
Recent Advances in Molecular Mechanisms of Cancer Immunotherapy

 ,
,  , , ,
, , ,
Abstract
Simple Summary
Abstract
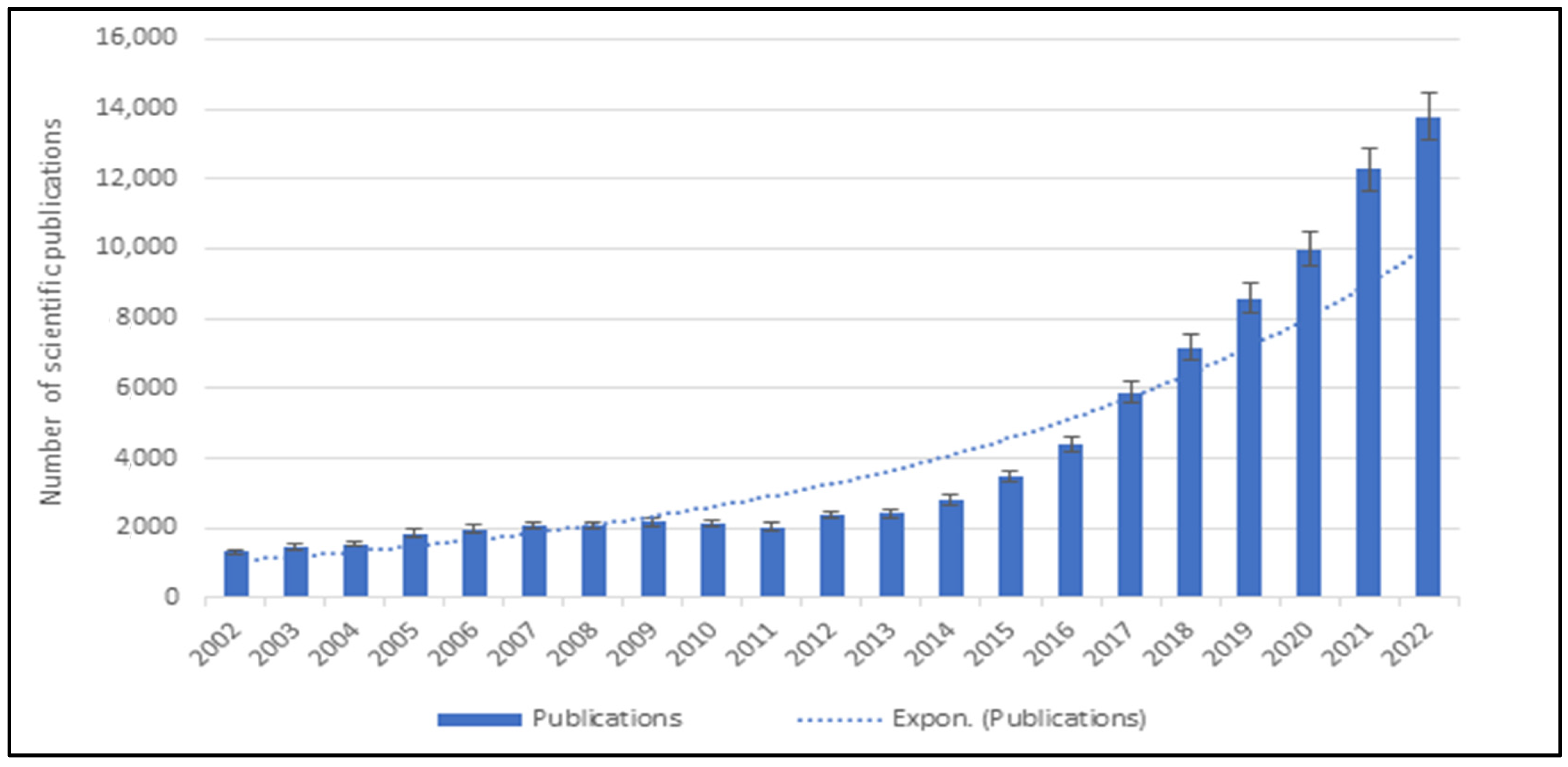
1. Introduction
2. Cancer and Immunotherapy
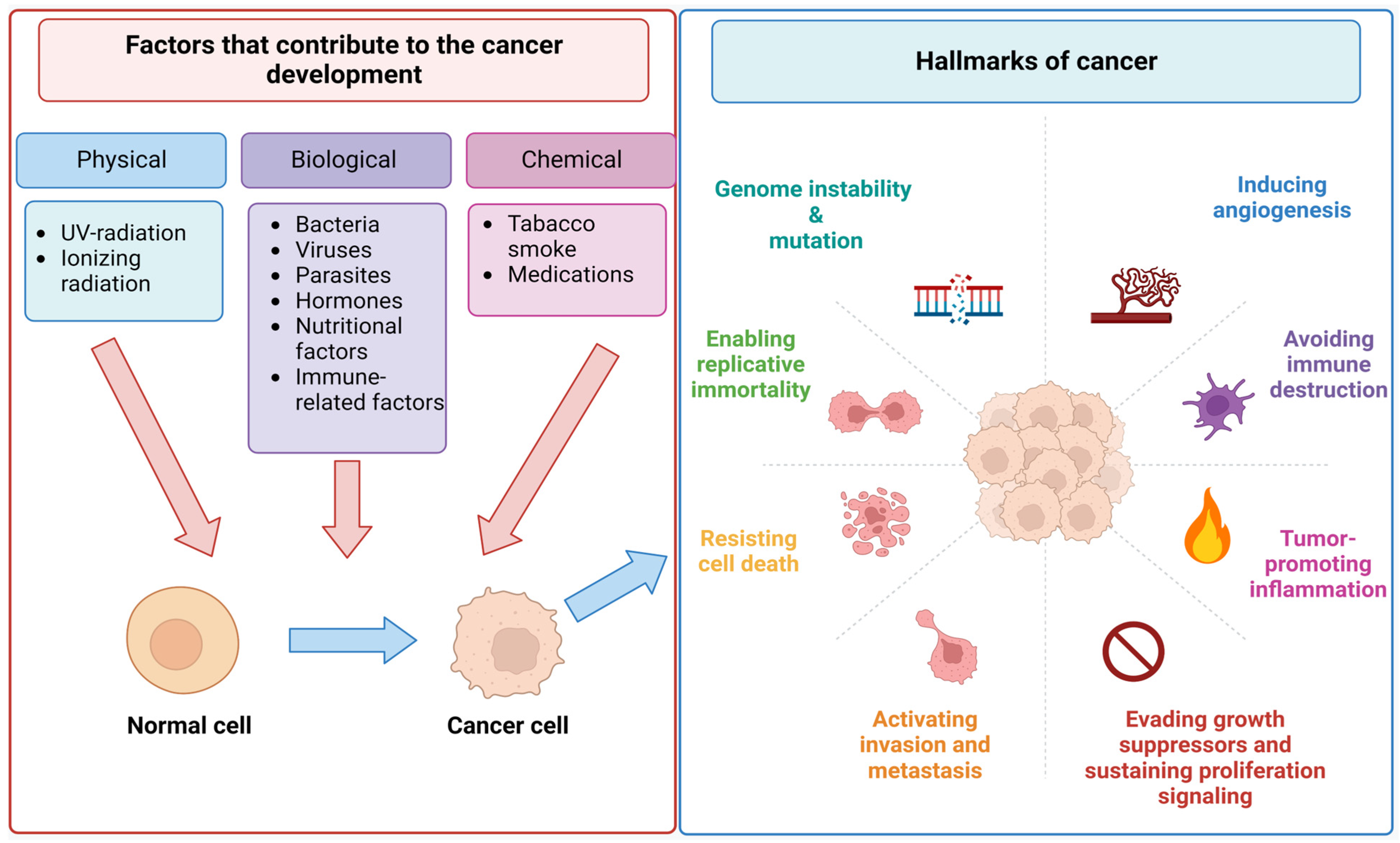
2.1. Incidence, Progression, and Metastasis of Cancer
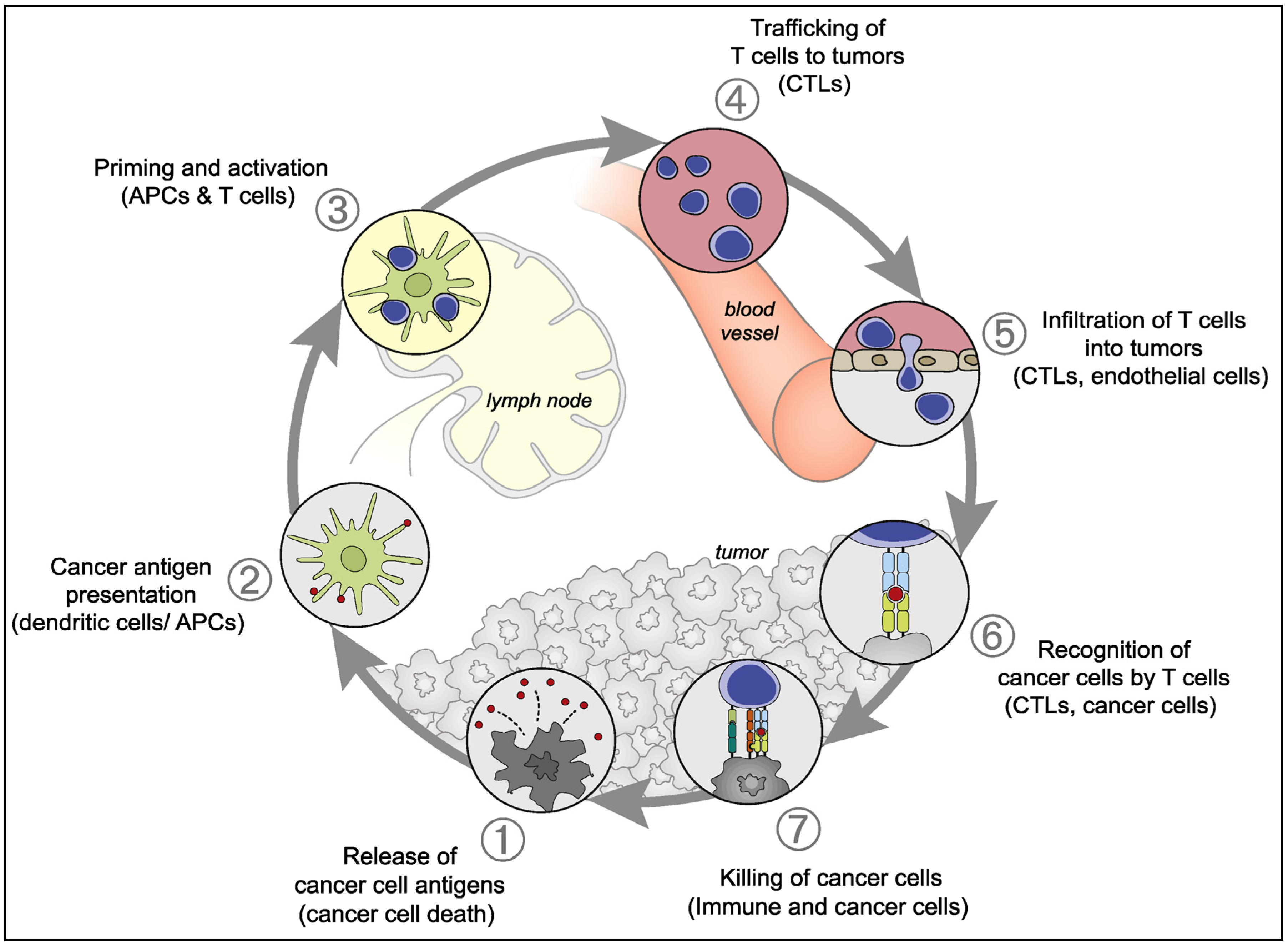
2.2. Antitumor Natural Immune Response
2.3. Chronological Development of Cancer Immunotherapy
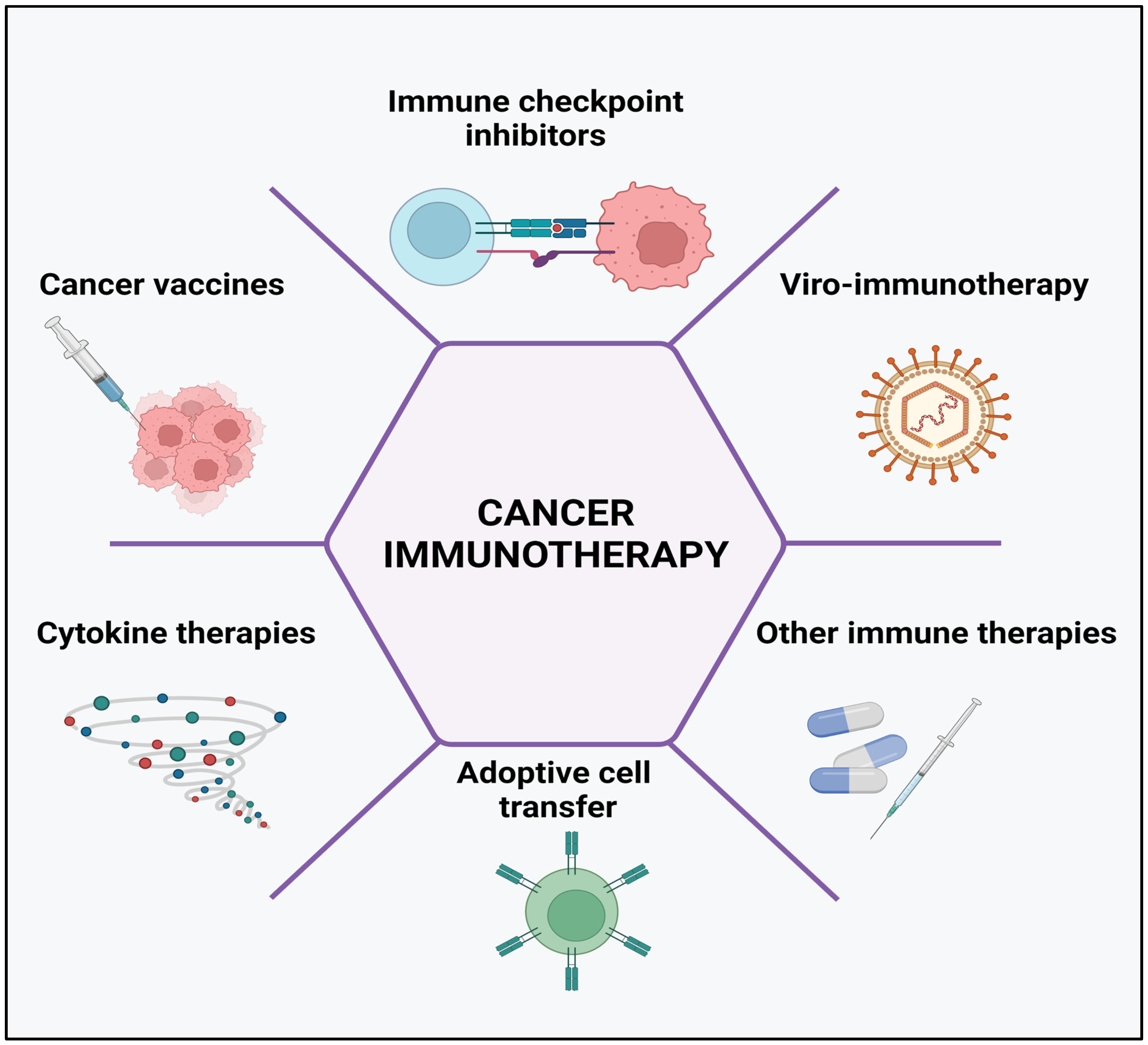
3. Mechanisms of Cancer Immunotherapy
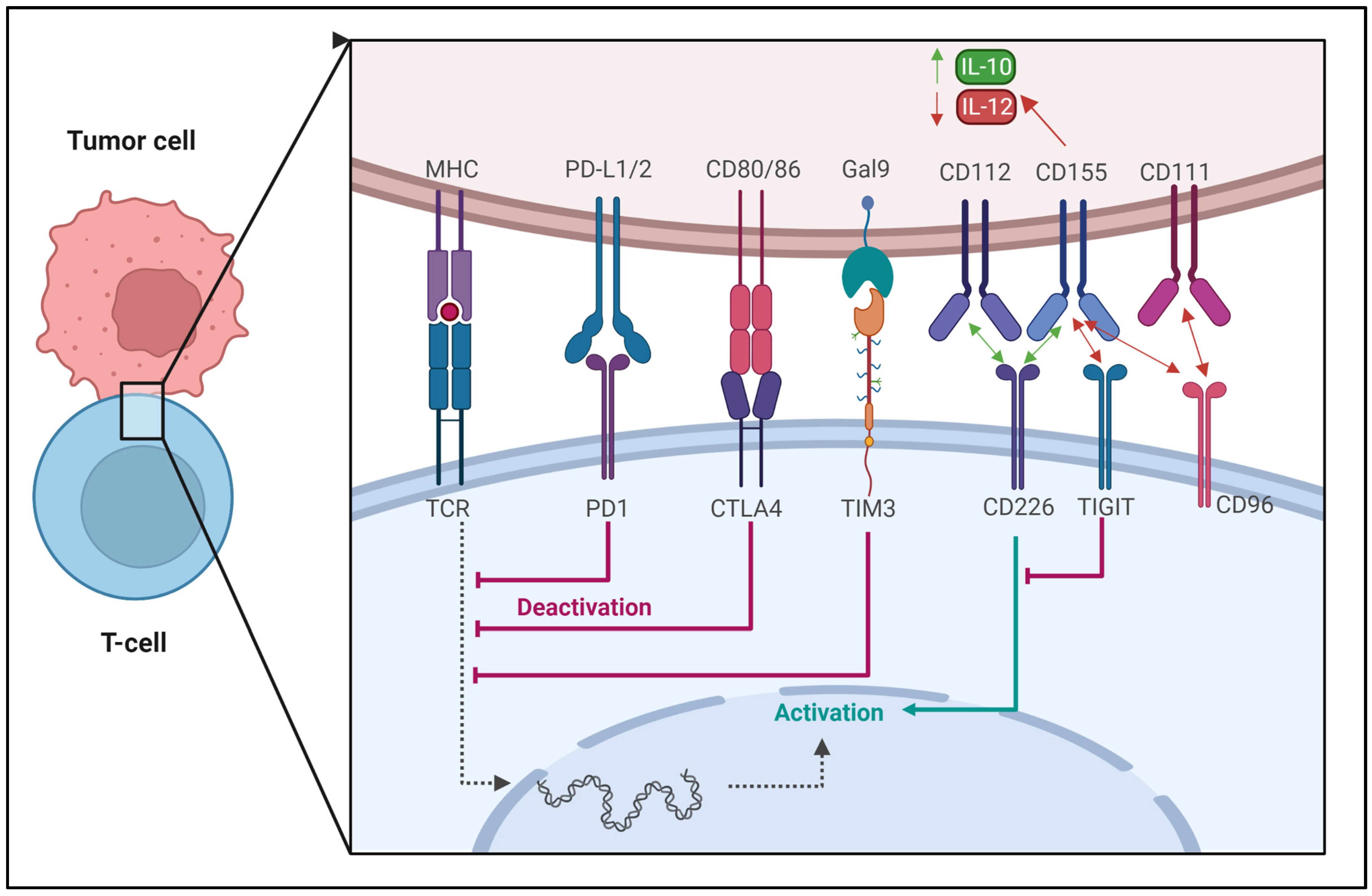
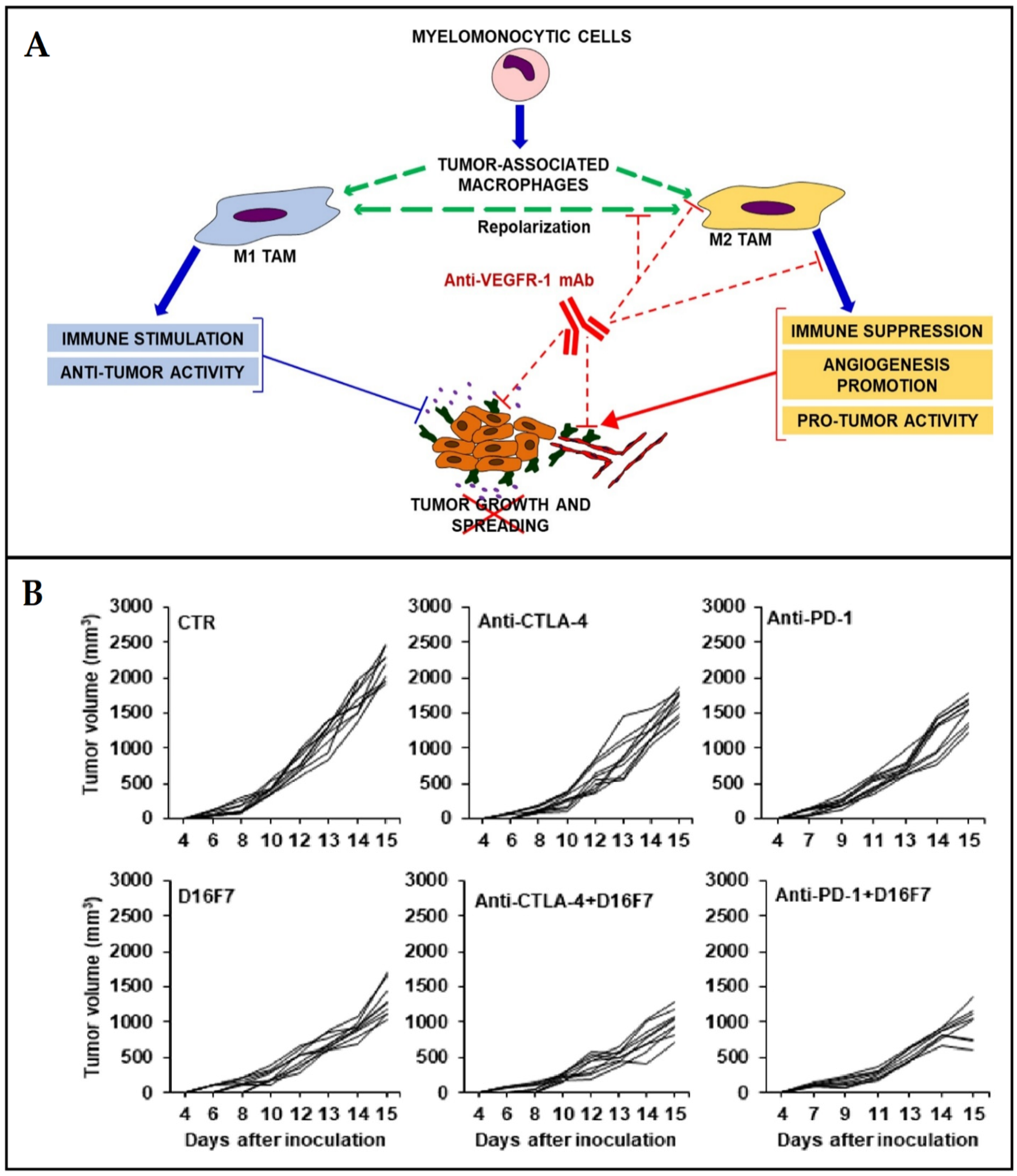
3.1. Regulation of Immune Checkpoints
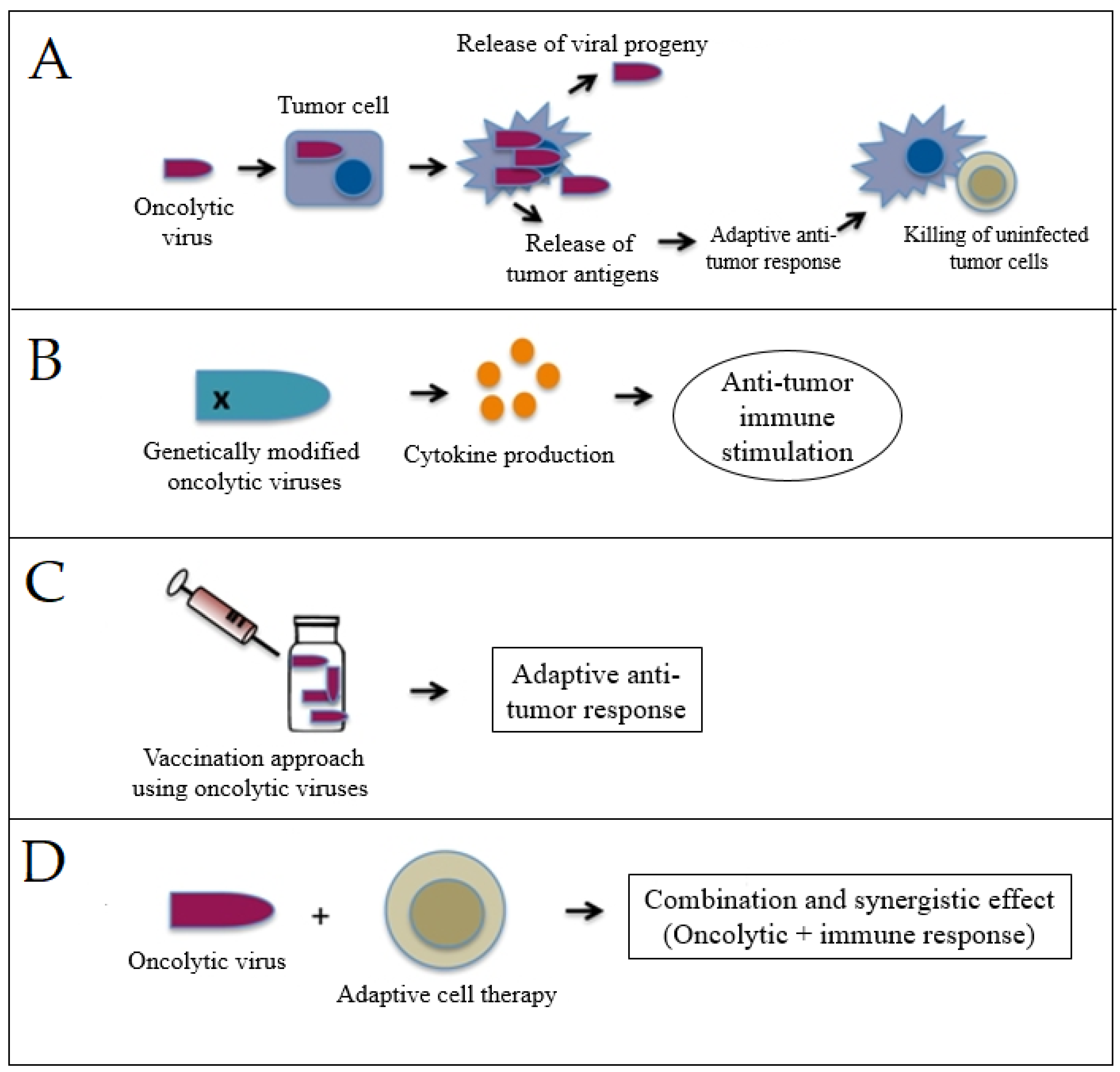
3.2. Viro-Immunotherapy
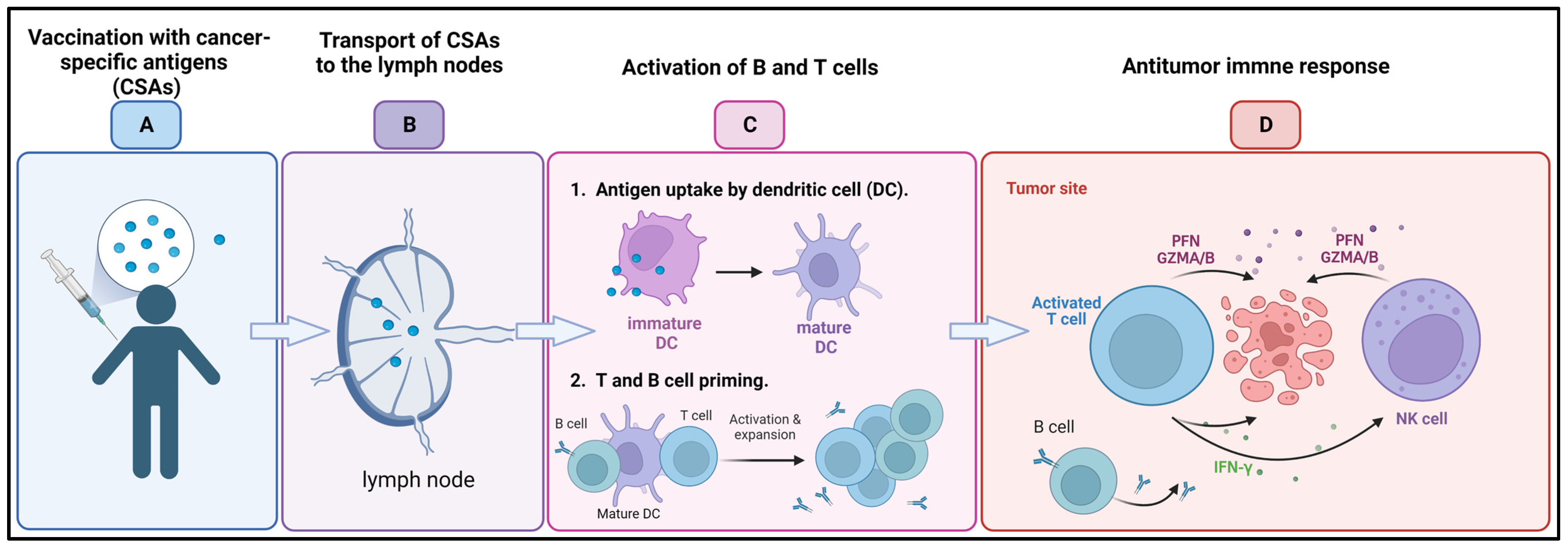
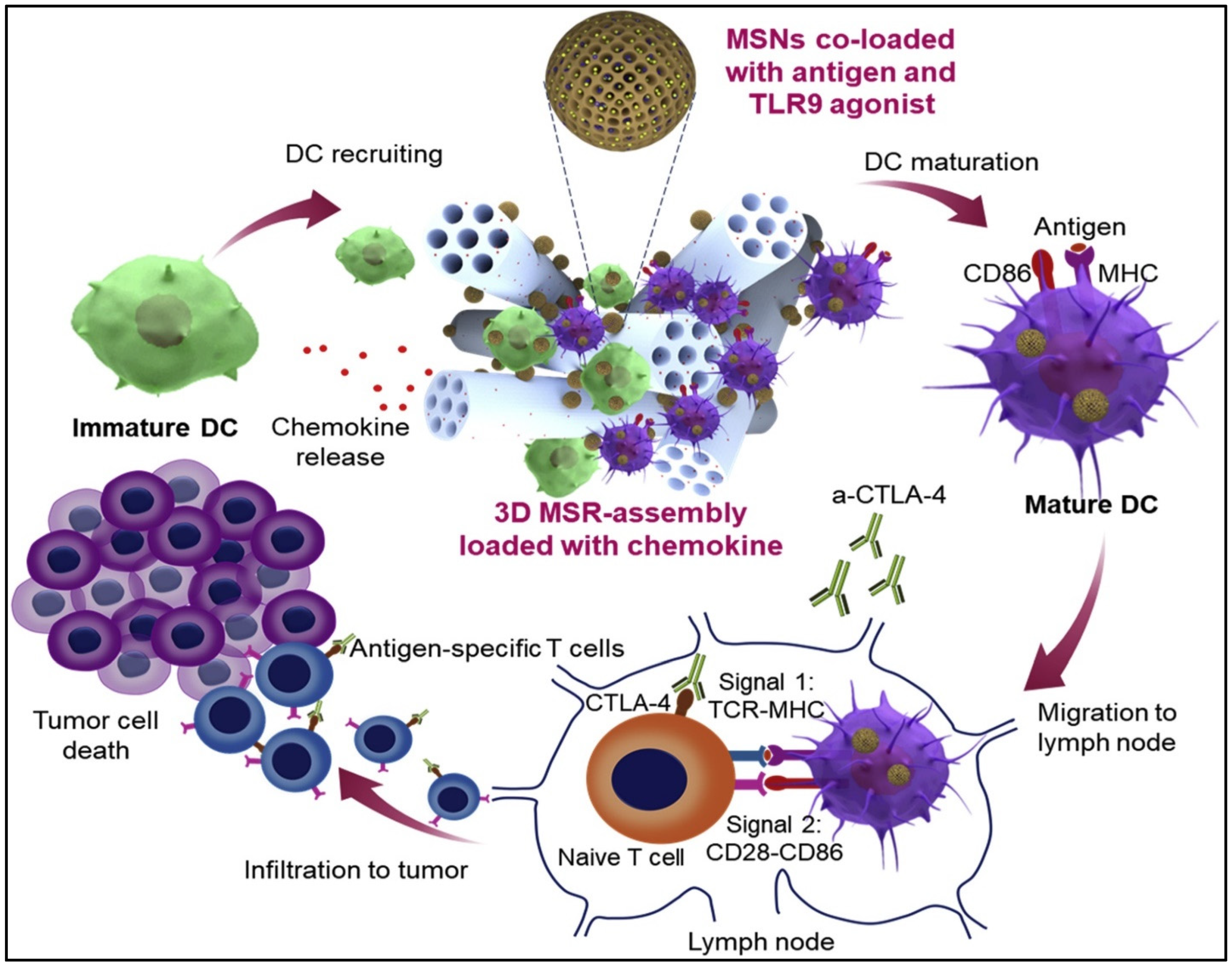
3.3. Cancer Vaccines
3.4. Cytokine Therapies
- Stimulation of the immune system: the immune system can be stimulated with cytokine treatments to better target and destroy cancer cells.
- Reduced toxicity: compared to conventional treatments such as chemotherapy and radiation, cytokine therapies are safer and have fewer adverse effects.
- Versatility: cytokine treatments are adaptable because they can be used to treat a wide variety of cancers.
- May provide long-term benefits: some cytokine therapies have been demonstrated to produce lasting advantages in cancer patients, increasing the likelihood of a long-lasting response to treatment.
- Can be used in combination with other treatments: combining cytokine therapies with standard cancer treatments such as chemotherapy, radiation therapy, and immunotherapy has been shown to improve patient outcomes.
- May induce remission: some cytokine therapies, such as high-dose IL-2 and IFN-α, have been shown to induce complete or partial remission in some cancer patients.
- Long-term immune memory: long-term immunological memory can be induced by some cytokine therapies, such as IL-2, meaning that the immune system will continue to detect and fight cancer cells even after treatment has ended.
- Fewer treatment sessions: patients may find cytokine therapies more convenient because they often need fewer treatment sessions than treatments such as chemotherapy.
- Limited effectiveness: some patients may only have a partial response to cytokine treatments, and they may not work for all forms of cancer.
- Side effects: side effects from cytokine therapies include fever, exhaustion, and muscle aches, but they are typically less severe than those from chemotherapy and radiation. Some cytokine therapies, such as high-dose IL-2, can cause damage to organs such as the kidneys and liver, which can be life-threatening.
- Expensive: some patients may not be able to afford cytokine therapy due to their high cost.
- Can cause autoimmune reactions: there are risks associated with cytokine treatments, including autoimmune reactions and tissue damage.
3.5. Adoptive Cell Transfer
3.6. Other Immune Therapies
4. Challenges and Prospects of Cancer Immunotherapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmed, S.A.; Parama, D.; Daimari, E.; Girisa, S.; Banik, K.; Harsha, C.; Dutta, U.; Kunnumakkara, A.B. Rationalizing the therapeutic potential of apigenin against cancer. Life Sci. 2020, 267, 118814. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, R.; Meskin, N.; Al Moustafa, A.-E. Control Strategies for Cancer Therapy. In Mathematical Models of Cancer and Different Therapies; Springer: Berlin/Heidelberg, Germany, 2021; pp. 215–247. [Google Scholar]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.F.; Sadelain, M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell 2015, 27, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325. [Google Scholar] [CrossRef]
- Bukhari, N.; Joseph, J.P.; Hussain, J.; Adeeb, M.A.M.; Wakim, M.J.Y.; Yahya, E.B.; Arif, A.; Saleem, A.; Sharif, N. Prevalence of human papilloma virus sub genotypes following head and neck squamous cell carcinomas in Asian continent, a systematic review Article. Asian Pac. J. Cancer Prev. APJCP 2019, 20, 3269. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Yahya, E.B.; Alqadhi, A.M. Recent trends in cancer therapy: A review on the current state of gene delivery. Life Sci. 2021, 119087. [Google Scholar] [CrossRef] [PubMed]
- Blackadar, C.B. Historical review of the causes of cancer. World J. Clin. Oncol. 2016, 7, 54. [Google Scholar] [CrossRef]
- Levine, M.S.; Bakker, B.; Boeckx, B.; Moyett, J.; Lu, J.; Vitre, B.; Spierings, D.C.; Lansdorp, P.M.; Cleveland, D.W.; Lambrechts, D. Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev. Cell 2017, 40, 313–322.e315. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016. [Google Scholar]
- Osisami, M.; Keller, E.T. Mechanisms of metastatic tumor dormancy. J. Clin. Med. 2013, 2, 136–150. [Google Scholar] [CrossRef]
- Loomans-Kropp, H.A.; Umar, A. Cancer prevention and screening: The next step in the era of precision medicine. NPJ Precis. Oncol. 2019, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Yahya, E.B.; Alfallous, K.A.; Wali, A.; Hameid, S.; Zwaid, H. Growth rate and antibiotic sensitivity effect of some natural and petroleum based materials on Staphylococcus aureus. Int. J. Res. Appl. Sci. Biotechnol. 2020, 7, 7–11. [Google Scholar] [CrossRef]
- Arneth, B. Tumor microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [PubMed]
- Caras, I.; Tucureanu, C.; Lerescu, L.; Pitica, R.; Melinceanu, L.; Neagu, S.; Salageanu, A. Influence of tumor cell culture supernatants on macrophage functional polarization: In vitro models of macrophage-tumor environment interaction. Tumori J. 2011, 97, 647–654. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef]
- Blonska, M.; Agarwal, N.K.; Vega, F. Shaping of the tumor microenvironment: Stromal cells and vessels. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2015; pp. 3–13. [Google Scholar]
- Abogmaza, A.F.; Keer, K.F.; Ayad, A.T.; Yahya, E.B. A Review on the Medicinal and Aromatic Plants Growing in Libya and Their Therapeutic Properties. Int. Res. J. Sci. Technol. 2020, 2. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Mellman, I. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 2005, 23, 975–1028. [Google Scholar] [CrossRef]
- Almashgab, A.M.; Yahya, E.B.; Banu, A. The Cytotoxicity Effects of Outer Membrane Vesicles Isolated from Hospital and Laboratory Strains of Pseudomonas Aeruginosa on Human Keratinocyte Cell Line. Malays. J. Sci. 2020, 39, 45–53. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef]
- Keenan, B.P.; Fong, L. Conditional Cancer Immunotherapy as a Safer Way to Step on the Gas. Cancer Discov. 2021, 11, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Rumolo, A.; Tapella, L.; Tanzi, M.; Metallo, A.; Conca, F.; Negri, S.; Lefkimmiatis, K.; Pedrazzoli, P.; Lim, D. Store-Operated Ca2+ Entry Is Up-Regulated in Tumour-Infiltrating Lymphocytes from Metastatic Colorectal Cancer Patients. Cancers 2022, 14, 3312. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Friedmann, K.S.; Lyrmann, H.; Zhou, Y.; Schoppmeyer, R.; Knörck, A.; Mang, S.; Hoxha, C.; Angenendt, A.; Backes, C.S. A calcium optimum for cytotoxic T lymphocyte and natural killer cell cytotoxicity. J. Physiol. 2018, 596, 2681–2698. [Google Scholar] [CrossRef] [PubMed]
- Espie, D.; Barrin, S.; Rajnpreht, I.; Vimeux, L.; Donnadieu, E. Live Imaging of CAR T Cell Ca2+ Signals in Tumor Slices Using Confocal Microscopy. In The Immune Synapse: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2023; pp. 453–462. [Google Scholar]
- Trebak, M.; Kinet, J.-P. Calcium signalling in T cells. Nat. Rev. Immunol. 2019, 19, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.W.; Chow, J.C.; Cho, W.C. Tumor mutational burden as a tissue-agnostic biomarker for cancer immunotherapy. Expert Rev. Clin. Pharmacol. 2020, 14, 141–143. [Google Scholar] [CrossRef]
- Bagheri, Y.; Barati, A.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Yousefi, M. Current progress in cancer immunotherapy based on natural killer cells. Cell Biol. Int. 2021, 45, 2–17. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Kunimasa, K.; Goto, T. Immunosurveillance and immunoediting of lung cancer: Current perspectives and challenges. Int. J. Mol. Sci. 2020, 21, 597. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Cha, B.G.; Choi, Y.; Im, J.; Kim, J. Injectable dual-scale mesoporous silica cancer vaccine enabling efficient delivery of antigen/adjuvant-loaded nanoparticles to dendritic cells recruited in local macroporous scaffold. Biomaterials 2020, 239, 119859. [Google Scholar] [CrossRef]
- Adamaki, M.; Zoumpourlis, V. Immunotherapy as a Precision Medicine Tool for the Treatment of Prostate Cancer. Cancers 2021, 13, 173. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.B. A Preliminary Note on the Treatment of Inoperable Sarcoma by the Toxic Products of Erysipelas; Meriden Gravure Company: Meriden, CT, USA, 1893. [Google Scholar]
- Romero, P.; Banchereau, J.; Bhardwaj, N.; Cockett, M.; Disis, M.L.; Dranoff, G.; Gilboa, E.; Hammond, S.A.; Hershberg, R.; Korman, A.J. The Human Vaccines Project: A roadmap for cancer vaccine development. Sci. Transl. Med. 2016, 8, 334ps339. [Google Scholar] [CrossRef] [PubMed]
- Hericourt, J.; Richet, C. Remarques a propos de la note de M. Boureau sur la serotherapie des neoplasms compte rend. Acad. Sc. 1895, 21, 373. [Google Scholar]
- Coley, W. The treatment of inoperable cancer. Practitioner 1899, 9, 510. [Google Scholar]
- Von Leyden, E.; Blumenthal, F. Attempts to immunize humans by inoculation of their own cancer. Dtsch. Med. Wochens. 1902, 28, 637–638. [Google Scholar]
- Murphy, J.B. Factors of resistance to heteroplastic tissue-grafting: Studies in Tissue Specificity. III. J. Exp. Med. 1914, 19, 513–522. [Google Scholar] [CrossRef]
- Pérez-Ibave, D.C.; Burciaga-Flores, C.H.; Elizondo-Riojas, M.-Á. Prostate-specific antigen (PSA) as a possible biomarker in non-prostatic cancer: A review. Cancer Epidemiol. 2018, 54, 48–55. [Google Scholar] [CrossRef]
- Gicobi, J.K.; Barham, W.; Dong, H. Immune resilience in response to cancer therapy. Cancer Immunol. Immunother. 2020, 69, 2165–2167. [Google Scholar] [CrossRef]
- Sjögren, H.; Hellström, I.; Bansal, S.; Hellström, K. Suggestive evidence that the “blocking antibodies” of tumor-bearing individuals may be antigen-antibody complexes. Proc. Natl. Acad. Sci. USA 1971, 68, 1372–1375. [Google Scholar] [CrossRef]
- Carswell, E.; Old, L.J.; Kassel, R.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef]
- Morgan, D.A.; Ruscetti, F.W.; Gallo, R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 1976, 193, 1007–1008. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, G.; Elliott, E.; Stevenson, F. Idiotypic determinants on the surface immunoglobulin of neoplastic lymphocytes: A therapeutic target. Fed. Proc. 1977, 36, 2268. [Google Scholar] [PubMed]
- Miller, R.; Levy, R. Response of cutaneous T cell lymphoma to therapy with hybridoma monoclonal antibody. Lancet 1981, 318, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Matsui, H.; Fujita, T.; Takaoka, C.; Kashima, N.; Yoshimoto, R.; Hamuro, J. Structure and expression of a cloned cDNA for human interleukin-2. Nature 1983, 302, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Haskins, K.; Kubo, R.; White, J.; Pigeon, M.; Kappler, J.; Marrack, P. The major histocompatibility complex-restricted antigen receptor on T cells. I. Isolation with a monoclonal antibody. J. Exp. Med. 1983, 157, 1149–1169. [Google Scholar] [CrossRef]
- Knuth, A.; Danowski, B.; Oettgen, H.F.; Old, L.J. T-cell-mediated cytotoxicity against autologous malignant melanoma: Analysis with interleukin 2-dependent T-cell cultures. Proc. Natl. Acad. Sci. USA 1984, 81, 3511–3515. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Weiner, L.M. Monoclonal antibody therapy of cancer. Semin. Oncol. 1999, 26, 43–51. [Google Scholar] [PubMed]
- Hurwitz, A.A.; Foster, B.A.; Kwon, E.D.; Truong, T.; Choi, E.M.; Greenberg, N.M.; Burg, M.B.; Allison, J.P. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res. 2000, 60, 2444–2448. [Google Scholar] [PubMed]
- Hirano, F.; Kaneko, K.; Tamura, H.; Dong, H.; Wang, S.; Ichikawa, M.; Rietz, C.; Flies, D.B.; Lau, J.S.; Zhu, G. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005, 65, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.B.; Rivière, I.; Sénéchal, B.; Wang, X.; Wang, Y.; Purdon, T.J.; Hsu, M.; Devlin, S.M.; Halton, E.; Lamanna, N. Autologous CD19-targeted CAR T cells in patients with residual CLL following initial purine analog-based therapy. Mol. Ther. 2018, 26, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.; Hu, R.; Mosbruger, T.L.; Dahlem, T.J.; Stephens, W.Z.; Rao, D.S.; Round, J.L.; O’Connell, R.M. Genome-wide CRISPR-Cas9 screen identifies microRNAs that regulate myeloid leukemia cell growth. PLoS ONE 2016, 11, e0153689. [Google Scholar] [CrossRef]
- Freedman, J.D.; Duffy, M.R.; Lei-Rossmann, J.; Muntzer, A.; Scott, E.M.; Hagel, J.; Campo, L.; Bryant, R.J.; Verrill, C.; Lambert, A. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. 2018, 78, 6852–6865. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, Y.; Zhang, Y.; Wang, Y.; Alotaibi, F.; Qiu, L.; Wang, H.; Peng, S.; Liu, Y.; Li, Q. miRNA-5119 regulates immune checkpoints in dendritic cells to enhance breast cancer immunotherapy. Cancer Immunol. Immunother. 2020, 69, 951–967. [Google Scholar] [CrossRef]
- Yahya, E.B.; Amirul, A.; HPS, A.K.; Olaiya, N.G.; Iqbal, M.O.; Jummaat, F.; AK, A.S.; Adnan, A. Insights into the Role of Biopolymer Aerogel Scaffolds in Tissue Engineering and Regenerative Medicine. Polymers 2021, 13, 1612. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Ma, Y.; Yu, L.; Jiang, J.; Shen, S.; Hou, Y.; Wang, T. Cancer immunotherapy: A focus on the regulation of immune checkpoints. Int. J. Mol. Sci. 2018, 19, 1389. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Witkowska, M.; Smolewski, P. Immune checkpoint inhibitors to treat malignant lymphomas. J. Immunol. Res. 2018, 2018, 1982423. [Google Scholar] [CrossRef]
- Zhang, C.; Peng, Y.; Hublitz, P.; Zhang, H.; Dong, T. Genetic abrogation of immune checkpoints in antigen-specific cytotoxic T-lymphocyte as a potential alternative to blockade immunotherapy. Sci. Rep. 2018, 8, 5549. [Google Scholar] [CrossRef]
- Akhbariyoon, H.; Azizpour, Y.; Esfahani, M.F.; Firoozabad, M.S.M.; Rad, M.R.; Esfahani, K.S.; Khoshavi, N.; Karimi, N.; Shirinisaz, A.; Abedi, F. Immune checkpoint inhibition for the treatment of cancers: An update and critical review of ongoing clinical trials. Clin. Immunol. 2021, 232, 108873. [Google Scholar] [CrossRef]
- Gadducci, A.; Guerrieri, M.E. Immune checkpoint inhibitors in gynecological cancers: Update of literature and perspectives of clinical research. Anticancer Res. 2017, 37, 5955–5965. [Google Scholar]
- Kudo, M. Immune checkpoint inhibition in hepatocellular carcinoma: Basics and ongoing clinical trials. Oncology 2017, 92, 50–62. [Google Scholar] [CrossRef]
- Munari, E.; Mariotti, F.R.; Quatrini, L.; Bertoglio, P.; Tumino, N.; Vacca, P.; Eccher, A.; Ciompi, F.; Brunelli, M.; Martignoni, G. PD-1/PD-L1 in cancer: Pathophysiological, diagnostic and therapeutic aspects. Int. J. Mol. Sci. 2021, 22, 5123. [Google Scholar] [CrossRef]
- Chang, E.; Pelosof, L.; Lemery, S.; Gong, Y.; Goldberg, K.B.; Farrell, A.T.; Keegan, P.; Veeraraghavan, J.; Wei, G.; Blumenthal, G.M. Systematic review of PD-1/PD-L1 inhibitors in oncology: From personalized medicine to public health. Oncol. 2021, 26, e1786–e1799. [Google Scholar] [CrossRef] [PubMed]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Z.; Li, Y.; Zhao, W.; Wu, J.; Zhang, Z. PD-1/PD-L1 checkpoint inhibitors in tumor immunotherapy. Front. Pharmacol. 2021, 12, 731798. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Kołat, D.; Kałuzińska-Kołat, Ż.; Gawrysiak, M.; Drozda, R.; Celik, I.; Kontek, R. PD-1/PD-L1 and DNA Damage Response in Cancer. Cells 2023, 12, 530. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood J. Am. Soc. Hematol. 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Sobhani, N.; Tardiel-Cyril, D.R.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in regulatory T cells for cancer immunotherapy. Cancers 2021, 13, 1440. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 2016, 39, 98. [Google Scholar] [CrossRef]
- De Silva, P.; Aiello, M.; Gu-Trantien, C.; Migliori, E.; Willard-Gallo, K.; Solinas, C. Targeting CTLA-4 in cancer: Is it the ideal companion for PD-1 blockade immunotherapy combinations? Int. J. Cancer 2021, 149, 31–41. [Google Scholar] [CrossRef]
- Wojtukiewicz, M.Z.; Rek, M.M.; Karpowicz, K.; Górska, M.; Polityńska, B.; Wojtukiewicz, A.M.; Moniuszko, M.; Radziwon, P.; Tucker, S.C.; Honn, K.V. Inhibitors of immune checkpoints—PD-1, PD-L1, CTLA-4—New opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev. 2021, 40, 949–982. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. OncoTargets Ther. 2018, 7005–7009. [Google Scholar] [CrossRef]
- Saleh, R.; Toor, S.M.; Elkord, E. Targeting TIM-3 in solid tumors: Innovations in the preclinical and translational realm and therapeutic potential. Expert Opin. Ther. Targets 2020, 24, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, M.; Tan, J.; Zeng, C.; Li, Y.; Ganjalikhani-Hakemi, M. TIM-3 in leukemia; immune response and beyond. Front. Oncol. 2021, 11, 753677. [Google Scholar] [CrossRef]
- Gomes de Morais, A.L.; Cerdá, S.; de Miguel, M. New checkpoint inhibitors on the road: Targeting TIM-3 in solid tumors. Curr. Oncol. Rep. 2022, 24, 651–658. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; Van den Eertwegh, A.J.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD 96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Seth, S.; Albrecht, J.; Maier, M.K.; du Pasquier, L.; Ravens, I.; Dreyer, L.; Burger, R.; Gramatzki, M.; Schwinzer, R. CD96 interaction with CD155 via its first Ig-like domain is modulated by alternative splicing or mutations in distal Ig-like domains. J. Biol. Chem. 2009, 284, 2235–2244. [Google Scholar] [CrossRef]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Pia Ciotti, G.M.; Chiavari, M.; Ruffini, F.; Lacal, P.M.; Graziani, G.; Navarra, P. Vascular endothelial growth factor receptor 1 in glioblastoma-associated microglia/macrophages. Oncol. Rep. 2020, 43, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Lacal, P.M.; Atzori, M.G.; Ruffini, F.; Scimeca, M.; Bonanno, E.; Cicconi, R.; Mattei, M.; Bernardini, R.; D’Atri, S.; Tentori, L. Targeting the vascular endothelial growth factor receptor-1 by the monoclonal antibody D16F7 to increase the activity of immune checkpoint inhibitors against cutaneous melanoma. Pharmacol. Res. 2020, 159, 104957. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.-h.T.; Maurer, M.; Korman, A.J. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018, 7, e1466769. [Google Scholar] [CrossRef]
- Yadav, M.; Green, C.; Ma, C.; Robert, A.; Glibicky, A.; Nakamura, R.; Sumiyoshi, T.; Meng, R.; Chu, Y.-W.; Wu, J. Tigit, CD226 and PD-L1/PD-1 are highly expressed by marrow-infiltrating T cells in patients with multiple myeloma. Blood 2016, 128, 2102. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Zhang, Y.; Chikata, T.; Kuse, N.; Murakoshi, H.; Gatanaga, H.; Oka, S.; Takiguchi, M. Immunological Control of HIV-1 Disease Progression by Rare Protective HLA Allele. J. Virol. 2022, 96, e01248-01222. [Google Scholar] [CrossRef] [PubMed]
- Naimi, A.; Mohammed, R.N.; Raji, A.; Chupradit, S.; Yumashev, A.V.; Suksatan, W.; Shalaby, M.N.; Thangavelu, L.; Kamrava, S.; Shomali, N. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun. Signal. 2022, 20, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.; Chester, C.; Melero, I.; Kohrt, H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann. Oncol. 2016, 27, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Dyer, A.; Frost, S.; Fisher, K.; Seymour, L. The role of cancer metabolism in defining the success of oncolytic viro-immunotherapy. Cytokine Growth Factor Rev. 2020, 56, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef]
- Choi, K.-J.; Zhang, S.-N.; Choi, I.-K.; Kim, J.-S.; Yun, C.-O. Strengthening of antitumor immune memory and prevention of thymic atrophy mediated by adenovirus expressing IL-12 and GM-CSF. Gene Ther. 2012, 19, 711–723. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef]
- Gong, J.; Sachdev, E.; Mita, A.C.; Mita, M.M. Clinical development of reovirus for cancer therapy: An oncolytic virus with immune-mediated antitumor activity. World J. Methodol. 2016, 6, 25. [Google Scholar] [CrossRef]
- Bergmann, M.; Romirer, I.; Sachet, M.; Fleischhacker, R.; García-Sastre, A.; Palese, P.; Wolff, K.; Pehamberger, H.; Jakesz, R.; Muster, T. A genetically engineered influenza A virus with ras-dependent oncolytic properties. Cancer Res. 2001, 61, 8188–8193. [Google Scholar]
- Eissa, I.R.; Naoe, Y.; Bustos-Villalobos, I.; Ichinose, T.; Tanaka, M.; Zhiwen, W.; Mukoyama, N.; Morimoto, T.; Miyajima, N.; Hitoki, H. Genomic signature of the natural oncolytic herpes simplex virus HF10 and its therapeutic role in preclinical and clinical trials. Front. Oncol. 2017, 7, 149. [Google Scholar] [CrossRef]
- Yu, W.; Fang, H. Clinical trials with oncolytic adenovirus in China. Curr. Cancer Drug Targets 2007, 7, 141–148. [Google Scholar] [CrossRef]
- Fujiyuki, T.; Amagai, Y.; Shoji, K.; Kuraishi, T.; Sugai, A.; Awano, M.; Sato, H.; Hattori, S.; Yoneda, M.; Kai, C. Recombinant SLAMblind Measles Virus Is a Promising Candidate for Nectin-4-Positive Triple Negative Breast Cancer Therapy. Mol. Ther. Oncolytics 2020, 19, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Fan, J.; Ding, Y.; Zhang, J.; Zhou, B.; Zhang, Y.; Huang, B.; Hu, Z. Oncolytic cancer therapy with a vaccinia virus strain. Oncol. Rep. 2019, 41, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.-L.; Lin, Y.-P.; Shieh, G.-S.; Su, C.-H.; Wu, W.-L.; Tsai, Y.-S.; Cheng, C.-W.; Lai, M.-D.; Wu, C.-L. Development of a conditionally replicating pseudorabies virus for HER-2/neu-overexpressing bladder cancer therapy. Mol. Ther. 2007, 15, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Smedberg, J.R.; Westcott, M.M.; Ahmed, M.; Lyles, D.S. Signaling pathways in murine dendritic cells that regulate the response to vesicular stomatitis virus vectors that express flagellin. J. Virol. 2014, 88, 777–785. [Google Scholar] [CrossRef]
- Melzer, M.K.; Lopez-Martinez, A.; Altomonte, J. Oncolytic vesicular stomatitis virus as a viro-immunotherapy: Defeating cancer with a “hammer” and “anvil”. Biomedicines 2017, 5, 8. [Google Scholar] [CrossRef]
- Pikor, L.A.; Bell, J.C.; Diallo, J.-S. Oncolytic viruses: Exploiting cancer’s deal with the devil. Trends Cancer 2015, 1, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Atherton, M.J.; Evgin, L.; Keller, B.A.; Shenouda, M.M.; Stephenson, K.B.; Vile, R.G.; Bell, J.C.; Evans, D.H.; Lichty, B.D. Infectious Optimism following the 10th International Oncolytic Virus Meeting. Mol. Ther. Oncolytics 2017, 7, 12–16. [Google Scholar] [CrossRef]
- Sultan, H.; Fesenkova, V.I.; Addis, D.; Fan, A.E.; Kumai, T.; Wu, J.; Salazar, A.M.; Celis, E. Designing therapeutic cancer vaccines by mimicking viral infections. Cancer Immunol. Immunother. 2017, 66, 203–213. [Google Scholar] [CrossRef]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef]
- Davola, M.E.; Mossman, K.L. Oncolytic viruses: How “lytic” must they be for therapeutic efficacy? Oncoimmunology 2019, 8, e1581528. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Burke, J.; Jonker, D.; Stephenson, J.; Haas, A.R.; Chow, L.Q.; Nieva, J.; Hwang, T.-H.; Moon, A.; Patt, R. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 2011, 477, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.; Scott, G.B.; Heaton, S.; Barr, T.; Askar, B.; Müller, L.M.; Jennings, V.A.; Ralph, C.; Burton, C.; Melcher, A. Efficacy of Coxsackievirus A21 against drug-resistant neoplastic B cells. Mol. Ther. Oncolytics 2023, 29, 17–29. [Google Scholar] [CrossRef]
- Enokida, T.; Moreira, A.; Bhardwaj, N. Vaccines for immunoprevention of cancer. J. Clin. Investig. 2021, 131, e146956. [Google Scholar] [CrossRef]
- Grimmett, E.; Al-Share, B.; Alkassab, M.B.; Zhou, R.W.; Desai, A.; Rahim, M.M.A.; Woldie, I. Cancer vaccines: Past, present and future; a review article. Discov. Oncol. 2022, 13, 31. [Google Scholar] [CrossRef]
- Hargadon, K.M. Tumor microenvironmental influences on dendritic cell and T cell function: A focus on clinically relevant immunologic and metabolic checkpoints. Clin. Transl. Med. 2020, 10, 374–411. [Google Scholar] [CrossRef]
- Kitadani, J.; Ojima, T.; Iwamoto, H.; Tabata, H.; Nakamori, M.; Nakamura, M.; Hayata, K.; Katsuda, M.; Miyajima, M.; Yamaue, H. Cancer vaccine therapy using carcinoembryonic antigen-expressing dendritic cells generated from induced pluripotent stem cells. Sci. Rep. 2018, 8, 4569. [Google Scholar] [CrossRef]
- Turriziani, M.; Fantini, M.; Benvenuto, M.; Izzi, V.; Masuelli, L.; Sacchetti, P.; Modesti, A.; Bei, R. Carcinoembryonic antigen (CEA)-based cancer vaccines: Recent patents and antitumor effects from experimental models to clinical trials. Recent Pat. Anti Cancer Drug Discov. 2012, 7, 265–296. [Google Scholar] [CrossRef]
- Adam, V.; Wauters, I.; Vansteenkiste, J. Melanoma-associated antigen-A3 vaccination in the treatment of non-small-cell lung cancer. Expert Opin. Biol. Ther. 2014, 14, 365–376. [Google Scholar] [CrossRef]
- Schäfer, P.; Paraschiakos, T.; Windhorst, S. Oncogenic activity and cellular functionality of melanoma associated antigen A3. Biochem. Pharmacol. 2021, 192, 114700. [Google Scholar] [CrossRef] [PubMed]
- Esfandiary, A.; Ghafouri-Fard, S. MAGE-A3: An immunogenic target used in clinical practice. Immunotherapy 2015, 7, 683–704. [Google Scholar] [CrossRef] [PubMed]
- Peled, N.; Oton, A.B.; Hirsch, F.R.; Bunn, P. MAGE A3 antigen-specific cancer immunotherapeutic. Immunotherapy 2009, 1, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; O’Herrin, S.M.; Wu, J.; Reagan-Shaw, S.; Ma, Y.; Bhat, K.M.; Gravekamp, C.; Setaluri, V.; Peters, N.; Hoffmann, F.M. MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res. 2007, 67, 9954–9962. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Cen, Q.; Lei, H. A review on development of MUC1-based cancer vaccine. Biomed. Pharmacother. 2020, 132, 110888. [Google Scholar] [CrossRef]
- Guo, M.; You, C.; Dou, J. Role of transmembrane glycoprotein mucin 1 (MUC1) in various types of colorectal cancer and therapies: Current research status and updates. Biomed. Pharmacother. 2018, 107, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 based immunotherapy of cancer: Current perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef]
- Madan, R.A.; Gulley, J.L.; Arlen, P.M. PSA-based vaccines for the treatment of prostate cancer. Expert Rev. Vaccines 2006, 5, 199–209. [Google Scholar] [CrossRef]
- Karan, D. Formulation of the bivalent prostate cancer vaccine with surgifoam elicits antigen-specific effector T cells in PSA-transgenic mice. Vaccine 2017, 35, 5794–5798. [Google Scholar] [CrossRef]
- Doehn, C.; Böhmer, T.; Kausch, I.; Sommerauer, M.; Jocham, D. Prostate cancer vaccines: Current status and future potential. BioDrugs 2008, 22, 71–84. [Google Scholar] [CrossRef]
- Oka, Y.; Udaka, K.; Tsuboi, A.; Elisseeva, O.A.; Ogawa, H.; Aozasa, K.; Kishimoto, T.; Sugiyama, H. Cancer immunotherapy targeting Wilms’ tumor gene WT1 product. J. Immunol. 2000, 164, 1873–1880. [Google Scholar] [CrossRef] [PubMed]
- Hein, K.Z.; Yao, S.; Fu, S. Wilms’ Tumor 1 (WT1): The Vaccine for Cancer. J. Immunother. Precis. Oncol. 2020, 3, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, B.; Van den Eynde, B.; van der Bruggen, P.; Romero, P.; Gaforio, J.J.; De Plaen, E.; Lethe, B.; Brasseur, F.; Boon, T. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J. Exp. Med. 1994, 179, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef]
- Khalil, H.A.; Adnan, A.; Yahya, E.B.; Olaiya, N.; Safrida, S.; Hossain, M.; Balakrishnan, V.; Gopakumar, D.A.; Abdullah, C.; Oyekanmi, A. A Review on plant cellulose nanofibre-based aerogels for biomedical applications. Polymers 2020, 12, 1759. [Google Scholar] [CrossRef]
- Yahya, E.B.; Jummaat, F.; Amirul, A.; Adnan, A.; Olaiya, N.; Abdullah, C.; Rizal, S.; Mohamad Haafiz, M.; Khalil, H. A review on revolutionary natural biopolymer-based aerogels for antibacterial delivery. Antibiotics 2020, 9, 648. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, Y.; Huang, L. Exosomes from M1-polarized macrophages potentiate the cancer vaccine by creating a pro-inflammatory microenvironment in the lymph node. Mol. Ther. 2017, 25, 1665–1675. [Google Scholar] [CrossRef]
- Emens, L.A. Cancer vaccines: On the threshold of success. Expert Opin. Emerg. Drugs 2008, 13, 295–308. [Google Scholar] [CrossRef]
- Donninger, H.; Li, C.; Eaton, J.W.; Yaddanapudi, K. Cancer vaccines: Promising therapeutics or an unattainable dream. Vaccines 2021, 9, 668. [Google Scholar] [CrossRef]
- Waldmann, T.A. Cytokines in cancer immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028472. [Google Scholar] [CrossRef]
- Yron, I.; Wood, T.; Spiess, P.; Rosenberg, S. In vitro growth of murine T cells. V. The isolation and growth of lymphoid cells infiltrating syngeneic solid tumors. J. Immunol. 1980, 125, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Leitman, S.; Chang, A.E.; Ettinghausen, S.E.; Matory, Y.L.; Skibber, J.M.; Shiloni, E.; Vetto, J.T. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N. Engl. J. Med. 1985, 313, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Mulé, J.J.; Spiess, P.J.; Reichert, C.M.; Schwarz, S.L. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high-dose recombinant interleukin 2. J. Exp. Med. 1985, 161, 1169–1188. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Cauwels, A.; Van Lint, S.; Paul, F.; Garcin, G.; De Koker, S.; Van Parys, A.; Wueest, T.; Gerlo, S.; Van der Heyden, J.; Bordat, Y. Delivering type I interferon to dendritic cells empowers tumor eradication and immune combination treatments. Cancer Res. 2018, 78, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, H.; Tao, L.; Eguchi, R.; Sato, A.; Honda, M.; Kaneko, S.; Iwata, Y.; Nishikawa, H.; Imanishi, H.; Iijima, H. The in vivo antitumor effects of type I-interferon against hepatocellular carcinoma: The suppression of tumor cell growth and angiogenesis. Sci. Rep. 2017, 7, 12189. [Google Scholar] [CrossRef]
- Cox, M.A.; Harrington, L.E.; Zajac, A.J. Cytokines and the inception of CD8 T cell responses. Trends Immunol. 2011, 32, 180–186. [Google Scholar] [CrossRef]
- Yan, W.-L.; Shen, K.-Y.; Tien, C.-Y.; Chen, Y.-A.; Liu, S.-J. Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 2017, 9, 347–360. [Google Scholar] [CrossRef]
- Kumar, A.; Khani, A.T.; Ortiz, A.S.; Swaminathan, S. GM-CSF: A double-edged sword in cancer immunotherapy. Front. Immunol. 2022, 13, 901277. [Google Scholar] [CrossRef]
- Bohlius, J.; Reiser, M.; Schwarzer, G.; Engert, A. Impact of granulocyte colony-stimulating factor (CSF) and granulocyte–macrophage CSF in patients with malignant lymphoma: A systematic review. Br. J. Haematol. 2003, 122, 413–423. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Z.; Chen, Y.; Lv, Z.; Ding, X.; Wang, R.; Xiao, H.; Hou, C.; Shen, B.; Feng, J. An epithelial-to-mesenchymal transition-inducing potential of granulocyte macrophage colony-stimulating factor in colon cancer. Sci. Rep. 2017, 7, 8265. [Google Scholar] [CrossRef] [PubMed]
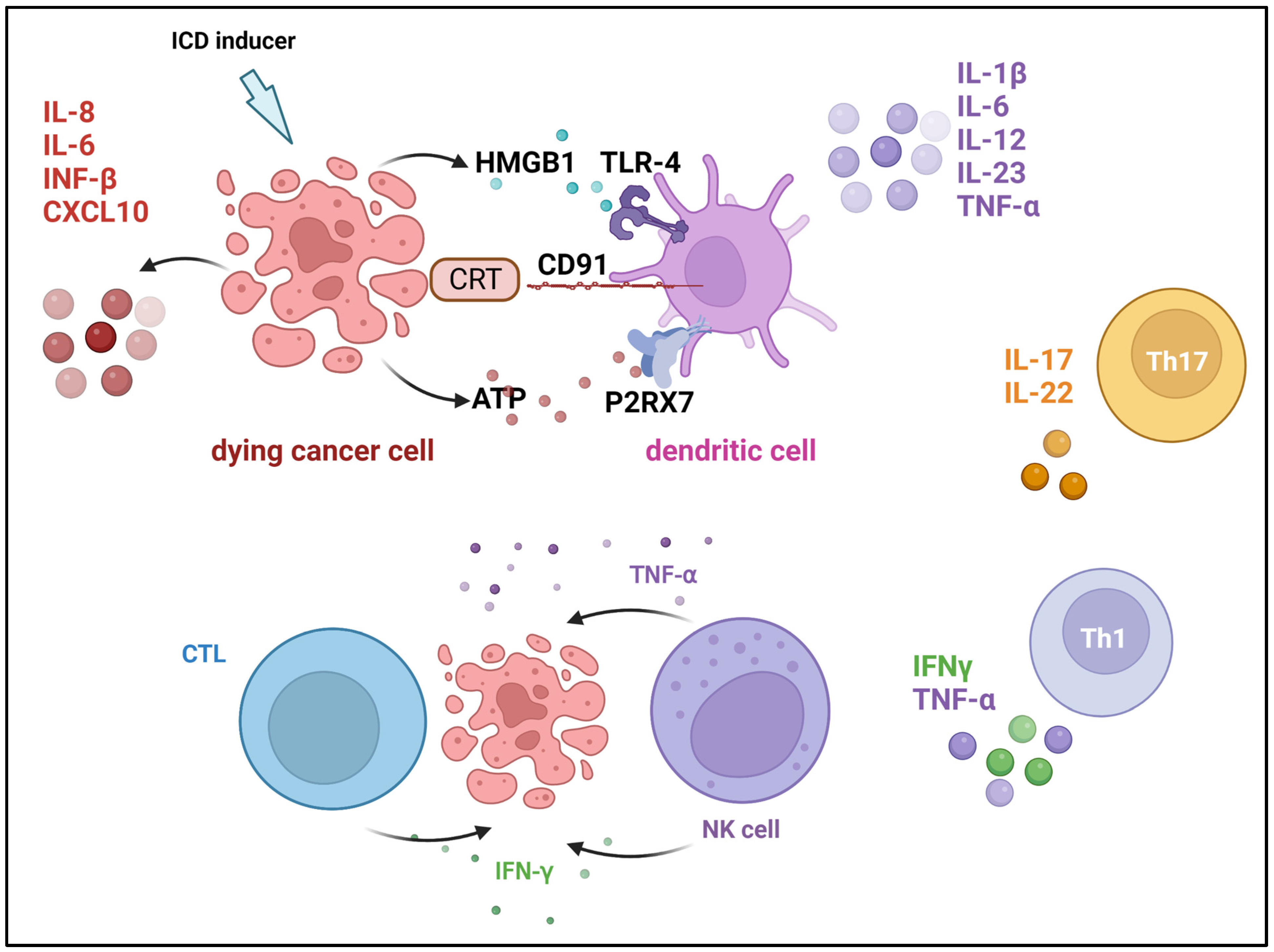
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Mi, Y.; Guo, N.; Xu, H.; Xu, L.; Gou, X.; Jin, W. Cytokine-induced killer cells as pharmacological tools for cancer immunotherapy. Front. Immunol. 2017, 8, 774. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.R.; Marone, G.; Mantovani, A. Cancer inflammation and cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028662. [Google Scholar] [CrossRef]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef]
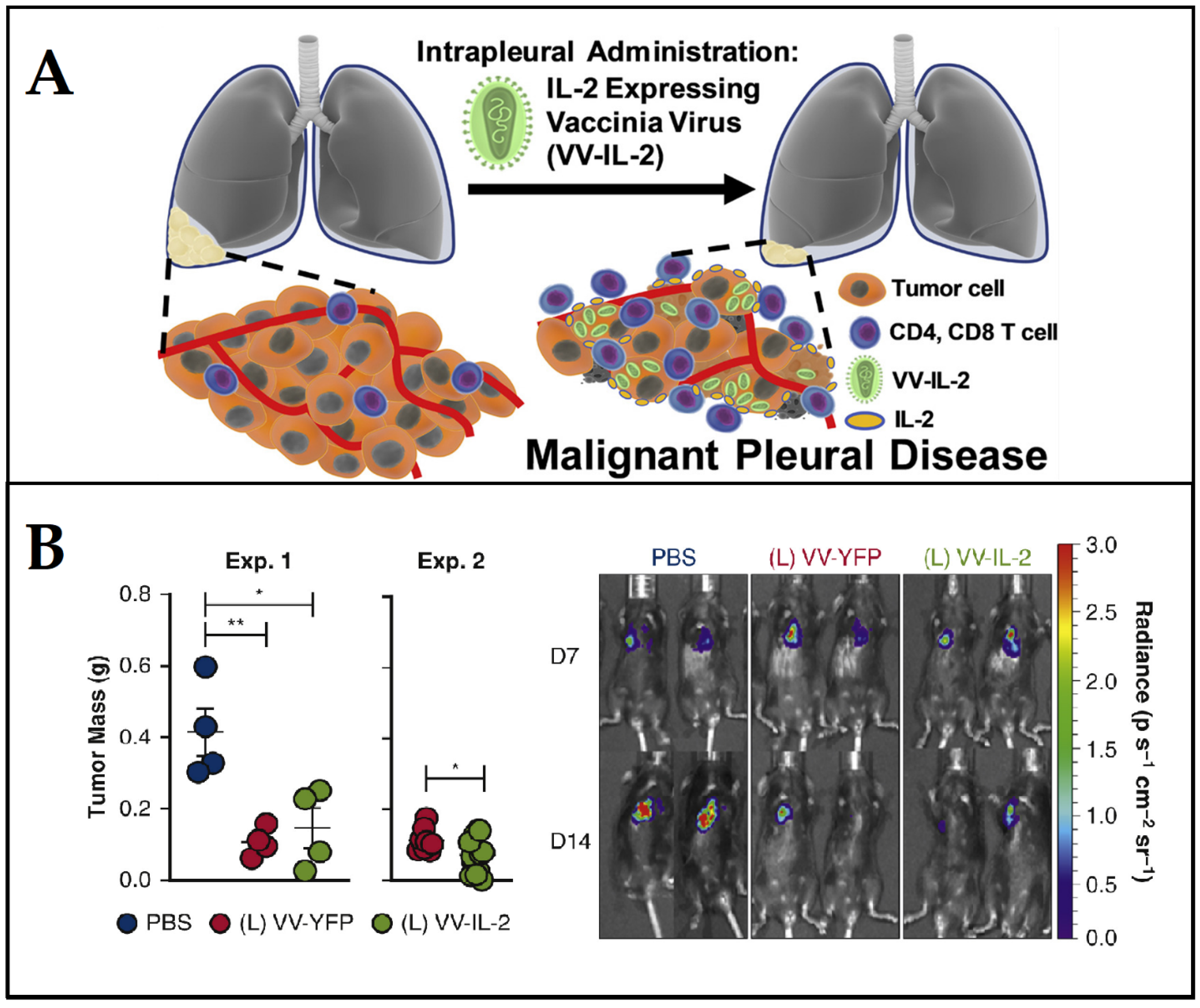
- Ekeke, C.N.; Russell, K.L.; Murthy, P.; Guo, Z.S.; Soloff, A.C.; Weber, D.; Pan, W.; Lotze, M.T.; Dhupar, R. Intrapleural IL-2 Expressing Oncolytic Virotherapy Enhances Acute Antitumor Effects and T Cell Receptor Diversity in Malignant Pleural Disease. J. Thorac. Cardiovasc. Surg. 2020, 163, e313–e328. [Google Scholar] [CrossRef]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the treatment of cancer. J. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef]
- Silk, A.W.; Margolin, K. Cytokine therapy. Hematol. Oncol. Clin. 2019, 33, 261–274. [Google Scholar] [CrossRef]
- Xue, D.; Hsu, E.; Fu, Y.-X.; Peng, H. Next-generation cytokines for cancer immunotherapy. Antibody Ther. 2021, 4, 123–133. [Google Scholar] [CrossRef]
- Qiu, Y.; Su, M.; Liu, L.; Tang, Y.; Pan, Y.; Sun, J. Clinical application of cytokines in cancer immunotherapy. Drug Des. Dev. Ther. 2021, 2269–2287. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Young, H.A.; Rosenberg, A.S. An overview of cytokines and cytokine antagonists as therapeutic agents. Ann. N. Y. Acad. Sci. 2009, 1182, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, M. Cytokine therapy for cancer. Curr. Pharm. Des. 2000, 6, 681–699. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef]
- Zhang, W.-y.; Wang, Y.; Guo, Y.-l.; Dai, H.-r.; Yang, Q.-m.; Zhang, Y.-j.; Zhang, Y.; Chen, M.-x.; Wang, C.-m.; Feng, K.-c. Treatment of CD20-directed chimeric antigen receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: An early phase IIa trial report. Signal Transduct. Target. Ther. 2016, 1, 16002. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, W.-y.; Han, Q.-w.; Liu, Y.; Dai, H.-r.; Guo, Y.-l.; Bo, J.; Fan, H.; Zhang, Y.; Zhang, Y.-j. Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin. Immunol. 2014, 155, 160–175. [Google Scholar] [CrossRef]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol. Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef]
- Siegler, E.L.; Kenderian, S.S. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: Insights into mechanisms and novel therapies. Front. Immunol. 2020, 11, 1973. [Google Scholar] [CrossRef]
- Murthy, H.; Iqbal, M.; Chavez, J.C.; Kharfan-Dabaja, M.A. Cytokine release syndrome: Current perspectives. ImmunoTargets Ther. 2019, 8, 43–52. [Google Scholar] [CrossRef]
- Lythgoe, M.P.; Liu, D.S.K.; Annels, N.E.; Krell, J.; Frampton, A.E. Gene of the month: Lymphocyte-activation gene 3 (LAG-3). J. Clin. Pathol. 2021, 74, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Huang, X.; Chen, X.; Liu, J.; Wu, C.; Pu, Q.; Wang, Y.; Kang, X.; Zhou, L. Characterization of a novel anti-human lymphocyte activation gene 3 (LAG-3) antibody for cancer immunotherapy. mAbs 2019, 11, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, V.; Khattak, A.; Haydon, A.; Eastgate, M.; Roy, A.; Prithviraj, P.; Mueller, C.; Brignone, C.; Triebel, F. Eftilagimod alpha, a soluble lymphocyte activation gene-3 (LAG-3) protein plus pembrolizumab in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8, e001681. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, H.; Xu, K.; Liu, X.; He, Y. Update on lymphocyte-activation gene 3 (LAG-3) in cancers: From biological properties to clinical applications. Chin. Med. J. 2022, 135, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef]
- Ji, G.; Zhang, Y.; Si, X.; Yao, H.; Ma, S.; Xu, Y.; Zhao, J.; Ma, C.; He, C.; Tang, Z. Biopolymer Immune Implants’ Sequential Activation of Innate and Adaptive Immunity for Colorectal Cancer Postoperative Immunotherapy. Adv. Mater. 2021, 33, 2004559. [Google Scholar] [CrossRef]
- Deng, J.; Zhao, S.; Zhang, X.; Jia, K.; Wang, H.; Zhou, C.; He, Y. OX40 (CD134) and OX40 ligand, important immune checkpoints in cancer. OncoTargets Ther. 2019, 12, 7347. [Google Scholar] [CrossRef]
- Sun, H.; Dong, Y.; Feijen, J.; Zhong, Z. Peptide-decorated polymeric nanomedicines for precision cancer therapy. J. Control. Release 2018, 290, 11–27. [Google Scholar] [CrossRef]
- Song, H.; Yang, P.; Huang, P.; Zhang, C.; Kong, D.; Wang, W. Injectable polypeptide hydrogel-based co-delivery of vaccine and immune checkpoint inhibitors improves tumor immunotherapy. Theranostics 2019, 9, 2299. [Google Scholar] [CrossRef]
- Shen, K.-Y.; Song, Y.-C.; Chen, I.-H.; Chong, P.; Liu, S.-J. Depletion of tumor-associated macrophages enhances the anti-tumor immunity induced by a Toll-like receptor agonist-conjugated peptide. Hum. Vaccines Immunother. 2014, 10, 3241–3250. [Google Scholar] [CrossRef]
- Choi, Y.J.; Park, S.-J.; Park, Y.-S.; Park, H.S.; Yang, K.M.; Heo, K. EpCAM peptide-primed dendritic cell vaccination confers significant anti-tumor immunity in hepatocellular carcinoma cells. PLoS ONE 2018, 13, e0190638. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Lacal, P.M.; Martire, M.; Navarra, P.; Graziani, G. Clinical experience with CTLA-4 blockade for cancer immunotherapy: From the monospecific monoclonal antibody ipilimumab to probodies and bispecific molecules targeting the tumor microenvironment. Pharmacol. Res. 2022, 175, 105997. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.-W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Mesa, C.; Fernández, L.E. Challenges facing adjuvants for cancer immunotherapy. Immunol. Cell Biol. 2004, 82, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yu, K.H.; Palmer, N.; Fox, K.; Kou, S.; Kohane, I.S. Autoimmune effects of lung cancer immunotherapy revealed by data-driven analysis on a nationwide cohort. Clin. Pharmacol. Ther. 2020, 107, 388–396. [Google Scholar] [CrossRef]
- Amos, S.M.; Duong, C.P.; Westwood, J.A.; Ritchie, D.S.; Junghans, R.P.; Darcy, P.K.; Kershaw, M.H. Autoimmunity associated with immunotherapy of cancer. Blood 2011, 118, 499–509. [Google Scholar] [CrossRef]
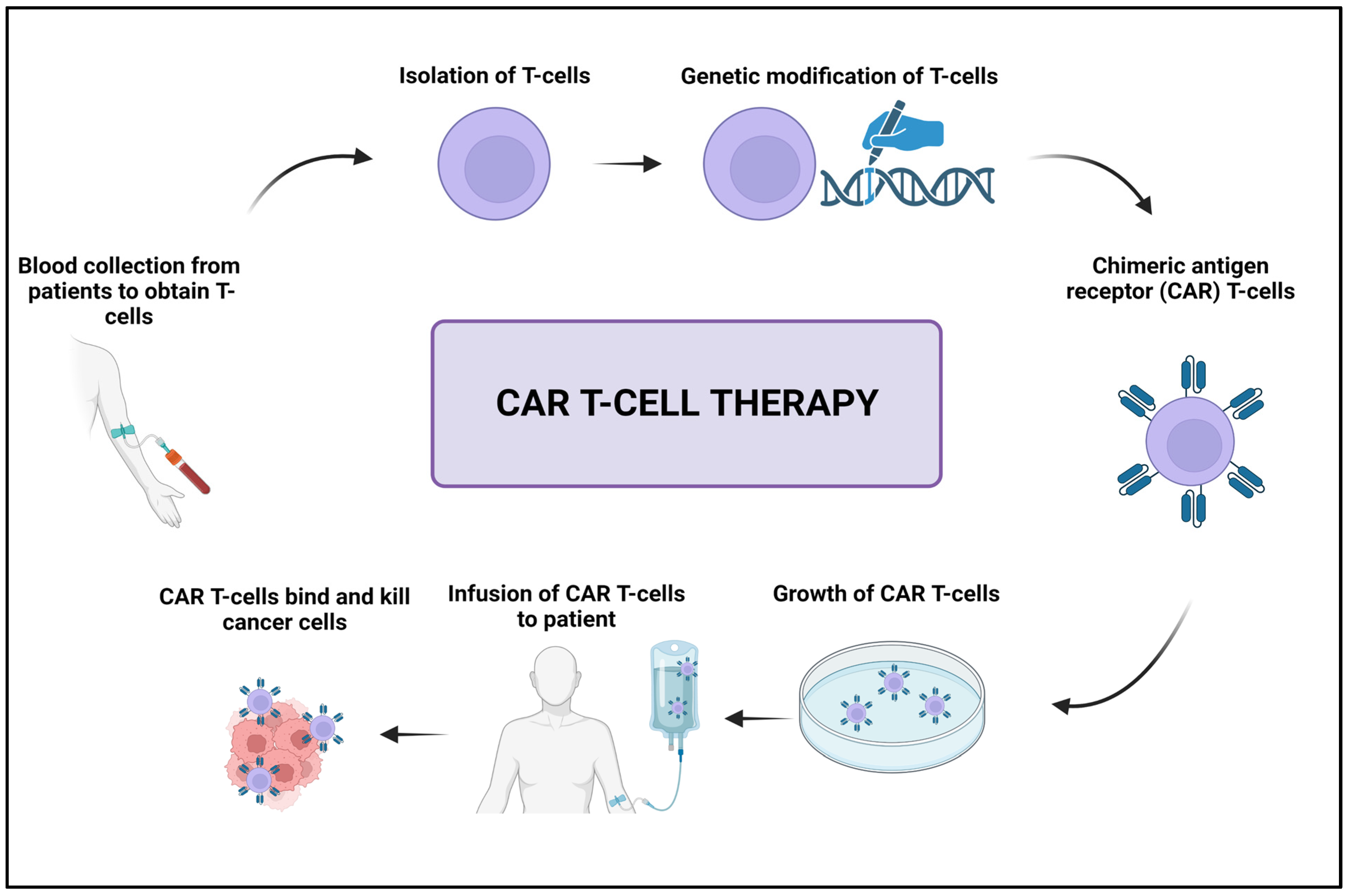
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell therapy: A new era in cancer immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Emens, L.A.; Ascierto, P.A.; Darcy, P.K.; Demaria, S.; Eggermont, A.M.; Redmond, W.L.; Seliger, B.; Marincola, F.M. Cancer immunotherapy: Opportunities and challenges in the rapidly evolving clinical landscape. Eur. J. Cancer 2017, 81, 116–129. [Google Scholar] [CrossRef]
- Quinn, C.; Garrison, L.P.; Pownell, A.K.; Atkins, M.B.; De Pouvourville, G.; Harrington, K.; Ascierto, P.A.; McEwan, P.; Wagner, S.; Borrill, J. Current challenges for assessing the long-term clinical benefit of cancer immunotherapy: A multi-stakeholder perspective. J. ImmunoTherapy Cancer 2020, 8, e000648. [Google Scholar] [CrossRef] [PubMed]
- Berz, A.M.; Dromain, C.; Vietti-Violi, N.; Boughdad, S.; Duran, R. Tumor response assessment on imaging following immunotherapy. Front. Oncol. 2022, 12, 982983. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
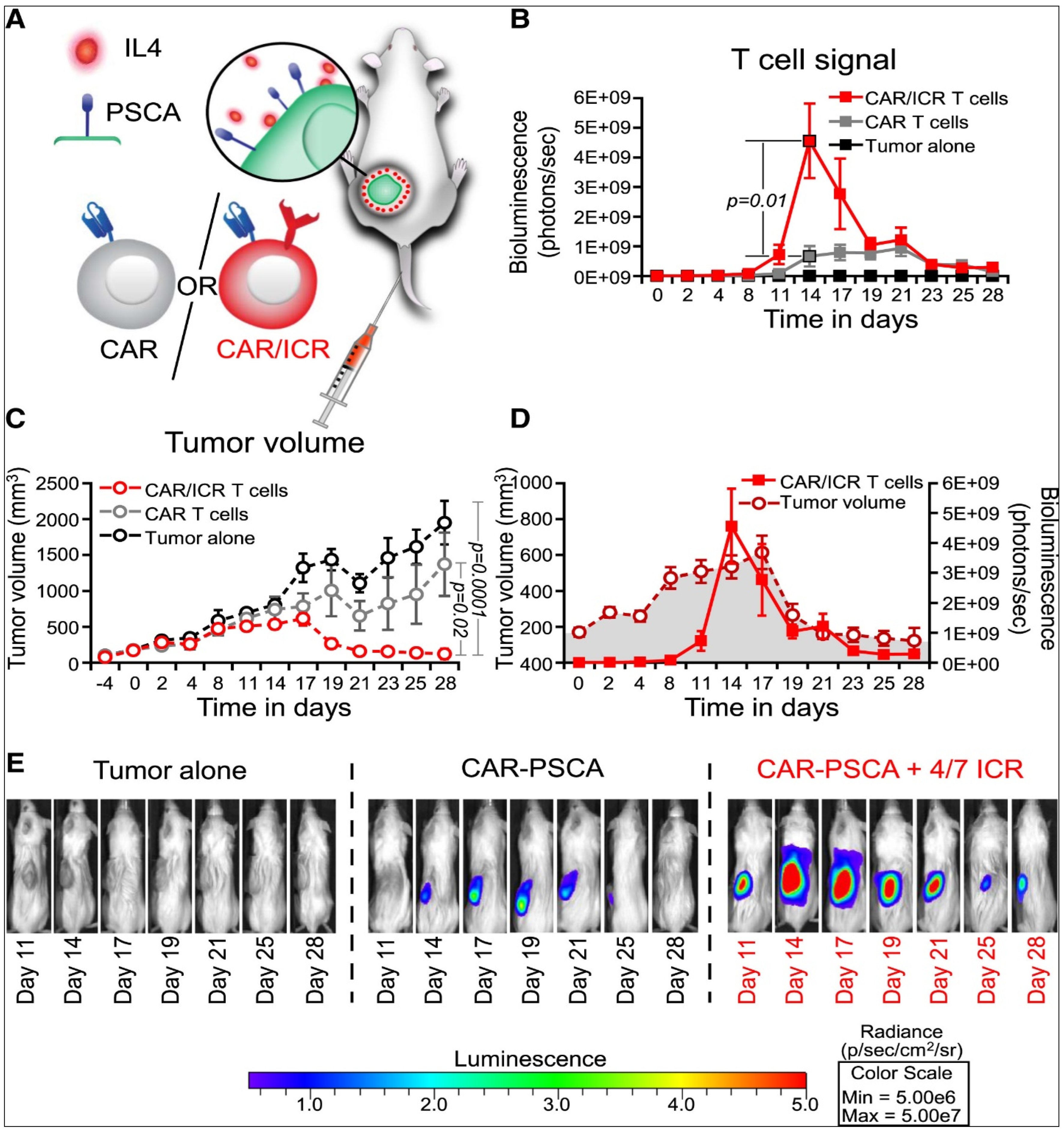{kind=link}
| Scientist/s and Year | Remark | Ref |
|---|---|---|
| W. B. Coley in 1893 | The first use of immunotherapy to treat a cancer patient. | [45] |
| Hericourt and Richet in 1895 | Serotherapy in cancer treatment. | [47] |
| W. B. Coley in 1899 | The first vaccine against sarcoma. | [48] |
| Leyden et al. in 1902 | The first cancer vaccine was created using a patient’s tumor cells. | [49] |
| J. B. Murphy in 1914 | Understanding the function of lymphocytes in the rejection of grafted tumors. | [50] |
| Ernest Witebsky in 1929 | Tumor molecular markers are used as antigens. | [51] |
| Burnet and Lewis in 1957 | The concept of the natural anticancer immune response. | [52] |
| Sjögren et al. in 1971 | The role of antibodies in shielding tumor cells from the anticancer immune reaction. | [53] |
| Carswell et al. in 1975 | Discovery of tumor necrosis factor (TNF-α) with antitumor effects. | [54] |
| Morgan et al. in 1976 | The first discovery of IL-2 as a T-cell growth factor. | [55] |
| Stevenson and Elliott in 1977 | First anti-idiotype antibodies activate immune cells against the tumor. | [56] |
| Ronald Levy in 1981 | First successful cancer treatment with anti-idiotype antibody. | [57] |
| Taniguchi et al. in 1983 | First cloning of T-cell growth factor (IL-2). | [58] |
| Kappler et al. in 1983 | First identification of T-cell antigen receptor. | [59] |
| Knuth et al. in 1984 | First clinical trial on the potential use of T cells to fight off tumors. | [60] |
| S A Rosenberg in 1988 | Successful application of IL-2-based cancer therapy. | [61] |
| Gross et al. in 1989 | The first use of genetically engineered T cells for tumor targeting. | [62] |
| Dranoff et al. in 1993 | Using macrophage colony-stimulating factor (M-CSF) to improve cancer immunity. | [63] |
| Leach et al. in 1996 | Inhibition of the immune cells CTLA-4 molecule in cancer treatment. | [64] |
| Levy and Rastetter in 1997 | First FDA-approved monoclonal antibody as an anticancer drug. | [65] |
| J. P. Allison in 2000 | First immune-checkpoint inhibitor drug against CTLA-4. | [66] |
| Hirano et al. in 2005 | Development of fully human anti-PD-1 antibody for cancer therapy. | [67] |
| Kershaw et al. in 2006 | Development of adoptive cell-mediated immunotherapy. | [68] |
| Rosenberg et al. in 2008 | Anti-PD-1 antibody started phase 1 of the clinical trial for cancer | [69] |
| June and Whitehead in 2012 | First leukemia treatment by adoptive cell (CAR-T based therapy). | [70] |
| Quezada et al. in 2016 | Genetic modification of immune cells to prompt antitumor response. | [71] |
| Wallace et al. in 2016 | A first clinical trial of gene editing tool CRISPR/Cas 9 for cancer. | [72] |
| Freedman et al. in 2018 | Genetically engineered virus to selectively kill cancer cells. | [73] |
| Zhang et al. in 2020 | The use of miRNA to regulate immune checkpoints. | [74] |
| Virus (Family) | Nucleic Acid Type (Group) | Study Remarks | Ref |
|---|---|---|---|
| Reovirus (Reoviridae) | dsRNA (G III) | Viruses exhibited a safe and tolerable toxicity profile in early clinical trials, with minimal viral shedding, replication, localization, and cytotoxic effects. | [122] |
| Influenza A Virus (Orthomyxoviridae) | ssRNA-ve (G V) | Engineered viruses exhibited tumor-ablative potential and do not replicate in normal cell lines. | [123] |
| Herpes Simplex Virus1 (Herpesviridae) | dsDNA (G I) | A virus exhibited high antitumor efficacy, has a high safety margin, is well tolerated and its use is associated with a low frequency of adverse effects in most of the treated patients. | [124] |
| Adenovirus (Adenoviridae) | dsDNA (G I) | Clinical data revealed that the virus was tolerable and effective even when combined with other therapeutic approaches. | [125] |
| Measles virus (Paramyxoviridae) | ssRNA-ve (G V) | The virus replicated selectively in the tumor and significantly suppressed its development. It was completely safe and did not result in any measles-like symptoms. | [126] |
| Vaccinia virus (Poxviridae) | dsDNA (G I) | The virus replicated rapidly in tumor cells with significant antitumor effects, but its cytotoxicity varied based on cell lines. | [127] |
| Pseudorabies (Herpesviridae) | dsDNA (G I) | Selective replication of viruses has been observed in many kinds of cancers, with tumor growth being significantly slowed by more than 50%. | [128] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kciuk, M.; Yahya, E.B.; Mohamed Ibrahim Mohamed, M.; Rashid, S.; Iqbal, M.O.; Kontek, R.; Abdulsamad, M.A.; Allaq, A.A. Recent Advances in Molecular Mechanisms of Cancer Immunotherapy. Cancers 2023, 15, 2721. https://doi.org/10.3390/cancers15102721
Kciuk M, Yahya EB, Mohamed Ibrahim Mohamed M, Rashid S, Iqbal MO, Kontek R, Abdulsamad MA, Allaq AA. Recent Advances in Molecular Mechanisms of Cancer Immunotherapy. Cancers. 2023; 15(10):2721. https://doi.org/10.3390/cancers15102721
Chicago/Turabian StyleKciuk, Mateusz, Esam Bashir Yahya, Montaha Mohamed Ibrahim Mohamed, Summya Rashid, Muhammad Omer Iqbal, Renata Kontek, Muhanad A. Abdulsamad, and Abdulmutalib A. Allaq. 2023. "Recent Advances in Molecular Mechanisms of Cancer Immunotherapy" Cancers 15, no. 10: 2721. https://doi.org/10.3390/cancers15102721
APA StyleKciuk, M., Yahya, E. B., Mohamed Ibrahim Mohamed, M., Rashid, S., Iqbal, M. O., Kontek, R., Abdulsamad, M. A., & Allaq, A. A. (2023). Recent Advances in Molecular Mechanisms of Cancer Immunotherapy. Cancers, 15(10), 2721. https://doi.org/10.3390/cancers15102721