsh, M. Neurofibromatosis: A Review of NF1, NF2, and Schwannomatosis. J. Pediatr. Genet. 2016, 5, 98–104. [Google Scholar] [CrossRef]
- Yap, Y.S.; McPherson, J.R.; Ong, C.K.; Rozen, S.G.; Teh, B.T.; Lee, A.S.; Callen, D.F. The NF1 gene revisited—From bench to bedside. Oncotarget 2014, 5, 5873–5892. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.N.; John, A.M.; Handler, M.Z.; Schwartz, R.A. Neurofibromatosis type 1: New developments in genetics and treatment. J. Am. Acad. Dermatol. 2021, 84, 1667–1676. [Google Scholar] [CrossRef]
- Shapira, S.; Barkan, B.; Fridman, E.; Kloog, Y.; Stein, R. The tumor suppressor neurofibromin confers sensitivity to apoptosis by Ras-dependent and Ras-independent pathways. Cell Death Differ. 2007, 14, 895–906. [Google Scholar] [CrossRef]
- Arima, Y.; Hayashi, H.; Kamata, K.; Goto, T.M.; Sasaki, M.; Kuramochi, A.; Saya, H. Decreased expression of neurofibromin contributes to epithelial–mesenchymal transition in neurofibromatosis type 1. Exp. Dermatol. 2010, 19, e136–e141. [Google Scholar] [CrossRef]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet. Med. 2021, 23, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M. Neurofibromatosis 1. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1109/ (accessed on 8 February 2023).
- Wang, S.; Liechty, B.; Patel, S.; Weber, J.S.; Hollmann, T.J.; Snuderl, M.; Karajannis, M.A. Programmed death ligand 1 expression and tumor infiltrating lymphocytes in neurofibromatosis type 1 and 2 associated tumors. J. Neurooncol. 2018, 138, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Cipolletta, S.; Spina, G.; Spoto, A. Psychosocial functioning, self-image, and quality of life in children and adolescents with neurofibromatosis type 1. Child Care Health Dev. 2018, 44, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Leidger, A.; Vosschulte, M.; Nieder, T.O.; Mautner, V.F. Sexual Self-Esteem and Psychological Burden of Adults With Neurofibromatosis Type 1. Front. Psychol. 2022, 13, 883019. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.R.; Korf, B.R.; Nathanson, K.L.; Stevenson, D.A.; Yohay, K. Care of adults with neurofibromatosis type 1: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Brenaut, E.; Nizery-Guermeur, C.; Audebert-Bellanger, S.; Ferkal, S.; Wolkenstein, P.; Misery, L.; Abasq-Thomas, C. Clinical Characteristics of Pruritus in Neurofibromatosis 1. Acta Derm. Venereol. 2016, 96, 398–399. [Google Scholar] [CrossRef]
- Dugoff, L.; Sujansky, E. Neurofibromatosis type 1 and pregnancy. Am. J. Med. Genet. 1996, 66, 7–10. [Google Scholar] [CrossRef]
- McLaughlin, M.E.; Jacks, T. Progesterone receptor expression in neurofibromas. Cancer Res. 2003, 63, 752–755. [Google Scholar] [PubMed]
- Lammert, M.; Mautner, V.F.; Kluwe, L. Do hormonal contraceptives stimulate growth of neurofibromas? A survey on 59 NF1 patients. BMC Cancer 2005, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Well, L.; Jaeger, A.; Kehrer-Sawatzki, H.; Farschtschi, S.; Avanesov, M.; Sauer, M.; De Sousa, M.T.; Bannas, P.; Derlin, T.; Adam, G.; et al. The effect of pregnancy on growth-dynamics of neurofibromas in Neurofibromatosis type 1. PLoS ONE 2020, 15, e0232031. [Google Scholar] [CrossRef]
- Slopis, J.M.; Arevalo, O.; Bell, C.S.; Hebert, A.A.; Northrup, H.; Riascos, R.F.; Samuels, J.A.; Smith, K.C.; Tate, P.; Koenig, M.K. Treatment of Disfiguring Cutaneous Lesions in Neurofibromatosis-1 with Everolimus: A Phase II, Open-Label, Single-Arm Trial. Drugs RD 2018, 18, 295–302. [Google Scholar] [CrossRef]
- Chhabra, G.; Verma, P. Successful treatment of cutaneous (dermal) neurofibromas with imatinib mesylate. Dermatol. Ther. 2020, 33, e13262. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.; Zehou, O.; Ducassou, S.; Minard-Colin, V.; Hamel-Teillac, D.; Wolkenstein, P.; Valeyrie-Allanore, L. Sirolimus Improves Pain in NF1 Patients with Severe Plexiform Neurofibromas. Pediatrics 2014, 133, e1792–e1797. [Google Scholar] [CrossRef]
- Casey, D.; Demko, S.; Sinha, A.; Mishra-Kalyani, P.S.; Shen, Y.-L.; Khasar, S.; Goheer, M.A.; Helms, W.S.; Pan, L.; Xu, Y.; et al. FDA Approval Summary: Selumetinib for Plexiform Neurofibroma. Clin. Cancer Res. 2021, 27, 4142–4146. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.M.; Wolters, P.; Baldwin, A.; Dombi, E.; Fisher, M.J.; Weiss, B.D.; Kim, A.; Blakeley, J.O.; Whitcomb, P.; Holmblad, M.; et al. SPRINT: Phase II study of the MEK 1/2 inhibitor selumetinib (AZD6244, ARRY-142886) in children with neurofibromatosis type 1 (NF1) and inoperable plexiform neurofibromas (PN). JCO 2018, 36 (Suppl. 15), 10503. [Google Scholar] [CrossRef]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of Selumetinib in Neurofibromatosis Type 1–Related Plexiform Neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef]
- National Cancer Institute (NCI). Pilot Study of the MEK1/2 Inhibitor Selumetinib (AZD6244 Hydrogen Sulfate) for Adults with Neurofibromatosis Type 1 (NF1) and Cutaneous Neurofibromas (CNF). 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT02839720 (accessed on 23 March 2023).
- PhD SRP MD. Pilot Study of Topical Imiquimod 5% Cream for Treatment of Cutaneous Neurofibromas in Adults with Neurofibromatosis 1. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT00865644 (accessed on 20 February 2023).
- Chamseddin, B.H.; Le, L.Q. Management of cutaneous neurofibroma: Current therapy and future directions. Neurooncol. Adv. 2020, 2 (Suppl. 1), i107–i116. [Google Scholar] [CrossRef] [PubMed]
- NFlection Therapeutics, Inc. A Randomized, Double-Blind, Vehicle-Controlled, Parallel Group Phase 2a Study to Determine Safety, Tolerability, Pharmacokinetics, and Pharmacodynamic Activity of NFX-179 Gel in Subjects with Cutaneous Neurofibromas. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04435665 (accessed on 13 April 2023).
- NFlection Therapeutics, Inc. A Randomized, Double-Blind, Vehicle-Controlled, Parallel Group Phase 2 Dose-Response Study to Determine Safety and Effectiveness of Two Concentrations of NFX-179 Gel in Subjects with Cutaneous Neurofibromas. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT05005845 (accessed on 13 April 2023).
- Gulati, N. A Phase I, Open Label Study Employing the Topical Immunomodulator Diphencyprone to Treat Cutaneous Neurofibromas Associated with NF1. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT05438290 (accessed on 7 February 2023).
- Chamseddin, B.H.; Hernandez, L.; Solorzano, D.; Vega, J.; Le, L.Q. Robust surgical approach for cutaneous neurofibroma in neurofibromatosis type 1. JCI Insight 2019, 4, e128881. [Google Scholar] [CrossRef]
- Lutterodt, C.G.; Mohan, A.; Kirkpatrick, N. The use of electrodessication in the treatment of cutaneous neurofibromatosis: A retrospective patient satisfaction outcome assessment. J. Plast. Reconstr. Aesthet. Surg. 2016, 69, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Roenigk, R.K.; Ratz, J.L. CO2 laser treatment of cutaneous neurofibromas. J. Dermatol. Surg. Oncol. 1987, 13, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.W.J. Use of the Carbon Dioxide Laser in Treating Multiple Cutaneous Neurofibromas. Ann. Plast. Surg. 1991, 26, 582. [Google Scholar] [CrossRef]
- Chiang, Y.Z.; Al-Niaimi, F.; Ferguson, J.; August, P.J.; Madan, V. Carbon Dioxide Laser Treatment of Cutaneous Neurofibromas. Dermatol. Ther. 2012, 2, 7. [Google Scholar] [CrossRef]
- Grossman, A.R.; Majidian, A.M.; Grossman, P.H. Thermal injuries as a result of CO2 laser resurfacing. Plast. Reconstr. Surg. 1998, 102, 1247–1252. [Google Scholar] [CrossRef]
- Moreno, J.C.; Mathoret, C.; Lantieri, L.; Zeller, J.; Revuz, J.; Wolkenstein, P. Carbon dioxide laser for removal of multiple cutaneous neurofibromas. Br. J. Dermatol. 2001, 144, 1096–1097. [Google Scholar] [CrossRef] [PubMed]
- Kriechbaumer, L.K.; Susani, M.; Kircher, S.G.; Distelmaier, K.; Happak, W. Comparative study of CO2− and Er:YAG laser ablation of multiple cutaneous neurofibromas in von Recklinghausen’s disease. Lasers Med. Sci. 2014, 29, 1083–1091. [Google Scholar] [CrossRef]
- Yumeen, S.; Hohman, M.H.; Khan, T. Laser Erbium-Yag Resurfacing. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK560931/ (accessed on 21 February 2023).
- Kim, H.J.; Lee, K.G.; Yi, S.M.; Kim, J.H.; Kim, I.H. Successful Treatment of Multiple Cutaneous Neurofibromas Using a Combination of Shave Excision and Laser Photothermocoagulation with a 1,444-nm Neodymium-Doped Yttrium Aluminum Garnet Laser. Dermatol. Surg. 2012, 38, 960. [Google Scholar] [CrossRef]
- Anderson, R.R. Tolerability of Device Based Therapies for Neurofibromatosis Type 1 Cutaneous Neurofibromas. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04730583 (accessed on 23 March 2023).
- Quirk, B.; Olasz, E.; Kumar, S.; Basel, D.; Whelan, H. Photodynamic Therapy for Benign Cutaneous Neurofibromas Using Aminolevulinic Acid Topical Application and 633 nm Red Light Illumination. Photobiomodul. Photomed. Laser Surg. 2021, 39, 411–417. [Google Scholar] [CrossRef]
- Verma, N.; Yumeen, S.; Raggio, B.S. Ablative Laser Resurfacing. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK557474/ (accessed on 24 February 2023).
- Elwakil, T.F.; Samy, N.A.; Elbasiouny, M.S. Non-excision treatment of multiple cutaneous neurofibromas by laser photocoagulation. Lasers Med. Sci. 2008, 23, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Klesse, L.J.; Jordan, J.T.; Radtke, H.B.; Rosser, T.; Schorry, E.; Ullrich, N.; Viskochil, D.; Knight, P.; Plotkin, S.R.; Yohay, K. The Use of MEK Inhibitors in Neurofibromatosis Type 1–Associated Tumors and Management of Toxicities. Oncologist 2020, 25, e1109–e1116. [Google Scholar] [CrossRef]
- Mohamad, T.; Plante, C.; Brosseau, J.P. Toward Understanding the Mechanisms of Malignant Peripheral Nerve Sheath Tumor Development. Int. J. Mol. Sci. 2021, 22, 8620. [Google Scholar] [CrossRef] [PubMed]
- Ortonne, N.; Wolkenstein, P.; Blakeley, J.O.; Korf, B.; Plotkin, S.R.; Riccardi, V.M.; Miller, D.C.; Huson, S.; Peltonen, J.; Rosenberg, A.; et al. Cutaneous neurofibromas: Current clinical and pathologic issues. Neurology 2018, 91 (Suppl. 1), S5–S13. [Google Scholar] [CrossRef] [PubMed]
- Ly, K.I.; Blakeley, J.O. The Diagnosis and Management of Neurofibromatosis Type 1. Med. Clin. N. Am. 2019, 103, 1035–1054. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Fletcher, C.D.M. Malignant peripheral nerve sheath tumor (MPNST) arising in diffuse-type neurofibroma: Clinicopathologic characterization in a series of 9 cases. Am. J. Surg. Pathol. 2015, 39, 1234–1241. [Google Scholar] [CrossRef]
- Akshintala, S.; Baldwin, A.; Liewehr, D.J.; Goodwin, A.; Blakeley, J.O.; Gross, A.M.; Steinberg, S.M.; Dombi, E.; Widemann, B.C. Longitudinal evaluation of peripheral nerve sheath tumors in neurofibromatosis type 1: Growth analysis of plexiform neurofibromas and distinct nodular lesions. Neuro Oncol. 2020, 22, 1368–1378. [Google Scholar] [CrossRef]
- Röhrich, M.; Koelsche, C.; Schrimpf, D.; Capper, D.; Sahm, F.; Kratz, A.; Reuss, J.; Hovestadt, V.; Jones, D.T.W.; Bewerunge-Hudler, M.; et al. Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol. 2016, 131, 877–887. [Google Scholar] [CrossRef]
- Somatilaka, B.N.; Sadek, A.; McKay, R.M.; Le, L.Q. Malignant Peripheral Nerve Sheath Tumor: Models, Biology, and Translation. Oncogene 2022, 41, 2405–2421. [Google Scholar] [CrossRef]
- Pemov, A.; Li, H.; Presley, W.; Wallace, M.R.; Miller, D.T. Genetics of human malignant peripheral nerve sheath tumors. Neurooncol. Adv. 2019, 2 (Suppl. 1), i50–i61. [Google Scholar] [CrossRef]
- Brohl, A.S.; Kahen, E.; Yoder, S.J.; Teer, J.K.; Reed, D.R. The genomic landscape of malignant peripheral nerve sheath tumors: Diverse drivers of Ras pathway activation. Sci. Rep. 2017, 7, 14992. [Google Scholar] [CrossRef]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef]
- Evans, D.G.R.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Ozarslan, B.; Russo, T.; Argenziano, G.; Santoro, C.; Piccolo, V. Cutaneous Findings in Neurofibromatosis Type 1. Cancers 2021, 13, 463. [Google Scholar] [CrossRef]
- Lipner, S.R.; Scher, R.K. Subungual glomus tumors: Underrecognized clinical findings in neurofibromatosis 1. J. Am. Acad. Dermatol. 2021, 84, e269. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.R.; Sloan, J.L.; Yao, L.; Mannes, A.J.; Moshyedi, A.; Lee, C.-C.; Sciot, R.; De Smet, L.; Mautner, V.-F.; Legius, E. Diagnosis, management, and complications of glomus tumours of the digits in neurofibromatosis type 1. J. Med. Genet. 2010, 47, 525–532. [Google Scholar] [CrossRef]
- Sacchetti, F.; De Gori, M.; Grossi, S.; Bonadio, G.A.; Capanna, R. An exceptional case of malignant glomus tumor and a review of the literature. Acta Orthop. Traumatol. Turc. 2019, 53, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Mravic, M.; LaChaud, G.; Nguyen, A.; Scott, M.A.; Dry, S.M.; James, A.W. Clinical and Histopathological Diagnosis of Glomus Tumor: An Institutional Experience of 138 Cases. Int. J. Surg. Pathol. 2015, 23, 181–188. [Google Scholar] [CrossRef]
- Aqil, N.; Gallouj, S.; Moustaide, K.; Mernissi, F.Z. Painful tumors in a patient with neurofibromatosis type 1: A case report. J. Med. Case Rep. 2018, 12, 319. [Google Scholar] [CrossRef] [PubMed]
- Scaravilli, G.; Rossi, R.; Artiaco, S.; Merolla, G. Glomus Tumor of the Thenar Eminence in Neurofibromatosis Type 1: Case Report and Literature Review. Transl. Med. UniSa. 2014, 11, 63–68. [Google Scholar]
- Kumar, M.G.; Emnett, R.J.; Bayliss, S.J.; Gutmann, D.H. Glomus tumors in individuals with neurofibromatosis type 1. J. Am. Acad. Dermatol. 2014, 71, 44–48. [Google Scholar] [CrossRef]
- Harrison, B.; Moore, A.M.; Calfee, R.; Sammer, D.M. The association between glomus tumors and neurofibromatosis. J. Hand Surg. Am. 2013, 38, 1571–1574. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.; Sammer, D. Glomus tumors and neurofibromatosis: A newly recognized association. Plast. Reconstr. Surg. Glob. Open 2014, 2, e214. [Google Scholar] [CrossRef]
- Brems, H.; Park, C.; Maertens, O.; Pemov, A.; Messiaen, L.; Upadhyaya, M.; Claes, K.; Beert, E.; Peeters, K.; Mautner, V.; et al. Glomus tumors in neurofibromatosis type 1: Genetic, functional and clinical evidence of a novel association. Cancer Res. 2009, 69, 7393–7401. [Google Scholar] [CrossRef]
- Xu, J.; Ismat, F.A.; Wang, T.; Yang, J.; Epstein, J.A. NF1 Regulates a Ras-Dependent Vascular Smooth Muscle Proliferative Injury Response. Circulation 2007, 116, 2148–2156. [Google Scholar] [CrossRef]
- Wood, T.R.; McHugh, J.B.; Siegel, G.W. Glomus tumors with malignant features of the extremities: A case series. Clin. Sarcoma Res. 2020, 10, 20. [Google Scholar] [CrossRef]
- Sanagoo, A.; Jouybari, L.; Koohi, F.; Sayehmiri, F. Evaluation of QoL in neurofibromatosis patients: A systematic review and meta-analysis study. BMC Neurol. 2019, 19, 123. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.; Masurel, A.; Olivier-Faivre, L.; Vabres, P. Juvenile xanthogranuloma and nevus anemicus in the diagnosis of neurofibromatosis type 1. JAMA Dermatol. 2014, 150, 42–46. [Google Scholar] [CrossRef]
- Collie, J.S.; Harper, C.D.; Fillman, E.P. Juvenile Xanthogranuloma. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK526103/ (accessed on 8 February 2023).
- Liy-Wong, C.; Mohammed, J.; Carleton, A.; Pope, E.; Parkin, P.; Lara-Corrales, I. The relationship between neurofibromatosis type 1, juvenile xanthogranuloma, and malignancy: A retrospective case-control study. J. Am. Acad. Dermatol. 2017, 76, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Cosgarea, I.; Ugurel, S.; Sucker, A.; Livingstone, E.; Zimmer, L.; Ziemer, M.; Utikal, J.; Mohr, P.; Pfeiffer, C.; Pföhler, C.; et al. Targeted next generation sequencing of mucosal melanomas identifies frequent NF1 and RAS mutations. Oncotarget 2017, 8, 40683–40692. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef]
- Tod, B.M.; Schneider, J.W.; Bowcock, A.M.; Visser, W.I.; Kotze, M.J. The tumor genetics of acral melanoma: What should a dermatologist know? JAAD Int. 2020, 1, 135–147. [Google Scholar] [CrossRef]
- Trinh, P.; Li, S.; Sarin, K.Y. Neurofibromatosis Type 1 and Risk of Skin Cancer. JAMA Dermatol. 2022, 158, 1214–1216. [Google Scholar] [CrossRef]
- Seminog, O.O.; Goldacre, M.J. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: Population-based record-linkage study. Br. J. Cancer 2013, 108, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Landry, J.P.; Schertz, K.L.; Chiang, Y.-J.; Bhalla, A.D.; Yi, M.; Keung, E.Z.; Scally, C.P.; Feig, B.W.; Hunt, K.K.; Roland, C.L.; et al. Comparison of Cancer Prevalence in Patients with Neurofibromatosis Type 1 at an Academic Cancer Center vs in the General Population From 1985 to 2020. JAMA Netw. Open 2021, 4, e210945. [Google Scholar] [CrossRef]
- Miraglia, E.; Moliterni, E.; Iacovino, C.; Roberti, V.; Laghi, A.; Moramarco, A.; Giustini, S. Cutaneous manifestations in neurofibromatosis type 1. Clin Ther. 2020, 171, e371–e377. [Google Scholar] [CrossRef]
- Guillot, B.; Dalac, S.; Delaunay, M.; Baccard, M.; Chevrant-Breton, J.; Dereure, O.; Machet, L.; Sassolas, B.; Zeller, J.; Bernard, P.; et al. Cutaneous malignant melanoma and neurofibromatosis type 1. Melanoma Res. 2004, 14, 159–163. [Google Scholar] [CrossRef]
- Curti, B.D.; Faries, M.B. Recent Advances in the Treatment of Melanoma. N. Engl. J. Med. 2021, 384, 2229–2240. [Google Scholar] [CrossRef]
- Hernandez-Martin, A.; Duat-Rodriguez, A. An Update on Neurofibromatosis Type 1: Not Just Cafe-au-Lait Spots, Freckling, and Neurofibromas. An Update. Part I. Dermatological Clinical Criteria Diagnostic of the Disease. Actas Dermosifiliogr. 2016, 107, 454–464. [Google Scholar] [CrossRef]
- Hernandez-Martin, A.; Duat-Rodriguez, A. An Update on Neurofibromatosis Type 1: Not Just Cafe-au-Lait Spots and Freckling. Part II. Other Skin Manifestations Characteristic of NF1. NF1 and Cancer. Actas Dermosifiliogr. 2016, 107, 465–473. [Google Scholar] [CrossRef]
- Bergqvist, C.; Servy, A.; Valeyrie-Allanore, L.; Ferkal, S.; Combemale, P.; Wolkenstein, P. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J. Rare Dis. 2020, 15, 37. [Google Scholar] [CrossRef] [PubMed]
- Pollack, L.A.; Li, J.; Berkowitz, Z.; Weir, H.K.; Wu, X.-C.; Ajani, U.A.; Ekwueme, D.U.; Li, C.; Pollack, B.P. Melanoma survival in the United States, 1992 to 2005. J. Am. Acad. Dermatol. 2011, 65 (Suppl. 1), S78–S86. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.; Jefferson, I.S.; Robinson, S.K.; Griffin, D.; Adams, W.; Speiser, J.; Winterfield, L.; Peterson, A.; Tung-Hahn, E.; Lee, K.; et al. Skin cancer discovery during total body skin examinations. Int. J. Womens Dermatol. 2021, 7, 411–414. [Google Scholar] [CrossRef]
- Shamsuyarova, A.; Kamil, Z.; Delabie, J.; Al-Faraidy, N.; Ghazarian, D. Primary Cutaneous Follicular Helper T-cell Lymphoma in a Patient With Neurofibromatosis Type 1: Case Report and Review of the Literature. Am. J. Dermatopathol. 2017, 39, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Braam, P.; Sanders, C.J.G.; Canninga-Van Dijk, M.R. Neurofibromatosis type 1 and mycosis fungoides. Int. J. Dermatol. 2002, 41, 236–238. [Google Scholar] [CrossRef]
- Herbert, C.R.; McBurney, E.I. Cutaneous T-cell lymphoma in a patient with neurofibromatosis type 1. Cutis 2003, 72, 27–30. [Google Scholar]
- Park, J.; Yang, J.; Wenzel, A.T.; Ramachandran, A.; Lee, W.J.; Daniels, J.C.; Kim, J.; Martinez-Escala, E.; Amankulor, N.; Pro, B.; et al. Genomic analysis of 220 CTCLs identifies a novel recurrent gain-of-function alteration in RLTPR (p.Q575E). Blood 2017, 130, 1430–1440. [Google Scholar] [CrossRef]
- Koivunen, J.; Karvonen, S.L.; Ylä-Outinen, H.; Aaltonen, V.; Oikarinen, A.; Peltonen, J. NF1 tumor suppressor in epidermal wound healing with special focus on wound healing in patients with type 1 neurofibromatosis. Arch. Dermatol. Res. 2005, 296, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, T.; Billings, B.; Har-Shai, Y.; Agbenorku, P.; Kokuba, E.; Moreira-Gonzalez, A.; Tsukuno, M.; Kurihara, K.; Jackson, I.T. Multicenter study of wound healing in neurofibromatosis and neurofibroma. J. Craniofac. Surg. 2007, 18, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- McGinty, S.; Siddiqui, W.J. Keloid. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK507899/ (accessed on 24 February 2023).
- Alhady, S.M.; Sivanantharajah, K. Keloids in various races. A review of 175 cases. Plast. Reconstr. Surg. 1969, 44, 564–566. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug | Mechanism of Action | Study Population | Results | Side Effects | Status | |
|---|---|---|---|---|---|---|
| Systemic Treatments | ||||||
| Imatinib mesylate 400 mg daily [18] | C-kit receptor tyrosine kinase inhibitor | 1 adult patient | Small reduction in visual burden | Fluid retention, gastrointestinal symptoms, hepatotoxicity, fatigue, rash | N/A a | |
| Everolimus 5–15 ng/mL serum concentration [17] | mTOR b inhibitor | 17 adult patients | Significant reduction in absolute value of paired lesion height and significant reduction in lesion surface volume in 3/17 patients | Gastrointestinal symptoms, upper respiratory infections, skin irritation | Complete (NCT02332902) | |
| Selumetinib c [23] | MEK d 1/2 inhibitor | Adult patients | N/A | N/A | Active, not recruiting (NCT02839720) | |
| Topical Treatments | ||||||
| Imiquimod 5% [24,25] | Toll-like receptor 7 agonist | 11 adult patients | No significant reduction in tumor volume | Skin irritation and erythema | Complete (NCT00865644) | |
| Diphen-cyprone 0.04% [28] | Hapten that induces a delayed-type hypersensitivity reacion | Adult patients | N/A | Skin irritation and erythema | Active and recruiting (NCT05438290) | |
| NFX-179 0.05%, 0.15%, 0.50% [26] | MEK inhibitor | 47 adult patients (35 in the treatment arms) | −1.6, −11.9, and −16.7, percent changes in CN volume for 0.05%, 0.15%, and 0.50% concentrations, respectively | Pruritis, stinging, erythema | Complete (NCT04435665) | |
| NFX-179 0.5%, 1.5% [27] | MEK inhibitor | Adult patients | N/A | N/A | Active, not recruiting (NCT05005845) | |
| Mechanism | Amount of CNs a That Can Be Removed in One Session | Anesthesia | Benefits | Risks and Limitations | |
|---|---|---|---|---|---|
| Electrodessication | Radiofrequency ablation | 100–1000 s | Local or general anesthesia | Time-sparing, low cost, does not require specialized providers, low rates of post-op bleeding, deep penetration, ideal for small tumors | Thermal necrosis of surrounding normal skin, increased scarring [30] |
| Traditional Surgical Removal | Excision | 10–100 s | Often requires general anesthesia | Good cosmetic outcomes, linear scarring, ideal for large tumors > 4 cm | Expensive, time-intensive, requires trained surgical specialists, higher risk of adverse events, requires sterile technique, higher risk of bleeding, difficult to remove small CNs, requires suture removal [25,29] |
| Modified Biopsy Removal | Excision | 100 s | Local anesthesia | Good cosmetic outcomes, low cost, does not require specialized providers, outpatient setting, nonsterile technique, prevents tumor regrowth due to dermal removal | Requires suture removal [29,30] |
| CO2 b Laser | Thermal ablation | 100 s | Local or general anesthesia | Time-sparing, immediate hemostasis, healing by secondary intention, improved symptoms of CNs, ideal for small tumors | Expensive, requires trained specialists, scarring (often hypopigmented, atrophic, or hypertrophic), less penetration (better-suited for superficial CNs) [33,35,41] |
| Er:YAG c Laser | Thermal ablation | 100 s | Local or general anesthesia | Time-sparing, rapid re-epithelialization, decreased duration of post-op erythema, shorter recovery, less edema, decreased thermal necrosis, greater precision, good cosmetic outcomes, ideal for small tumors | Expensive, requires trained specialists, less hemostasis than CO2 laser [41] |
| Nd:YAG d Laser | Thermal ablation | 100 s | Local or general anesthesia | Time-sparing, decreased post-op hypopigmentation, preserves epidermis, high penetration, ideal for small tumors | Expensive, requires trained specialists [42] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poplausky, D.; Young, J.N.; Tai, H.; Rivera-Oyola, R.; Gulati, N.; Brown, R.M. Dermatologic Manifestations of Neurofibromatosis Type 1 and Emerging Treatments. Cancers 2023, 15, 2770. https://doi.org/10.3390/cancers15102770
Poplausky D, Young JN, Tai H, Rivera-Oyola R, Gulati N, Brown RM. Dermatologic Manifestations of Neurofibromatosis Type 1 and Emerging Treatments. Cancers. 2023; 15(10):2770. https://doi.org/10.3390/cancers15102770
Chicago/Turabian StylePoplausky, Dina, Jade N. Young, Hansen Tai, Ryan Rivera-Oyola, Nicholas Gulati, and Rebecca M. Brown. 2023. "Dermatologic Manifestations of Neurofibromatosis Type 1 and Emerging Treatments" Cancers 15, no. 10: 2770. https://doi.org/10.3390/cancers15102770
APA StylePoplausky, D., Young, J. N., Tai, H., Rivera-Oyola, R., Gulati, N., & Brown, R. M. (2023). Dermatologic Manifestations of Neurofibromatosis Type 1 and Emerging Treatments. Cancers, 15(10), 2770. https://doi.org/10.3390/cancers15102770