
Intratumoral Heterogeneity in Lung Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Genetic ITH in LC
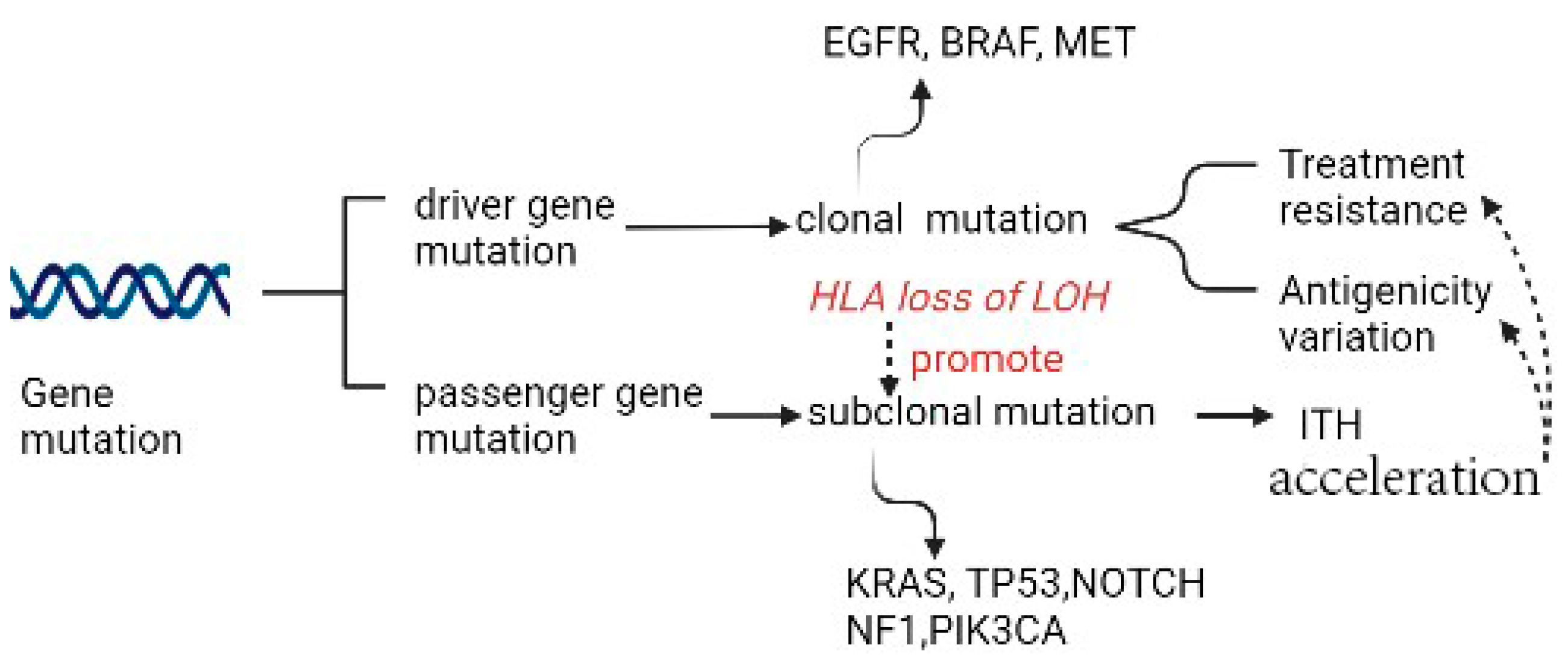
2.1. Gene Mutation as a Source of Genetic ITH
2.2. Treatment Resistance Due to Specific Gene Mutation
2.3. Antigenicity Variation Due to Specific Gene Mutation
2.4. Genomic Instability Due to Specific Gene Mutation
2.5. Relationship between Genetic ITH and Immunological ITH
3. ITH of Pulmonary Neoplastic Microenvironment
3.1. Immunological ITH and Pulmonary Neoplastic Microenvironment
3.2. Other Neoplastic Microenvironmental Factors and TMH
3.3. TIME ITH Affected by TME and Genetic ITH
3.4. Immunoescape Affected by TIME
3.5. Relationship of Immunoediting and ITH in LC
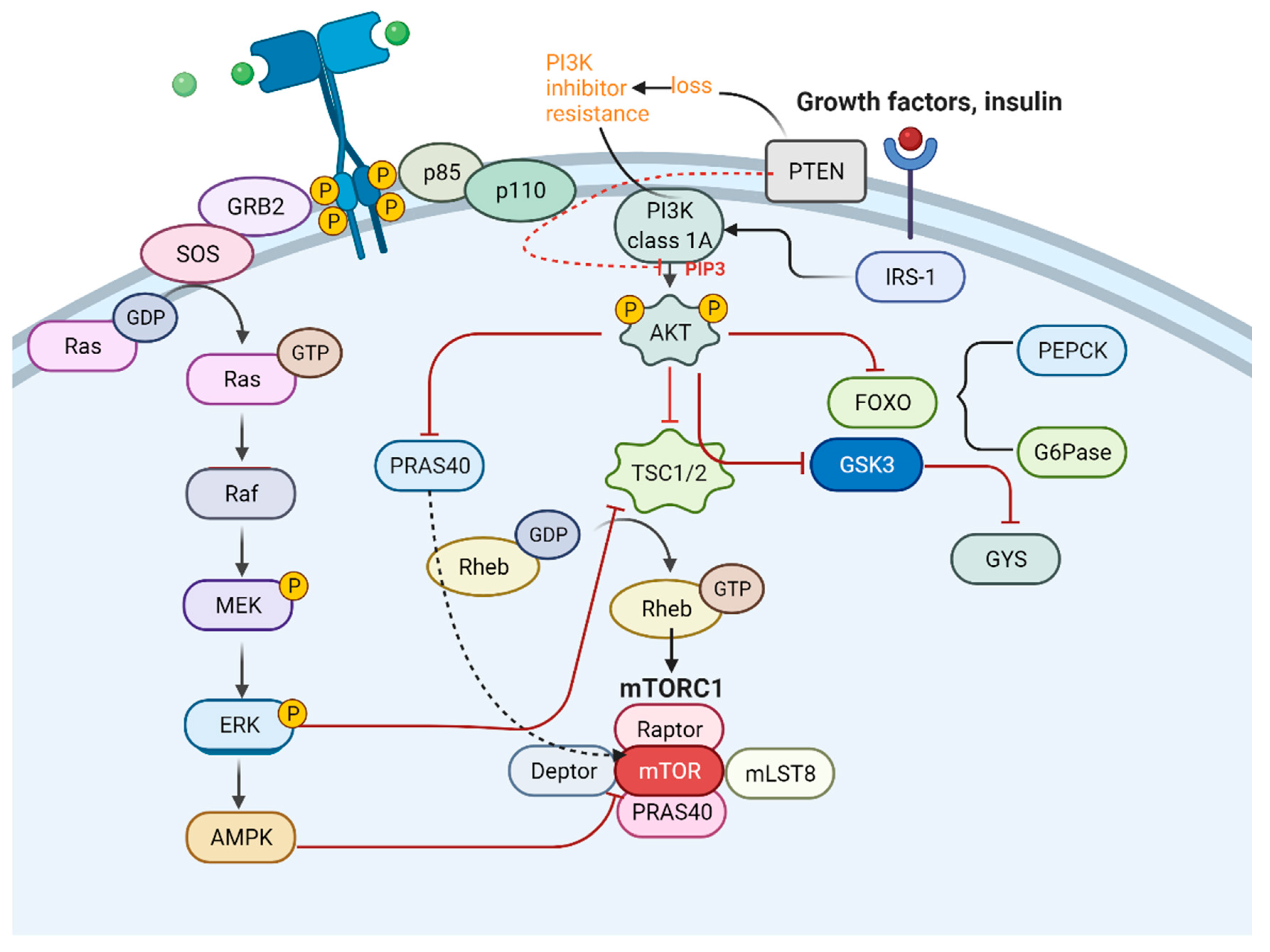
4. Metabolic ITH in LC
4.1. Relationship across Metabolic ITH, Genetic ITH, and TMH
4.2. Main Forms of Metabolic ITH in LC
4.3. ITH in Glucose Metabolism
4.4. ITH in Lipid Metabolism
4.5. Typical Signal Pathways Influencing Metabolic ITH
5. Spatial and Temporal ITH in LC
5.1. Temporal ITH in LC
5.1.1. Lung Cancer Reappearance Due to Genetic Temporal ITH
5.1.2. Drug Resistance Due to Genetic and TME Temporal ITH
5.1.3. A Landscape Continuum of Functional Cells Due to Temporal ITH in TME
5.1.4. Epigenetic ITH Due to Metabolic Temporal ITH
5.2. Spatial ITH in LC
5.2.1. Difference Related to Regions Due to Genetic Spatial ITH
5.2.2. The Classification and Diversity in TME Due to Spatial ITH
6. ITH of LC and Novel Research Methods and Tools
7. Therapeutic Strategies for Treating LC with Elevated ITH
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LC | Lung cancer |
| ITH | Intratumoral heterogeneity |
| TME | Tumor microenvironment |
| TIME | Tumor immune microenvironment |
| NSCLC | Non-small cell lung cancer |
| HLA | Human leukocyte antigen |
| LOH | Loss of heterozygosity |
| SqCC | Squamous cell carcinoma |
| ADC | Adenocarcinoma |
| SCLC | Small cell lung cancer |
| PTEN | Phosphatase and tensin homolog |
| EGFR | Growth factor receptor |
| ALK | Anaplastic lymphoma kinase |
| TKI | Tyrosine kinase inhibitor |
| MHC | Major histocompatibility complex |
| CTLs | Cytotoxic T lymphocytes |
| TNAs | Tumor neoantigens |
| MSI | Microsatellite instability |
| MMR | Mismatch repair |
| TMH | Tumor microenvironment heterogeneity |
| ICB | Immune check-point blockade |
| CTLA-4 | Cytotoxic T lymphocyte-associated antigen-4 |
| PD-1 | Programmed cell death protein |
| ICT | Immune check-point therapy |
| DCs | Dendritic cells |
| GM-CSF | Granulocyte monocyte colony-stimulating factor |
| Ccl4 | CC motif ligand4 |
| TAM | Tumor-associated macrophage |
| TILs | Tumor-infiltrating lymphocytes |
| Treg | Regulatory T cell |
| RCD | Regulated cell death |
| PRRs | Pattern recognition receptors |
| TLRs | Toll-like receptors |
| DAMPs | Damage-associated molecular patterns |
| IRF3 | Interfon regulatory factor 3 |
| GSDMD | Gasdermin D |
| INAM | IRF3-dependent NK-activating molecule |
| ME | Malic enzymes |
| METTL3 | Methyltransferase like 3 |
| ATG-5 | Autophagy-related gene-5 |
| TCA | Tricarboxylic acid cycle |
| ROS | Reactive oxygen species |
| HK2 | Hexokinase 2 |
| PFK | Phosphofructokinase |
| PKM | Pyruvate kinase |
| LDH | Lactate dehydrogenase |
| EGFR-TKI | Epidermal growth factor receptor-tyrosine kinase inhibitor |
| BCC | Breast cancer cells |
| PDHE1α | Pyruvate dehydrogenase complex E1α |
| EMT | Epithelial-mesenchymal-transition |
| LPA | Lysophosphatidic acid |
| PGE2 | Prostaglandin E2 |
| S1P | Sphingosine-1-phosphate |
| CREB | Cyclic AMP-responsive element binding |
| CTCs | Circulating tumor cells |
| DTCs | Disseminated tumor cells |
| RTK | Receptor tyrosine kinases |
| PDGFR | Platelet-derived growth factor receptor |
| IRS | Insulin receptor substrate |
| FOXO | Forkhead box O |
| G6Pase | Glucose-6-phosphatase |
| PTKs | Protein tyrosine kinase |
| CAFs | Cancer-associated fibroblasts |
| TEFF | Effector T cell |
| IFN-γ | Interferon gamma |
| TIL-Bs | Tumor-infiltrating B cells |
| TANs | Tumor-associated neutrophils |
| MMPs | Matrix metalloproteinases |
| KLKs | Kallikrein-related peptidases |
| PARs | Protease-activated receptors |
| CAA | Cancer-associated adipocytes |
| TAAs | Tumor-associated antigens |
| ICIs | Immune checkpoint inhibitors |
| TECs | Tumor endothelial cells |
| ICD | Immunogenic cell death |
| CAR T cells | Chimeric antigen receptor T cells |
References
- Raynaud, F.; Mina, M.; Tavernari, D.; Ciriello, G. Pan-cancer inference of intra-tumor heterogeneity reveals associations with different forms of genomic instability. PLoS Genet. 2018, 14, e1007669. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Minchom, A.; Larkin, J. BRIM-1, -2 and -3 trials: Improved survival with vemurafenib in metastatic melanoma patients with a BRAFV600E mutation. Futur. Oncol. 2012, 8, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Devarakonda, S.; Rotolo, F.; Tsao, M.; Lanc, I.; Brambilla, E.; Masood, A.; Olaussen, K.A.; Fulton, R.; Sakashita, S.; McLeer-Florin, A.; et al. Tumor Mutation Burden as a Biomarker in Resected Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2995–3006. [Google Scholar] [CrossRef]
- Zito Marino, F.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular Heterogeneity in Lung Cancer: From Mechanisms of Origin to Clinical Implications. Int. J. Med. Sci. 2019, 16, 981–989. [Google Scholar] [CrossRef]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef]
- Fathi, Z.; Mousavi, S.A.J.; Roudi, R.; Ghazi, F. Distribution of KRAS, DDR2, and TP53 gene mutations in lung cancer: An analysis of Iranian patients. PLoS ONE 2018, 13, e0200633. [Google Scholar] [CrossRef]
- La Fleur, L.; Falk-Sörqvist, E.; Smeds, P.; Berglund, A.; Sundström, M.; Mattsson, J.S.; Brandén, E.; Koyi, H.; Isaksson, J.; Brunnström, H. Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11. Lung Cancer 2019, 130, 50–58. [Google Scholar] [CrossRef]
- Karachaliou, N.; Mayo, C.; Costa, C.; Magrí, I.; Gimenez-Capitan, A.; Molina-Vila, M.A.; Rosell, R. KRAS Mutations in Lung Cancer. Clin. Lung Cancer 2013, 14, 205–214. [Google Scholar] [CrossRef]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non–Small Cell Lung CancerCo-occurring Genomic Alterations in KRAS-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef]
- Van Gool, I.C.; Eggink, F.A.; Freeman-Mills, L.; Stelloo, E.; Marchi, E.; de Bruyn, M.; Palles, C.; Nout, R.A.; de Kroon, C.D.; Osse, E.M. POLE proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin. Cancer Res. 2015, 21, 3347–3355. [Google Scholar] [CrossRef] [PubMed]
- Caron, P.; Choudjaye, J.; Clouaire, T.; Bugler, B.; Daburon, V.; Aguirrebengoa, M.; Mangeat, T.; Iacovoni, J.S.; Álvarez-Quilón, A.; Cortés-Ledesma, F.; et al. Non-redundant Functions of ATM and DNA-PKcs in Response to DNA Double-Strand Breaks. Cell Rep. 2015, 13, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Pierce, L.M.; Sivaraman, L.; Chang, W.; Lum, A.; Donlon, T.; Seifried, A.; Wilkens, L.R.; Lau, A.F.; Le Marchand, L. Relationships of TP53 codon 72 and HRAS1 polymorphisms with lung cancer risk in an ethnically diverse population. Cancer Epidemiol. Biomark. Prev. 2000, 9, 1199–1204. [Google Scholar]
- Dong, Z.-Y.; Zhong, W.-Z.; Zhang, X.-C.; Su, J.; Xie, Z.; Liu, S.-Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.-L.; Zhou, Q. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Cai, D.W.; Georges, R.N.; Mukhopadhyay, T.; Grimm, E.A.; Roth, J.A. Therapeutic Effect of a Retroviral Wild-Type p53 Expression Vector in an Orthotopic Lung Cancer Model. Gynecol. Oncol. J. Natl. Cancer Inst. 1994, 86, 1458–1462. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.; Nguyen, D.; Lawrence, D.; Kemp, B.; Carrasco, C.; Ferson, D.; Hong, W.; Komaki, R.; Lee, J.; Nesbitt, J. Retrovirus–mediated wild–type P53 gene transfer to tumors of patients with lung cancer. Nat. Med. 1996, 2, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Ferrara, R.; Signorelli, D.; Ghidini, A.; Proto, C.; Roudi, R.; Sabet, M.N.; Facelli, S.; Garassino, M.C.; Luciani, A.; et al. Immune checkpoint inhibitors and chemotherapy in first-line NSCLC: A meta-analysis. Immunotherapy 2021, 13, 621–631. [Google Scholar] [CrossRef]
- Zhu, J.; Yuan, Y.; Wan, X.; Yin, D.; Li, R.; Chen, W.; Suo, C.; Song, H. Immunotherapy (excluding checkpoint inhibitors) for stage I to III non-small cell lung cancer treated with surgery or radiotherapy with curative intent. Cochrane Database Syst. Rev. 2017, 12, CD011300. [Google Scholar] [CrossRef]
- Tartarone, A.; Roviello, G.; Lerose, R.; Roudi, R.; Aieta, M.; Zoppoli, P.; Reck, M.; Borghaei, H.; O’byrne, K.J.; Santoni, M.; et al. Anti-PD-1 versus anti-PD-L1 therapy in patients with pretreated advanced non-small-cell lung cancer: A meta-analysis. Futur. Oncol. 2019, 15, 2423–2433. [Google Scholar] [CrossRef]
- Montesion, M.; Murugesan, K.; Jin, D.X.; Sharaf, R.; Sanchez, N.; Guria, A.; Minker, M.; Li, G.; Fisher, V.; Sokol, E.S.; et al. Somatic HLA Class I Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discov. 2021, 11, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.-C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; von Felden, J.; Labgaa, I.; Dʹavola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef]
- Senovilla, L.; Vitale, I.; Martins, I.; Tailler, M.; Pailleret, C.; Michaud, M.; Galluzzi, L.; Adjemian, S.; Kepp, O.; Niso-Santano, M.; et al. An Immunosurveillance Mechanism Controls Cancer Cell Ploidy. Science 2012, 337, 1678–1684. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Labrie, M.; Brugge, J.S.; Mills, G.B.; Zervantonakis, I.K. Therapy resistance: Opportunities created by adaptive responses to targeted therapies in cancer. Nat. Rev. Cancer 2022, 22, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Duan, Y.; Xiao, Y.; Sun, K.; Qi, Y.; Zhang, Y.; Ahmed, Z.; Moiani, D.; Yao, J.; Li, H.; et al. Vitamin E Enhances Cancer Immunotherapy by Reinvigorating Dendritic Cells via Targeting Checkpoint SHP. Cancer Discov. 2022, 12, 1742–1759. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Bonavita, E.; Gentile, S.; Rubino, M.; Maina, V.; Papait, R.; Kunderfranco, P.; Greco, C.; Feruglio, F.; Molgora, M.; Laface, I.; et al. PTX3 Is an Extrinsic Oncosuppressor Regulating Complement-Dependent Inflammation in Cancer. Cell 2015, 160, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Khalili, J.S.; Liu, S.; Rodriguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- O’Sullivan, T.; Saddawi-Konefka, R.; Vermi, W.; Koebel, C.M.; Arthur, C.; White, J.M.; Uppaluri, R.; Andrews, D.; Ngiow, S.F.; Teng, M.; et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med. 2012, 209, 1869–1882. [Google Scholar] [CrossRef]
- Joshi, K.; TRACERx Consortium; De Massy, M.R.; Ismail, M.; Reading, J.; Uddin, I.; Woolston, A.; Hatipoglu, E.; Oakes, T.; Rosenthal, R.; et al. Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat. Med. 2019, 25, 1549–1559. [Google Scholar] [CrossRef]
- Nguyen, P.H.D.; Ma, S.; Phua, C.Z.J.; Kaya, N.A.; Lai, H.L.H.; Lim, C.J.; Lim, J.Q.; Wasser, M.; Lai, L.; Tam, W.L.; et al. Intratumoural immune heterogeneity as a hallmark of tumour evolution and progression in hepatocellular carcinoma. Nat. Commun. 2021, 12, 227. [Google Scholar] [CrossRef]
- Zhao, M.; Yao, P.; Mao, Y.; Wu, J.; Wang, W.; Geng, C.; Cheng, J.; Du, W.; Jiang, P. Malic enzyme 2 maintains protein stability of mutant p53 through 2-hydroxyglutarate. Nat. Metab. 2022, 4, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, Q.; Li, G.; Zhang, Q.; Zhuo, L.; Han, X.; Zhang, M.; Chen, X.; Pan, T.; Yan, L.; et al. The mechanism of m(6)A methyltransferase METTL3-mediated autophagy in reversing gefitinib resistance in NSCLC cells by beta-elemene. Cell Death Dis. 2020, 11, 969. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, T.; Herbig, J.; Kohl, I.; Las, G.; Cancilla, J.C.; Torrecilla, J.S.; Ilouze, M.; Haick, H.; Peled, N. Cancer metabolism: The volatile signature of glycolysis—In vitro model in lung cancer cells. J. Breath Res. 2017, 11, 016008. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef]
- Hoang-Minh, L.B.; Siebzehnrubl, F.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Massari, M.S.; Patel, J.; Amin, K.; et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018, 37, e98772. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Parlo, R.A.; Coleman, P.S. Continuous pyruvate carbon flux to newly synthesized cholesterol and the suppressed evolution of pyruvate-generated CO2 in tumors: Further evidence for a persistent truncated Krebs cycle in hepatomas. Biochim. Biophys. Acta BBA Mol. Cell Res. 1986, 886, 169–176. [Google Scholar] [CrossRef]
- Vaitheesvaran, B.; Xu, J.; Yee, J.; Lu, Q.-Y.; Go, V.L.; Xiao, G.G.; Lee, W.-N. The Warburg effect: A balance of flux analysis. Metabolomics 2014, 11, 787–796. [Google Scholar] [CrossRef]
- Wu, R.; Galan-Acosta, L.; Norberg, E. Glucose metabolism provide distinct prosurvival benefits to non-small cell lung carcinomas. Biochem. Biophys. Res. Commun. 2015, 460, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-B.; Gu, J.-D.; Zhou, Q.-H. Review of aerobic glycolysis and its key enzymes—New targets for lung cancer therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef] [PubMed]
- IGA. Inhibition of Stearoyl-CoA Desaturase 1 expression in human lung adenocarcinoma cells impairs tumorigenesis. Int. J. Oncol. 1992, 33, 839–850. [Google Scholar]
- Takeda, K. [Overcoming resistance to EGFR-TKI in lung cancer]. Gan Kagaku Ryoho. 2009, 36, 552–556. [Google Scholar]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.F.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, M.; Gao, H.; Yu, G.; Zhao, Y.; Yao, F.; Yang, W. MAPK signalling-induced phosphorylation and subcellular translocation of PDHE1α promotes tumour immune evasion. Nat. Metab. 2022, 4, 374–388. [Google Scholar] [CrossRef]
- Yano, K. Lipid metabolic pathways as lung cancer therapeutic targets: A computational study. Int. J. Mol. Med. 2012, 29, 519–529. [Google Scholar] [CrossRef]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro-o, M.; Deberardinis, R.J.; Boothman, D.A. Metabolic reprogramming during TGFbeta1-induced epithelial-to-mesenchymal transition. Oncogene 2015, 34, 3908–3916. [Google Scholar] [CrossRef]
- Kazlauskas, A. Lysophosphatidic acid contributes to angiogenic homeostasis. Exp. Cell Res. 2015, 333, 166–170. [Google Scholar] [CrossRef]
- Mendelson, K.; Evans, T.; Hla, T. Sphingosine 1-phosphate signalling. Development 2014, 141, 5–9. [Google Scholar] [CrossRef]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-F.; Liu, Y.; Huang, H.; Shan, L.; Han, Z.-G.; Liu, J.-Y.; Li, Y.-L.; Dong, X.; Zeng, W. MicroRNA-133b/EGFR axis regulates esophageal squamous cell carcinoma metastases by suppressing anoikis resistance and anchorage-independent growth. Cancer Cell Int. 2018, 18, 193. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, T.; Huang, H.; Jiang, Y.; Yang, L.; Lin, Z.; He, H.; Liu, T.; Wu, B.; Chen, J.; et al. miR-363-3p inhibits tumor growth by targeting PCNA in lung adenocarcinoma. Oncotarget 2017, 8, 20133–20144. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, J.; Dearden, S.; Ratcliffe, M.; Walker, J. Mutation status concordance between primary lesions and metastatic sites of advanced non-small-cell lung cancer and the impact of mutation testing methodologies: A literature review. J. Exp. Clin. Cancer Res. 2015, 34, 92. [Google Scholar] [CrossRef] [PubMed]
- Schmid, K.; Oehl, N.; Wrba, F.; Pirker, R.; Pirker, C.; Filipits, M. Faculty Opinions recommendation of EGFR/KRAS/BRAF mutations in primary lung adenocarcinomas and corresponding locoregional lymph node metastases. Clin. Cancer Res. 2009, 15, 4554–4560. [Google Scholar] [CrossRef]
- Yatabe, Y.; Matsuo, K.; Mitsudomi, T. Heterogeneous Distribution of EGFR Mutations Is Extremely Rare in Lung Adenocarcinoma. J. Clin. Oncol. 2011, 29, 2972–2977. [Google Scholar] [CrossRef]
- Badalian, G.; Barbai, T.; Rásó, E.; Derecskei, K.; Szendrõi, M.; Tímár, J. Phenotype of bone metastases of nonsmall cell lung cancer: Epidermal growth factor receptor expression and KRAS mutational status. Pathol. Oncol. Res. 2007, 13, 99–104. [Google Scholar] [CrossRef]
- Alsdorf, W.H.; Clauditz, T.S.; Hoenig, T.; Quaas, A.; Sirma, H.; Koenig, A.M.; Izbicki, J.; Sauter, G.; Marx, A.H.; Grob, T.J. Intratumoral heterogeneity of KRAS mutation is rare in non-small-cell lung cancer. Exp. Mol. Pathol. 2013, 94, 155–159. [Google Scholar] [CrossRef]
- Li, S.; Rosell, R.; Urban, A.; Font, A.; Ariza, A.; Armengol, P.; Abad, A.; Navas, J.; Monzo, M. K-ras gene point mutation: A stable tumor marker in non-small cell lung carcinoma. Lung Cancer 1994, 11, 19–27. [Google Scholar] [CrossRef]
- Cheng, X.; Chen, H. Tumor heterogeneity and resistance to EGFR-targeted therapy in advanced nonsmall cell lung cancer: Challenges and perspectives. OncoTargets Ther. 2014, 7, 1689–1704. [Google Scholar] [CrossRef]
- Renovanz, M.; Kim, E.L. Intratumoral Heterogeneity, Its Contribution to Therapy Resistance and Methodological Caveats to Assessment. Front. Oncol. 2014, 4, 142. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-H.; Chen, Y.-M. Influence of chemotherapy on EGFR mutation status. Transl. Lung Cancer Res. 2013, 2, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Wang, Z.; Wang, Y.; Zhuo, M.; Zhou, Q.; Duan, J.; Yang, L.; Wu, M.; An, T.; Zhao, J.; et al. Detection and Clinical Significance of Intratumoral EGFR Mutational Heterogeneity in Chinese Patients with Advanced Non-Small Cell Lung Cancer. PLoS ONE 2013, 8, e54170. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Wang, Z.; Chen, K.; Zhao, J.; Lee, J.J.; Wang, S.; Zhou, Q.; Zhuo, M.; Mao, L.; An, T.; et al. Influence of Chemotherapy on EGFR Mutation Status Among Patients with Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2012, 30, 3077–3083. [Google Scholar] [CrossRef]
- Thatcher, N.; Chang, A.; Parikh, P.; Rodrigues Pereira, J.; Ciuleanu, T.; von Pawel, J.; Thongprasert, S.; Tan, E.H.; Pemberton, K.; Archer, V.; et al. Faculty Opinions recommendation of Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: Results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005, 366, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Okami, J.; Kodama, K.; Higashiyama, M.; Kato, K. Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci. 2008, 99, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.B.; Salama, A.K.S. A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef]
- Upadhaya, S.; Neftelino, S.T.; Hodge, J.P.; Oliva, C.; Campbell, J.R.; Yu, J.X. Combinations take centre stage in PD1/PDL1 inhibitor clinical trials. Nat. Rev. Drug Discov. 2021, 20, 168–170. [Google Scholar] [CrossRef]
- Madden, E.C.; Gorman, A.M.; Logue, S.E.; Samali, A. Tumour cell secretome in chemoresistance and tumour recurrence. Trends Cancer 2020, 6, 489–505. [Google Scholar] [CrossRef]
- Maynard, A.; McCoach, C.E.; Rotow, J.K.; Harris, L.; Haderk, F.; Kerr, D.L.; Yu, E.A.; Schenk, E.L.; Tan, W.; Zee, A.; et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 2020, 182, 1232–1251.e22. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2019, 176, 404. [Google Scholar] [CrossRef]
- Laumont, C.M.; Banville, A.C.; Gilardi, M.; Hollern, D.P.; Nelson, B.H. Tumour-infiltrating B cells: Immunological mechanisms, clinical impact and therapeutic opportunities. Nat. Rev. Cancer 2022, 22, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Pittet, M.J.; Michielin, O.; Migliorini, D. Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 2022, 19, 402–421. [Google Scholar] [CrossRef] [PubMed]
- Mascaux, C.; Angelova, M.; Vasaturo, A.; Beane, J.; Hijazi, K.; Anthoine, G.; Buttard, B.; Rothe, F.; Willard-Gallo, K.; Haller, A.; et al. Faculty Opinions recommendation of Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature 2019, 571, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Filippou, P.S.; Karagiannis, G.S.; Musrap, N.; Diamandis, E.P. Kallikrein-related peptidases (KLKs) and the hallmarks of cancer. Crit. Rev. Clin. Lab. Sci. 2016, 53, 277–291. [Google Scholar] [CrossRef]
- Dong, Y.; Harrington, B.S.; Adams, M.N.; Wortmann, A.; Stephenson, S.-A.; Lisle, J.; Herington, A.; Hooper, J.D.; Clements, J.A. Activation of membrane-bound proteins and receptor systems: A link between tissue kallikrein and the KLK-related peptidases. Biol. Chem. 2014, 395, 977–990. [Google Scholar] [CrossRef]
- Haeger, A.; Krause, M.; Wolf, K.; Friedl, P. Cell jamming: Collective invasion of mesenchymal tumor cells imposed by tissue confinement. Biochim. Biophys. Acta BBA Gen. Subj. 2014, 1840, 2386–2395. [Google Scholar] [CrossRef]
- Pietilä, E.A.; Gonzalez-Molina, J.; Moyano-Galceran, L.; Jamalzadeh, S.; Zhang, K.; Lehtinen, L.; Turunen, S.P.; Martins, T.A.; Gultekin, O.; Lamminen, T.; et al. Co-evolution of matrisome and adaptive adhesion dynamics drives ovarian cancer chemoresistance. Nat. Commun. 2021, 12, 3904. [Google Scholar] [CrossRef] [PubMed]
- Young, J.L.; Hua, X.; Somsel, H.; Reichart, F.; Kessler, H.; Spatz, J.P. Integrin Subtypes and Nanoscale Ligand Presentation Influence Drug Sensitivity in Cancer Cells. Nano Lett. 2020, 20, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Protease-activated receptors (PARs)—Biology and role in cancer invasion and metastasis. Cancer Metastasis Rev. 2015, 34, 775–796. [Google Scholar] [CrossRef] [PubMed]
- Michel, N.; Heuzé-Vourc’h, N.; Lavergne, E.; Parent, C.; Jourdan, M.-L.; Vallet, A.; Iochmann, S.; Musso, O.; Reverdiau, P.; Courty, Y. Growth and survival of lung cancer cells: Regulation by kallikrein-related peptidase 6 via activation of proteinase-activated receptor 2 and the epidermal growth factor receptor. Biol. Chem. 2014, 395, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Schrader, C.H.; Kolb, M.; Zaoui, K.; Flechtenmacher, C.; Grabe, N.; Weber, K.-J.; Hielscher, T.; Plinkert, P.K.; Hess, J. Kallikrein-related peptidase 6 regulates epithelial-to-mesenchymal transition and serves as prognostic biomarker for head and neck squamous cell carcinoma patients. Mol. Cancer 2015, 14, 107. [Google Scholar] [CrossRef]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA A Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef]
- Au Yeung, C.L.; Co, N.-N.; Tsuruga, T.; Yeung, T.-L.; Kwan, S.-Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.-K. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef]
- Teixeira, V.H.; Pipinikas, C.P.; Pennycuick, A.; Lee-Six, H.; Chandrasekharan, D.; Beane, J.; Morris, T.J.; Karpathakis, A.; Feber, A.; Breeze, C.E.; et al. Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions. Nat. Med. 2019, 25, 517–525. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, D.; Gimeno, J.; Clavé, S.; Taus, Á.; Pijuan, L.; Arumí, M.; Lorenzo, M.; Menéndez, S.; Cañadas, I.; Albanell, J.; et al. MET expression and copy number heterogeneity in nonsquamous non-small cell lung cancer (nsNSCLC). Oncotarget 2015, 6, 16215–16226. [Google Scholar] [CrossRef]
- Francis, G.; Stein, S. Circulating Cell-Free Tumour DNA in the Management of Cancer. Int. J. Mol. Sci. 2015, 16, 14122–14142. [Google Scholar] [CrossRef]
- Mateo, J.; Gerlinger, M.; Rodrigues, D.N.; de Bono, J.S. The promise of circulating tumor cell analysis in cancer management. Genome Biol. 2014, 15, 448. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-L.; Wei, F.; Wong, D.T.; Lin, C.-C.; Su, W.-C. The Emergent Landscape of Detecting EGFR Mutations Using Circulating Tumor DNA in Lung Cancer. BioMed Res. Int. 2015, 2015, 340732. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Hav, M.; Colombo, A.; Chavez, E.A.; Nissen, M.; Wang, X.; Miyata-Takata, T.; et al. Single-Cell Transcriptome Analysis Reveals Disease-Defining T-cell Subsets in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Cancer Discov. 2020, 10, 406–421. [Google Scholar] [CrossRef]
- Van Der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8+ T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef]
- Goveia, J.; Rohlenova, K.; Taverna, F.; Treps, L.; Conradi, L.C.; Pircher, A.; Geldhof, V.; de Rooij, L.; Kalucka, J.; Sokol, L.; et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell. 2020, 37, 21–36.e13. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Y.; Han, Y.; Zhang, Q.; Jiang, Z.; Zhang, X.; Huang, B.; Xu, X.; Zheng, J.; Cao, X. Tumor Exosomal RNAs Promote Lung Pre-Metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell 2016, 30, 243–256. [Google Scholar] [CrossRef]
- Bigagli, E.; Luceri, C.; Guasti, D.; Cinci, L. Exosomes secreted from human colon cancer cells influence the adhesion of neighboring metastatic cells: Role of microRNA-210. Cancer Biol. Ther. 2016, 17, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signaling in Breast Cancer Cell Migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- LeSavage, B.L.; Suhar, R.A.; Broguiere, N.; Lutolf, M.P.; Heilshorn, S.C. Next-generation cancer organoids. Nat. Mater. 2022, 21, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-J.; Chu, P.-Y. Current and Developing Liquid Biopsy Techniques for Breast Cancer. Cancers 2022, 14, 2052. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid BiopsyCTCs and ctDNA Clinical Applications. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef]
- Stewart, C.A.; Gay, C.M.; Xi, Y.; Sivajothi, S.; Sivakamasundari, V.; Fujimoto, J.; Bolisetty, M.; Hartsfield, P.M.; Balasubramaniyan, V.; Chalishazar, M.D.; et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat. Cancer 2020, 1, 423–436. [Google Scholar] [CrossRef]
- Gao, Q.; Niu, W.; Tse, A.; Lin, J.; Zhou, J.; Shung, S.; Fan, J. Abstract LB-223: Intratumoral heterogeneity and precise personalized therapy revealed by one patient panel in hepatocellular carcinoma. Cancer Res. 2014, 74 (Suppl. S19), LB-223. [Google Scholar] [CrossRef]
- Argyriadis, A.; Kalfoutzos, K.; Khodaverdi, S.; Braun, S.; Jackisch, C. Resistance to HER2 targeted therapy in early breast cancer (EBC): The clinical management of intratumoral heterogeneity (ITH) in HER2 overexpressing EBC. Geburtshilfe Und Frauenheilkd. 2022, 82, 21. [Google Scholar] [CrossRef]
- Foo, J.; Michor, F. Evolution of resistance to anti-cancer therapy during general dosing schedules. J. Theor. Biol. 2010, 263, 179–188. [Google Scholar] [CrossRef][Green Version]
- Beckman, R.A.; Schemmann, G.S.; Yeang, C.-H. Impact of genetic dynamics and single-cell heterogeneity on development of nonstandard personalized medicine strategies for cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 14586–14591. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Foo, J.; Oxnard, G.R.; Hutchinson, K.; Ohashi, K.; Somwar, R.; Wang, L.; Amato, K.R.; Arcila, M.; Sos, M.L.; et al. Optimization of Dosing for EGFR-Mutant Non–Small Cell Lung Cancer with Evolutionary Cancer Modeling. Sci. Transl. Med. 2011, 3, 90ra59. [Google Scholar] [CrossRef] [PubMed]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Faculty Opinions recommendation of Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic Instability in Cancer: Teetering on the Limit of Tolerance. Cancer Res. 2017, 77, 2179–2185. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.-N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150.e9. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Gujar, S.; Pol, J.G.; Kim, Y.; Lee, P.W.; Kroemer, G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018, 39, 209–221. [Google Scholar] [CrossRef]
- Yamazaki, T.; Pitt, J.M.; Vétizou, M.; Marabelle, A.; Flores, C.; Rekdal, O.; Kroemer, G.; Zitvogel, L. The oncolytic peptide LTX-315 overcomes resistance of cancers to immunotherapy with CTLA4 checkpoint blockade. Cell Death Differ. 2016, 23, 1004–1015. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Rodriguez-Ruiz, M.E.; Vitale, I.; Harrington, K.J.; Melero, I.; Galluzzi, L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat. Immunol. 2020, 21, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.N.; Kishton, R.J.; Restifo, N.P. Developing neoantigen-targeted T cell–based treatments for solid tumors. Nat. Med. 2019, 25, 1488–1499. [Google Scholar] [CrossRef]
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef]
- DeSelm, C.; Palomba, M.L.; Yahalom, J.; Hamieh, M.; Eyquem, J.; Rajasekhar, V.K.; Sadelain, M. Low-Dose Radiation Conditioning Enables CAR T Cells to Mitigate Antigen Escape. Mol. Ther. 2018, 26, 2542–2552. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Slaney, C.Y.; Wang, P.; Darcy, P.K.; Kershaw, M.H. CARs versus BiTEs: A Comparison between T Cell–Redirection Strategies for Cancer Treatment. Cancer Discov. 2018, 8, 924–934. [Google Scholar] [CrossRef]
- Fucikova, J.; Becht, E.; Iribarren, K.; Goc, J.; Remark, R.; Damotte, D.; Alifano, M.; Devi, P.; Biton, J.; Germain, C. Calreticulin Expression in Human Non–Small Cell Lung Cancers Correlates with Increased Accumulation of Antitumor Immune Cells and Favorable PrognosisPrognostic Impact of Calreticulin in NSCLC Patients. Cancer Res. 2016, 76, 1746–1756. [Google Scholar] [CrossRef]
- Yang, H.; Lundbäck, P.; Ottosson, L.; Erlandsson-Harris, H.; Venereau, E.; Bianchi, M.E.; Al-Abed, Y.; Andersson, U.; Tracey, K.J.; Antoine, D.J. Retracted Article: Redox Modification of Cysteine Residues Regulates the Cytokine Activity of High Mobility Group Box-1 (HMGB1). Mol. Med. 2012, 18, 250–259. [Google Scholar] [CrossRef]
- Liu, A.; Fang, H.; Dirsch, O.; Jin, H.; Dahmen, U. Oxidation of HMGB1 Causes Attenuation of Its Pro-Inflammatory Activity and Occurs during Liver Ischemia and Reperfusion. PLoS ONE 2012, 7, e35379. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Yao, B.; Dong, T.; Chen, Y.; Yao, J.; Liu, Y.; Li, H.; Bai, H.; Liu, X.; Zhang, Y.; et al. Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell 2022, 185, 1356–1372.e26. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, X.; Mao, Z.; Sun, X.; Liu, B. Intratumoral Heterogeneity in Lung Cancer. Cancers 2023, 15, 2709. https://doi.org/10.3390/cancers15102709
Lv X, Mao Z, Sun X, Liu B. Intratumoral Heterogeneity in Lung Cancer. Cancers. 2023; 15(10):2709. https://doi.org/10.3390/cancers15102709
Chicago/Turabian StyleLv, Xiaodi, Zixian Mao, Xianjun Sun, and Baojun Liu. 2023. "Intratumoral Heterogeneity in Lung Cancer" Cancers 15, no. 10: 2709. https://doi.org/10.3390/cancers15102709
APA StyleLv, X., Mao, Z., Sun, X., & Liu, B. (2023). Intratumoral Heterogeneity in Lung Cancer. Cancers, 15(10), 2709. https://doi.org/10.3390/cancers15102709