Abstract
Appendiceal neuroendocrine neoplasms (ANENs) usually present as incidental findings at the time of appendectomy for acute appendicitis. They are rare, accounting for only 0.5–1% of intestinal neoplasms; they are found in 0.3–0.9% of all appendectomy specimens. They are usually sporadic tumors. There are several histological types including well-differentiated neuroendocrine tumors (NETs), poorly differentiated neuroendocrine carcinomas (NECs), and mixed neuroendocrine-non-neuroendocrine neoplasms (MiNENs). Histologic differentiation and the grade of well-differentiated NETs correlate with clinical behavior and prognosis. Management varies based on differentiation, aggressiveness, and metastatic potential. There is debate about the optimal surgical management for localized appendiceal NETs that are impacted by many factors including the tumor size, the extent of mesoappendiceal spread, lymphovascular invasion and perineural involvement. In addition, the data to guide therapy in metastatic disease are limited due to the paucity of these tumors. Here, we review the current advances in the management of ANENs within the context of a multidisciplinary approach to these tumors.
1. Introduction
The most common appendiceal tumors are neuroendocrine neoplasms (ANENs), followed by mucinous neoplasms and adenocarcinomas [1]. ANENs account for 25–60% of primary malignancies in the appendix [2,3,4,5,6,7]. Most cases present as incidental findings; they are found in 0.16–2.3% of appendectomies performed for acute appendicitis [8,9,10]. The most common age at diagnosis is at the end of the second decade of life with an increased incidence in females [11,12,13,14,15,16]. According to the current WHO histological classification, ANENs include well-differentiated NETs, poorly differentiated NECs (large cell and small cell types), and mixed neuroendocrine-non-neuroendocrine neoplasms (MiNENs) [17,18,19]. The majority of ANENs are well-differentiated NETs (70–75%) that are further subdivided into different grades (G1, G2 and G3) according to their proliferative rate [19]. Grade 1 tumors are usually indolent tumors with relatively long median survival durations compared to high-grade tumors, which are more aggressive [20,21]. Poorly differentiated NECs, which resemble small-cell or large-cell neuroendocrine carcinomas of the lung, have aggressive behavior, and usually present with metastatic disease at diagnosis [18,19,22,23,24]. MiNENs are the rarest tumor type. ANENs are heterogeneous tumors, and their management depends on various factors including histological differentiation, disease stage, hormone production, somatostatin receptor expression, tumor burden, and hepatic versus extrahepatic disease. This review article focuses on the clinical presentation, staging workup, and current treatment guidelines of different types of NENs arising in the appendix.
2. Epidemiology
The incidence of NENs including ANENs has been increasing in the past decade, partly attributed to a better understanding of the pathophysiologic presentation, improved classification system, and availability of advanced imaging modalities [25]. The annual incidence of ANENs is reported to be around 3–9 cases per 1000 appendectomies [11,26,27]. The age at diagnosis ranges from 20 to 50 years in adults, and most patients are in their 40s, which is younger than the average age for other gastroenteropancreatic neuroendocrine neoplasms (GEP-NENs) and other primary malignant appendiceal neoplasms [3,5,28]. There is a relatively higher incidence in women; this has been attributed to the greater frequency of incidental appendectomies in women who undergo pelvic surgery [16,20,29].
ANENs are most often identified at the distal tip of the appendix; the next most common location is the body and they are only rarely present at the base of the structure [2], where they can cause complications due to obstruction [2,30,31,32]. Based on the SEER database of information collected between 1973 and 2004, 60% of ANENs are localized diseases at diagnosis, while 28% and 12% of cases have regional and distant metastasis, respectively [15]. About 50% of patients affected by poorly differentiated or undifferentiated tumors presented with synchronous distant metastasis at diagnosis [15].
3. Classification of Appendiceal Neuroendocrine Neoplasms
The 2019 World Health Organization (WHO) Classification of Tumors of the Digestive System categorizes neuroendocrine neoplasms into two broad subgroups: well-differentiated neuroendocrine tumors (NETs) and poorly differentiated neuroendocrine carcinomas (NECs) [19]. Well-differentiated NETs, formerly often called “carcinoid tumors”, are further subdivided into grades 1, 2, and 3 according to the proliferative rate determined by the Ki67 labeling index and/or the mitotic index (MI). Well-differentiated grade 1 tumors are considered relatively indolent, grade 3 tumors are more aggressive but still better than poorly differentiated NECs, which are high-grade aggressive carcinomas (small cell or large cell carcinomas) with poor outcome (median survival rate less than 2 years) [33]. Approximately 70 percent of NENs within the appendix are well-differentiated NETs [20,21]. A third category, mixed neuroendocrine-non-neuroendocrine neoplasms (MiNENs), includes tumors that are composed of a mixed population of neuroendocrine neoplasm and adenocarcinoma; currently, it is suggested that there must be a minimum of 30% of the tumor mass composed of the smaller component however this remains controversial [19]. Both the neuroendocrine and non-neuroendocrine components may have varying differentiation and grade. The median age at diagnosis of MiNENs is approximately 58 years. These tumors comprise up to 6.9% of ANENs and have a 5-year survival rate of 56.3% (95% CI, 42.1–68.4) which is lower than well-differentiated NETs but better than NECs [20,34]. Despite the fact that a large meta-analysis showed that the most common primary site of MiNEN is the appendix (60%), the data on these tumors is limited [35,36]. The tumor formerly known as “goblet cell carcinoid” is now recognized to be an adenocarcinoma with dominant mucin-secreting cells and a minor component of neuroendocrine cells; these are no longer considered among ANENs [37].
Localized well-differentiated NETs have median overall survival greater than 20 years, the best prognosis among all GEP-NENs; the overall prognosis is highly variable according to tumor morphology, size and stage. Small, low-grade ANENs localized to the appendix have a better prognosis than large ANENs with high-grade morphology, and tumors with extra-appendiceal invasion or metastasis [30]. Among well-differentiated NETs, grade 1 is the most common and many are smaller than 1 cm, which accounts for good survival rate [38]. The NCDB-based study has demonstrated that ANETs have a 5-year survival rate of 86.3% (95% confidence interval [CI], 81.4–89.9) and low rates of regional and distant metastases [20]. An association between tumor size and both 5-year survival rate and metastatic potential was reported in this study. The 5-year survival rate decreases as the lesion size increases 89.9%, 70.6% and 58.2% for tumor size ≤2 cm, 2–4 cm, and >4 cm, respectively [20]. In-addition, both regional and distant metastases are less commonly seen with tumors smaller than 2 cm. This information is the basis for the TNM-staging system for appendiceal NETs.
While the majority of appendiceal NETs are EC cell tumors producing serotonin, there are also less common L cell tumors that produce glucagon-like peptides, pancreatic polypeptides and Peptide YY [18,39,40]. L cell tumors may have unusual morphology, with predominant tubular or clear cell architecture. Accordingly, the product of the tumor can be important to be known for surveillance. However, it remains to be seen if the cell type affects prognosis as it does in rectal NETs [41].
4. Clinical Presentation
The majority of appendiceal NETs are asymptomatic and are found incidentally at the time of appendectomy. The reason for an appendectomy is usually acute appendicitis or chronic non-specific lower right quadrant abdominal pain. Because most of these tumors are located in the distal appendix, they are unlikely to cause obstruction; only 10% are located at the base of the appendix and can be implicated as the cause of obstruction and appendicitis [2,30,31,32]. Tumors larger than 2 cm and at the base of the appendix are associated with a higher incidence of appendicitis due to obstruction as well as higher rates of nodal and distant metastases [30,31,32]. Carcinoid features may be present in patients with tumors that have metastasized to the liver but they are very uncommon (<1%) [15,42]. As observed in midgut GEP-NET cases, symptomatic serotonin-producing NET is associated with mesenteric fibrosis, which could cause bowel obstruction. However, the evidence of relevant mechanisms underlying the rare obstruction identified in ANEN remains unavailable.
Patients with high-grade poorly differentiated NECs and MiNENs usually present with metastatic disease at the time of diagnosis [11,30,43]. They are hormonally inactive tumors with no associated symptoms of hormone hypersecretion. The most common signs and symptoms are non-specific and related to the size and location of the tumor; they include pain, nausea, fatigue, anorexia, weight loss, abnormal liver function test results, and bowel obstruction if associated with bowel metastasis [2,30,31,32].
5. Diagnostic Assessment
Most ANENs are found incidentally, therefore, the diagnostic work-up should focus on accurate histopathological assessment as well as biochemical and imaging evaluations to detect early recurrence and/or metastasis.
6. Circulating Biomarkers
Chromogranin A (CgA) is a protein that is stored and released with peptides and amines in NETs [44,45,46]. Several reports have shown that elevated levels are associated with recurrence, and levels twice the normal were associated with poor outcomes and shorter survival in NETs [47,48,49,50]. Some guidelines advocate surveillance with serum levels of CgA; this biomarker is often non-specific as it can be elevated in other medical conditions (renal and hepatic insufficiency, uncontrolled hypertension), and with certain medications (proton pump inhibitors and steroids) [51,52]. However, these comorbidities are less common in the population of younger patients with ANETs. Nevertheless, they must be considered when using surveillance with CgA.
Measurement of the serotonin metabolite 5-hydroxyindoleacetic acid (5-HIAA) in plasma or a 24 h urine collection can be considered, particularly in patients with liver metastasis and carcinoid features such as diarrhea, flushing, and wheezes. It is not indicated in tumors that do not produce serotonin and patients without carcinoid features [53,54,55].
7. Staging and Imaging
The role of staging anatomical imaging is mainly to identify nodal and distant metastasis. Given that tumor size is a strong factor in determining the likelihood of regional and distant metastases, the follow-up recommendations depend on tumor size, the status of resection margins and lymph nodes and mesoappendiceal invasion. Most patients with small well-differentiated appendiceal NETs <2 cm with negative margins and no deep mesoappendiceal invasion (<3 mm) will have a low risk of recurrence and most guidelines recommend no surveillance for them [26,56,57]. For patients with an appendiceal NET >2 cm, incomplete resection, and positive nodes or margins, the current guidelines recommend workup with contrast-enhanced, triple-phase computed tomography (CT) scan or magnetic resonance imaging (MRI). However, as discussed below, a more sensitive technique for this assessment is somatostatin receptor-based diagnostic positron emission tomography [PET] scanning, and it is important for patients with metastatic NETs. According to the North American Neuroendocrine Tumor Society (NANETS) and the European Neuroendocrine Tumor Society (ENETS) guidelines, somatostatin receptor (SSTR)-based imaging is not necessary for patients with localized disease but this is a position that might warrant reconsideration. Its role in advanced stages is to assess receptor status and other metastatic lesions that were not identified with anatomical scans [58,59]. Octreotide SPECT/CT (single-photon emission computed tomography) is less sensitive and SSTR-PET (somatostatin receptor-based PET) tracers (either gallium Ga-68 or copper CU-64 dotatate) are preferred. SSTR-PET is considered the gold standard in diagnosis and surveillance, and it is currently used for patients with symptoms suggestive of carcinoid syndrome and/or metastatic diseases.
Measurable lesions are considered SSTR-positive if the uptake is greater than that of the liver. SSTR-PET scan is usually combined with anatomical imaging (triple phase CT or MRI) to identify both SSTR-positive and -negative lesions.
In patients with high-grade, poorly differentiated NEC, 18F-FDG-PET combined with triple-phase CT or MRI is preferred, given that these de-differentiated carcinomas usually lack SSTR expression [60,61,62,63]. Several studies of functional and anatomical imaging for neuroendocrine tumors have shown that 18F-FDG PET/CT has sensitivity and specificity of 61.9% and 100%, while 68Ga-DOTATATE PET/CT has sensitivity and specificity of 100–81% and 90–80%, respectively [64,65,66,67]. The false positive rate of 68Ga-DOTATATE PET/CT varies between 0 to 38%, likely due to tracer accumulation in lymph nodes containing SSTR2+ macrophages, inflammatory cardiovascular tissues and other regions affected by chronic inflammatory conditions [65,68,69].
8. Management
Much of the data on the management of metastatic ANETs is based on large trials that include all GEP-NETs. There have been only a very few retrospective single-institution reports of ANETs [70,71,72].
8.1. Treatment of Localized Disease
The majority of patients are diagnosed after a simple appendectomy, and further surgical intervention will depend on tumor size and the presence of high-risk features (deep mesoappendiceal invasion >3 mm, positive/unclear margins, positive lymphovascular invasion, and a higher proliferative rate). The incidence of metastasis is associated with increases in the size of ANET lesions; 2 cm lesions are found to have nodal involvement in up to 33% of cases and distant metastasis in 12% [73]. Accordingly, the consensus-based guidelines from the NANETS and the ENETS recommend a simple appendectomy for any lesion smaller than 1 cm and for those tumors between 1.0 and 1.9 cm that lack high-risk features [74,75,76]. The optimal surgical approach for localized well-differentiated ANETs between 1–2 cm with high-risk features is controversial [77,78]. The NCCN guidelines consider appendectomy alone to be adequate for all tumors <2 cm, even with high-risk features [2,27]. Other reports demonstrate a higher potential for nodal and metastatic spread with small tumors ≥1.5 cm [57,79]. Analysis of the National Cancer Institute (NCI) Surveillance, Epidemiology, and End Results (SEER) database between 1988 and 2003 identified significantly higher rates of lymph node metastases in patients with appendiceal NETs larger than 2 cm compared with those with either between 1.0–1.9 cm or less than 1 cm (86% vs. 47% and 15%, respectively) [26]. The results were confirmed in another analysis of the SEER database between 1988 and 2013 that included 573 patients with well-differentiated ANETs. The results reported a 64% probability of nodal metastases in patients with tumors >2 cm vs. 31% in those with tumors measuring 1.1–2 cm [80].
Despite this higher risk of nodal metastasis in lesions measuring 1.0–1.9 cm compared to lesions smaller than 1 cm, a series from the National Cancer Database (NCDB) did not show a survival benefit with the addition of right hemicolectomy for patients with lesions smaller than 2 cm, even in those with high-risk features (5-year survival 88.7 versus 87.4%) [68,74,81] [Table 1]. Therefore, simple appendectomy is considered sufficient therapy for lesions smaller than 1 cm and right hemicolectomy only is recommended for lesions larger than 2 cm in most guidelines. For lesions between 1.0 and 2.0 cm, a multidisciplinary approach is essential to discuss several factors including high-risk features, lesion site (base vs. apex), patient age, comorbidities, and the likelihood of surgical complications. If a right hemicolectomy is planned, a full colonoscopy should be performed to rule out synchronous colorectal cancers and a full inspection of the bowel intraoperatively is suggested to identify other lesions [12,68,74,75,82,83] [Table 2]. Other retrospective studies and single institution experiences showed that following NANETs and ENETS guidelines may lead to overtreatment of patients RHC and suggested detailed counseling regarding the risk of over- and undertreatment needs in these patients [70,71,72,84].

Table 1.
Appendiceal Neuroendocrine Tumors-specific survival rates [68,74,81].
Table 2.
Management Strategies for localized Appendiceal Neuroendocrine Tumor According to Different Guidelines [12,68,74,75,82,83].
According to most of the available guidelines, patients with small tumors (<1 cm) with no aggressive features who were treated with appendectomy with clear margins (R0) do not need active surveillance and no surveillance imaging is required [82,83]. However, patients with symptoms of hormone hypersecretion warrant further evaluation for disease recurrence. Surveillance is also not required for patients with ANENs that measure between 1 and 2 cm if they received the right hemicolectomy and no residual disease was identified on histological examination [Figure 1]. NANETs guidelines recommend against surveillance imaging for all small well-differentiated ANENs (<2 cm); in contrast, other guidelines, including ENETS, recommend follow-up for patients with tumors measuring 1–2 cm that were incompletely resected, had lymph node involvement, exhibited lymphovascular invasion and/or were higher grade tumors (G2 or G3) [9,16,29,57]. The recommendation for these situations is surveillance with biochemical markers in selected patients and anatomical imaging with either a CT scan or MRI. Regarding biochemical markers, yearly chromogranin A (CgA) has been recommended with caution given its non-specificity. 24 h urinary or plasma 5HIAA can be used to monitor patients with serotonin-producing tumors and has been used for patients with clinical symptoms of carcinoid syndrome; its value in the surveillance of asymptomatic patients remains to be proven. There are no studies of the addition of other biomarkers such as glucagon, pancreatic polypeptide or PYY that are expressed by L cell tumors.
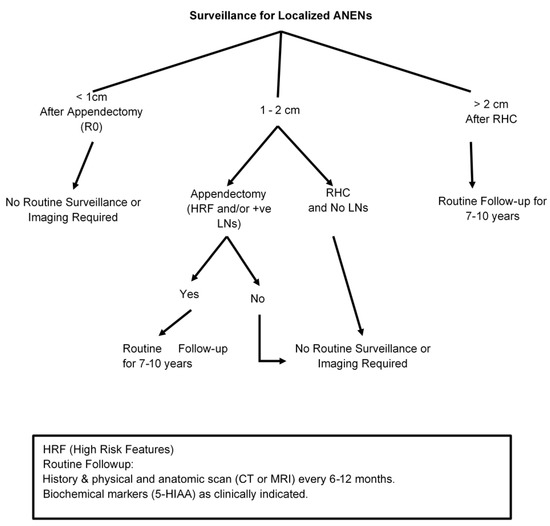
Figure 1.
Post-treatment Follow-up After Surgery for Appendiceal Neuroendocrine Neoplasms (ANENs).
A series of studies reported by Moertel et al. have had a great influence on the current treatment recommendations by reporting limited to no recurrence in ANET patients with tumor size ≥2 cm after appendectomy [2,85]. The larger tumor size of well-differentiated ANET is known to associate with a higher rate of lymph node involvement, 11.6–16.7% vs. 29.9–56.8% vs. 40.6% for tumor size <1 cm, 1–2 cm, and >2 cm, respectively [25,86]. The rationale of RHC in the treatment of ANET >2 cm or with high-risk features is to remove regional lymph nodes associated with the elevated risk of recurrence or distant metastasis to improve overall survival. However, Groth et al. has reported no significant differences in overall survival between appendectomy and RHC for ANET >2 cm [87]. Furthermore, unlike pancreatic and other GEP-NETs, various ANET studies have shown no differences in the rate of regional lymph node positivity, metastatic disease, and overall survival between different tumor sizes (>2 cm, 1–2 cm, or <1 cm). Worse survival was observed in patients with distant metastatic disease at diagnosis while regional lymph node involvement does not impact overall survival in ANET patients [77,78]. A recent meta-analysis has shown that 10-year disease-specific survival is not significantly different between patients with and without lymph node involvement, 95.6% and 99.2%, respectively (OR: 0.2, 95% CI: 0.02–2.4) [88]. ENETS and NCCN have recommended that simple appendectomy can be sufficient even for a tumor size great than 2 cm or any tumor size without the high-risk features mentioned above.
SSTR-based imaging scans (PET-Ga68 or CU-64) are not routinely recommended for the localized disease after complete resection. A reasonable follow-up strategy for selected patients involves a careful history and physical examination with biochemical markers when appropriate and anatomical imaging 6–12 months after surgery and yearly afterward for up to 10 years, given the risk of late recurrence [7,10,74,75,76,83,89,90] [Figure 1].
8.2. Treatment of Metastatic Disease
The first step in the management of patients with metastatic well-differentiated ANENs is to determine factors that may impact therapeutic strategies such as tumor grade, site of metastatic disease (hepatic vs. extra-hepatic), tumor burden, and the status of somatostatin expression [91,92]. This includes anatomical imaging as well as SSTR-PET scans for patients with well-differentiated NETs or 18F-FDG-PET scans for those with NEC, MiNEN or high-grade NET. There are several therapeutic options that can be considered, depending on the site and extent of disease; these include surgery, liver-directed therapy for localized liver disease, medical therapy with somatostatin, targeted agents or cytotoxic chemotherapy, and radiation therapy using radiolabeled somatostatin or external beam radiation. Most of the data from prospective trials are for patients with metastatic gastroenteropancreatic neuroendocrine tumors (GEP-NETs) with only a small number of ANENs in these trials.
8.3. Surgery
The presence of metastatic disease in NETs does not preclude surgical debulking of hepatic metastases. For patients with low and intermediate-grade tumors who are symptomatic and have mainly liver metastasis, surgical resection may palliate symptoms and improve long-term survival. The available data supporting surgical cytoreduction are exclusively retrospective, the level of evidence supporting this approach is limited and most data are for midgut and pancreatic NETs [93,94,95,96]. The degree of liver cytoreduction remains an area of debate, and most historical data adopt ≥90% threshold of cytoreduction to be associated with improved outcomes [97,98,99,100]. In contrast, other studies reported that ≥90% cytoreduction was not associated with the improved median OS when compared with at least a 70% threshold [99,101,102,103,104]. However, these are retrospective studies with potential selection bias; the current literature supports the hypothesis that debulking of liver metastases is associated with symptomatic relief leading to improved quality of life and improved survival. Therefore, surgical cytoreduction for patients with metastatic ANETs should be considered in a multidisciplinary discussion to appropriately select patients who may benefit from this approach, especially for those with grade 1 and 2 well-differentiated tumors [Figure 2]. Other sites of metastases such as peritoneal metastases are likely more common in mucinous appendiceal epithelial neoplasms as compared with ANETs. Based on data from other appendiceal neoplasms and colon cancer, debulking may improve the overall prognosis and survival of patients with exclusively peritoneal metastases but no current data are available specifically for ANETs. Due to the limited evidence of the overall course and outcome of surgical intervention in this subgroup of patients, there is no agreement or specific guideline established for the treatment of peritoneal metastasis of ANETs.
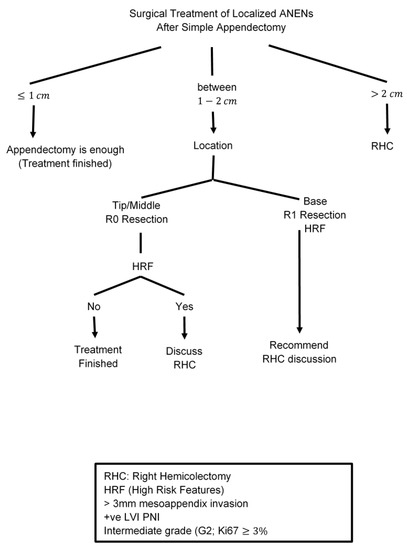
Figure 2.
Surgical Treatment of Localized ANENs after Simple Appendectomy.
8.4. Liver-Directed Therapy
The regional lymph nodes and liver are the dominant sites of metastases for ANETs, and liver-directed therapies are appropriate for patients whose tumors are predominantly metastatic to the liver and surgical resection is not feasible. Data are mainly retrospective and prospective trials are extremely limited [92,96,105,106,107]. Options for liver-directed therapies include bland hepatic artery embolization, chemoembolization with intra-arterial cytotoxic agents (doxorubicin or cisplatin), and radioembolization using 90Y embedded in either a resin or glass microsphere. Other ablative techniques include radiofrequency ablation (RFA), microwave ablation and cryoablation therapy. Current retrospective literature and single institution experiences demonstrate ORRS and symptomatic relief in approximately 40–50% of patients, but most of these data are for GEP-NETs with only a very small number of ANENs included [42,108].
There are no randomized prospective trials comparing the various liver-directed therapy techniques, therefore the choice of technique depends on various factors including tumor size, location, grade, patient comorbidities and, most importantly, institutional experience with the various embolization modalities. Generally, ablation techniques are reserved for small metastases (less than 5 cm in diameter) compared to embolization methods, which are recommended for larger, more extensive and multilobar metastases.
Toxicities associated with these techniques vary. The common side effects of chemoembolization and bland embolization include abdominal pain, nausea, pain, fever and elevated serum transaminases due to the induction of ischemic hepatitis [108,109,110]. 90Y radioembolization does not induce ischemic hepatitis so pain and other side effects are lower than with other embolization techniques, but it can be associated with radiation enteritis and delayed liver fibrosis [111]. Although surgical cytoreduction is the only potentially curative therapy for ANETs with exclusively liver metastasis, these liver-directed therapies may offer effective alternatives to alleviate symptoms and potentially impact the outcome in patients with unresectable tumors.
8.5. Somatostatin Analog Therapy
Somatostatin is a 14-amino acid peptide that inhibits the secretion of other hormones such as serotonin, insulin, glucagon, gastrin and VIP and also has potent antiproliferative actions [112,113]. Due to the short half-life of native somatostatin, synthetic agents with longer half-life have been developed for therapeutic applications. Long-acting somatostatin analogues (SSAs) are peptide hormones that bind to somatostatin receptors (SSTR, mainly subtypes 2 & 5) to inhibit the secretion of other hormones and alleviate hormonal symptoms such as diarrhea and flushing [113,114,115]. In addition, SSAs have also been shown to inhibit tumor growth in randomized phase III trials (PROMID and CLAIRNET) and therefore are considered the main foundation for treating metastatic well-differentiated NETs [116,117,118].
The PROMID phase III study showed that octreotide long-acting repeatable (LAR) 30 mg deep intramuscular every 4 weeks improves time to progression (TTP) compared to placebo in patients with metastatic midgut NETs (14.3 months versus 6 months, HR 0.34; p = 0.000072) [117]. The CLARINET phase III study demonstrated similar effects with lanreotide depot 120 mg deep subcutaneously every 4 weeks. Results have shown that lanreotide depot 120 mg every 4 weeks significantly prolonged median progression-free survival (PFS) compared to placebo (not reached versus 18 months, HR 0.47; p = 0.001) [117,119]. The PROMID study included mainly midgut NETs but the CLAIRNET trial included midgut, pancreas, hindgut and tumors of unknown primary site. There was no specific percentage of ANETs included in either trial. Generally, SSAs are well tolerated with few side effects that have been reported including fat malabsorption, diarrhea, increase risk of gallstones, hyperglycemia, and mild bradycardia [112,120]. The presence of somatostatin receptors determined by PET gallium Ga-68 DOTATATE or Cupper-64 [Ga-68 DOTATATE] is predictive of response to SSAs [121,122]. However, in some cases, these diagnostic imaging modalities may be negative even if the tumors express SSTRs, and the use of SSAs in these cases is controversial. Whether evidence of SSTR expression is required before starting SSAs is an area of debate that need further investigation.
8.6. Radiolabeled Somatostatin Analog Therapy
Radiolabeled somatostatin analogue therapy (also known as peptide receptor radionuclide therapy; PRRT), is a novel form of targeted radionuclide therapy. It involves the use of a radio-labelled somatostatin analogue to deliver a targeted cytotoxic amount of radiation by emitting radioactive particles to SSTR-expressing disseminated tumors. Recently, the FDA approved the use of Lutetium-177 (177Lu)-DOTA-TATE (LUTATHERA ®) for the treatment of metastatic somatostatin receptor-positive (SSTR) GEP-NETs). Data from the phase III NETTER-1 trial in patients with metastatic mid-gut NETs who progressed on first-line SSA therapy showed a 79% reduction in risk of progression or death (median PFS not reached versus 8.4 months, HR 0.21; p < 0.00001) compared to high dose octreotide [123]. The objective response rate was only 18% but was significantly higher compared to the control group (18% vs. 3%, p <.001). Only one patient had an ANET as the primary tumor site in that study. The side effects of PRRT include mild nausea and vomiting, fatigue, lymphopenia, and less than 2% risk of myelodysplastic syndrome (MDS) and acute leukemia. Based on the results of the NETTER-1 study, the current guidelines recommend PRRT for patients with metastatic well-differentiated GEP-NETs including ANETs who progress on first-line SSA therapy. These patients require positive SSTR-based imaging (PET-Ga68 or CU64) given that expression of SSTRs is predictive of PRRT benefit.
8.7. Targeted Therapies (mTORi and Anti-Angiogenesis)
Multiple targeted therapies including mTOR, tyrosine kinase and angiogenesis inhibitors have been approved for treating advanced NETs [124]. The mTOR inhibitor Everolimus has been shown to decrease NET cell growth and therefore was studied in patients with metastatic GEP-NETs [125,126]. Several phase III trials have demonstrated that Everolimus 10 mg significantly improves PFS in patients with well-differentiated low and intermediate-grade NETs [127,128,129]. However, RADIANT 2 trial did not show a promising clinical benefit of Everolimus versus placebo in functional midgut NETs (HR 0.77, p = 0.026), RADIANT 4 trial met its primary endpoint with the improvement om PFS in non-functional low and intermediate-grade gastrointestinal NETs (11 months versus 3.9 months, HR 0.48, p < 0.00001). Based on these results, Everolimus was FDA-approved for use in several metastatic GEP-NETs including ANETS. Comparing the results of RADIANT 2 and RADIANT 4, Everolimus had only marginal benefit in patients with functional midgut NETs including ANETs. Only 1% of the patients in RADIANT trials had ANET as the primary tumor site. The most commonly reported side effects of Everolimus are stomatitis with oral ulcers, diarrhea, pneumonitis, hyperglycemia and myelosuppression [130,131]. Whether decreased dosing of Everolimus (5 mg vs. 10 mg) will result in the same clinical benefit with fewer side effects is yet to be investigated.
Because NETs are highly vascular tumors and express vascular endothelial growth factor (VEGF), multi-tyrosine kinase receptor inhibitors including those that target VEGF, platelet-derived growth factor receptors (PDGFRs), and Fibroblast growth factor receptors (FGFRs), have been studied extensively in metastatic GEP-NETs [132,133,134]. Sunitinib, which targets VEGF receptors 1, 2, and 3, is the main approved tyrosine kinase inhibitor (TKI) for metastatic pancreatic NETs. No data are available for mid-gut NETs. Recently, other multi-TKIs have been investigated and have shown promising results for extra-pancreatic NETs.
Surufatinib is a novel, oral multi-kinase inhibitor that selectively inhibits the VEGFRs and FGFRs which both inhibit angiogenesis. SANET-ep, is a placebo-controlled phase III study that investigated the role of surufatinib in advanced extrapancreatic NETs [135,136]. The study performed in China enrolled 198 patients on surufatinib 300 mg daily (n = 129) or placebo (n = 69). The primary tumor was mainly gastrointestinal but only 1% were ANETs. The trial met the primary end point in which surufatinib was associated with improved PFS compared to placebo (9.2 vs. 3.8 months, HR 0·33; 95% CI 0.22–0.50; p < 0·0001) [137]. The most common side effects included hypertension, proteinuria, and less commonly headache, diarrhea, and myelosuppression. The results suggest that surufatinib might be a new treatment option for advanced metastatic well-differentiated NETs pending multiregional randomized clinical trials for FDA approval. Other multi-TKIs investigated in phase II trials for advanced NETs include lenvatinib, pazobanib, and cabozatinib; they have also shown promising results but the number of ANETs included in these trials is unknown [138,139,140]. Although anti-angiogenesis and mTOR inhibitors have potential clinical benefits in GEP-NETs including ANETs, less than 10% of patients will have a radiological response. Therefore, a better understanding of the disease biology with new predictive biomarkers may lead to improvement of our current therapeutic strategies for GEP-NET subtypes.
8.8. Cytotoxic Chemotherapy
The role of systemic chemotherapy in metastatic well differentiated NETs is debatable and restricted to patients with high tumor burden and high-grade NETs. The most common used chemotherapy in well-differentiated NETs is oral capecitabine plus the oral alkylating agent temozolomide (CAPTEM) and oxaliplatin based regimens. This regimen was shown to be effective in both cell lines and early phase I as well as recent phase II trial (ECOG 2211) mainly in patients with pancreatic NETs [141,142]. The current data for CAPTEM in midgut NETs are mainly retrospective and most have shown suboptimal responses compared to pancreatic neuroendocrine tumors (pNETs). Only one prospective study of CAPTEM has shown a response rate of 41% in patients with “carcinoids” gulati [143], however the primary tumor sites of the 28 patients in this study were unclear. Based on current data, there is no strong evidence to establish the benefit of CAPTEM in patients with ANETs, therefore, it should be only considered in a clinical trial setting or for selective ANET patients with high-grade tumors and high tumor burden who have exhausted other options of therapy.
Oxaliplatin-based combinations (FOLFOX and CAPOX) have shown anti-tumor activity in patients with advanced GI NETs [135,144]. The current data are based on small phase II trials and retrospective analyses with a small total number of treated patients. Data from combined analysis of two-phase II trials included 76 patients with metastatic “carcinoids” who were treated with either FOLFOX or CAPOX plus bevacizumab and demonstrated minimal clinical benefit. An objective response rate of 13.6% and median PFS of 19.3 months in FOLFOX plus bevacizumab and 18% with a median PFS of 16.7 months in CAPOX plus bevacizumab [136]. There was no clear description of how many patients had ANETs in this trial. Given limited data for oxaliplatin-based regimens in midgut NETs include ANETs, it should be considered only for patients with high-grade tumors and high tumor burden who do not have other options for treatment [Figure 3].
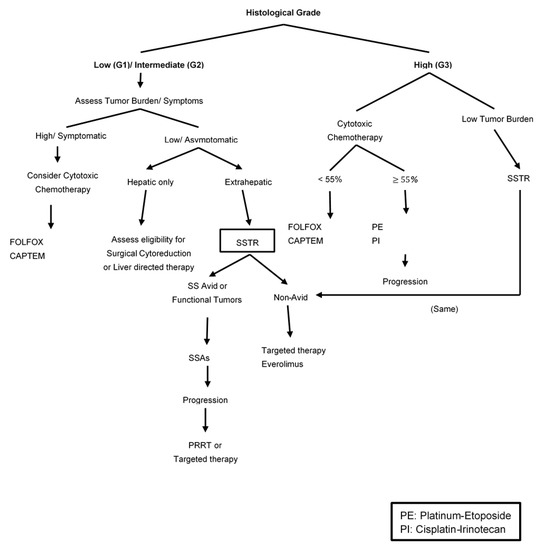
Figure 3.
Therapeutic Strategy for Metastatic Well-Differentiated ANENs based on histological grades.
Extra-pulmonary poorly differentiated neuroendocrine carcinomas (NECs) are extremely rare and the most common primary tumor locations are the pancreas, large bowel, and stomach [145]. Data regarding the prevalence of poorly differentiated appendiceal neuroendocrine carcinoma (ANEC) are scant and unreliable. Most patients with gastrointestinal NECs have metastatic disease at presentation and their main treatment is etoposide plus platinum (either cisplatin or carboplatin) which is similar to the treatment of small cell lung cancer [146,147,148]. The benefit of platinum etoposide in gastrointestinal NEC has been shown mainly in retrospective and early phase II studies. The Nordic consortium was a large retrospective study of 252 patients with advanced GEP-NEC [149]. The results demonstrated a response rate of 31%, 4 months PFS and 11 months median survival compared to those who had only supportive care. The most common site of the primary tumor was the stomach, pancreas, and colon, and 38% of patients had either “other GI” or an unknown primary site with no specific designation of ANECs. Another retrospective series included 123 GEP-NEC patients who received a platinum plus etoposide regimen for first-line treatment. The study showed an objective response rate (ORR) of 50% with a median PFS of 6.2 months and overall survival of 11.6 months [150]. It is not clear whether ANEC was part of the 20% “other GI and unknown primary” tumors included in this study. Overall, a platinum-etoposide regimen induces a response rate between 30% and 50% with a short duration (less than 9 months), poor overall survival of less than 2 years and a significant toxicity profile [151,152,153].
Recently, a phase III study evaluated Irinotecan-based regimens (irinotecan plus cisplatin doublet, IP) versus platinum etoposide (EP) in advanced NECs of the digestive system [154]. The study enrolled 170 patients with gastrointestinal and hepatobiliary NEC to either IP or EP. There were no significant differences in either PFS (5.6 vs. 5.1, HR 1.060, 95% CI, 0.777–1.445) or RR 54.5% vs. 52.5% in EP and IP, respectively. The study showed no superiority of one regimen over the other and both can be considered standard first-line therapy for metastatic gastroenteropancreatic neuroendocrine carcinomas (GEP-NEC.) There was no information about how many of the gastrointestinal NECs were ANECs.
Patients with gastrointestinal NEC who progress on platinum etoposide will have limited therapeutic options with poor outcome. There are limited data for second-line therapy and mostly derived by case series and small trials with no standard regimen has been established. Some of the available options include regimens that have been used in other GI malignancies such as Oxaliplatin- or irnotecan-based regimens (FOLFOX or FOLFIRI) [150,155,156]. Recently, several phase II trials (DART and CA209) have evaluated the benefit of dual anti-CTLA-4 and anti-PD-1 blockade in refractory extra-pulmonary NEC patients [145,153,157,158]. The results demonstrated significant clinical activity with a response rate range between 24–44%. The DART trial included only one patient with an appendiceal primary tumor, but the CA209 trial did not include patients with ANECs [Figure 4].
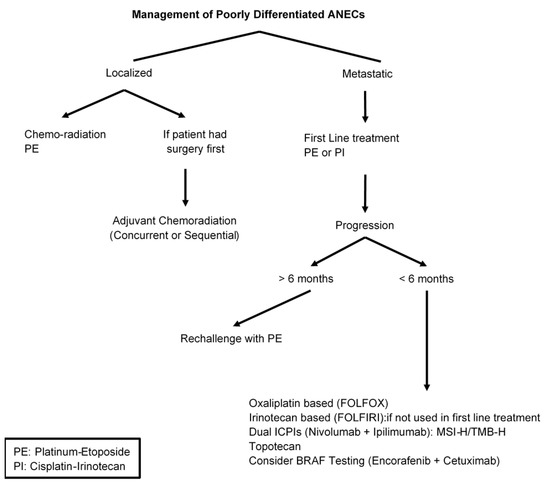
Figure 4.
Management of Poorly Differentiated ANEC.
MiNEN represents an extremely rare diagnosis, but a common primary site location is the appendix. The biological behavior is usually driven by the neuroendocrine component, which is most often a poorly differentiated NEC. Therefore, MiNENs are treated similarly to pure high-grade NEC. Surgery can be considered for potentially curable early-stage disease; palliation for symptom relief is the approach for advanced-stage diseases. In one retrospective study, the median recurrence-free survival for surgery in localized disease was 12.5 months [159]. There is very limited data about the benefit of neoadjuvant and/or adjuvant therapy and minimal guidance for the choice of regimen. Two of the most used regimens in clinical practice are either EP or oxaliplatin-based (FOLFOX), but neither is supported by randomized prospective evidence. Retrospective analyses and single-institution experience have shown that multimodal treatment (including chemotherapy and/or radiotherapy) vs. surgery alone was associated with a significantly prolonged OS (75.0 vs. 18.9 months, p = 0.0045) [159]. These results demonstrated the benefit of multimodal treatment over surgery alone in localized MiNEN.
For metastatic disease, the data are also limited. Treatment is similar to pure NEC with EP as the main regimen gave the aggressive nature of the disease [33,148,160]. The available results are exclusively retrospective and are similar to pure NEC with ORR between 30% and 40%, and median PFS of approximately 4–5 months [33,161]. There is a paucity of data regarding second-line therapy and most common regimens are irinotecan-based regimens (FOLFIRI, or Irinotecan plus cisplatin) with limited benefit. There are no similar data for dual ICPIs available for appendiceal MiNEN [Figure 5].
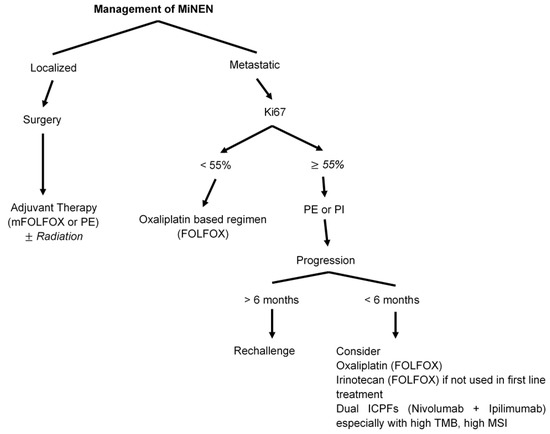
Figure 5.
Management of mixed neuroendocrine-non-neuroendocrine neoplasms (MiNENs).
9. Conclusions
Appendiceal NENs (ANENs) are a heterogeneous group of neoplasms with biological behaviors that range from indolent to highly aggressive. Due to limited data, every case requires a multi-disciplinary discussion to include clinical and pathological characteristics that will impact their management. Current surgical strategies for localized well-differentiated ANETs may vary from simple appendicectomy to right hemicolectomy depending on tumor size and patterns of invasion. Because the diagnosis is usually established at the time of histological examination of an appendectomy specimen, it is important to identify the subgroup of patients who will require further therapy. Recently, there have been significant advances in the management of NETs and novel therapeutic options are available for patients with metastatic disease; cytotoxic chemotherapy should be used only for patients with high-grade NETs and high tumor burden who have no other therapeutic options. Although poorly differentiated ANECs are extremely rare, their management involves platinum etoposide chemotherapy. Very few trials have included patients with ANENs and the data are scant. Therefore, it is crucial to evaluate many factors including surgical respectability, even in the metastatic setting, status of somatostatin avidity, histological grade, and tumor burden to select the appropriate personalized treatment for every patient through multidisciplinary discussion by NET experts.
Author Contributions
All authors have read and agreed to the published version of the manuscript.
Funding
This research received no specific grant from any funding agency.
Conflicts of Interest
None of the authors has a conflict of interest to declare.
Abbreviations
| Abbreviation | Description |
| NETs | neuroendocrine tumors |
| NECs | neuroendocrine carcinomas |
| ANENs | appendiceal neuroendocrine neoplasms |
| ANETs | Appendiceal neuroendocrine tumors |
| ANECs | appendiceal neuroendocrine carcinoma |
| MiNENs | mixed neuroendocrine-non-neuroendocrine neoplasms |
| pNETs | pancreatic neuroendocrine tumors |
| GEP-NENs | gastroenteropancreatic neuroendocrine neoplasms |
| G1 | low grade (Grade 1) |
| G2 | intermediate grade (Grade 2) |
| G3 | high grade (Grade 3) |
| WHO | World Health Organization |
| MI | mitotic index |
| CgA | Chromogranin A |
| 5-HIAA | 5-hydroxyindoleacetic acid |
| CT | computed tomography |
| MRI | magnetic resonance imaging |
| PET | positron emission tomography |
| SPECT | single-photon emission computed tomography |
| SSTR-PET | somatostatin receptor PET |
| NANETS | North American Neuroendocrine Tumor Society |
| ENETS | European Neuroendocrine Tumor Society |
| NCI | national cancer institute |
| RFA | radiofrequency ablation |
| RHC | Right hemicolectomy |
| SSAs | somatostatin analogues |
| SSTR | somatostatin receptor |
| LAR | long-acting release |
| TTP | time to progression |
| PFS | progression-free survival |
| PRRT | peptide receptor radionuclide therapy |
| HR | hazard ratio |
| MDS | myelodysplastic syndrome |
| VEGF | vascular endothelial growth factor |
| PDGFRs | platelet-derived growth factor receptors |
| FGFRs | Fibroblast growth factor receptors |
| TKIs | tyrosine kinase inhibitors |
| CAPTEM | capecitabine and temozolomide |
| ORR | objective response rate |
| IP | Irinotecan cisplatin doublet |
| EP | platinum etoposide |
| CI | confidence interval |
| RR | relative risk |
| OS | overall survival |
References
- Van De Moortele, M.; De Hertogh, G.; Sagaert, X.; Van Cutsem, E. Appendiceal cancer: A review of the literature. Acta Gastroenterol. Belg. 2020, 83, 441–448. [Google Scholar]
- Moertel, C.G.; Weiland, L.H.; Nagorney, D.M.; Dockerty, M.B. Carcinoid Tumor of the Appendix: Treatment and Prognosis. N. Eng. J. Med. 1987, 317, 1699–1701. [Google Scholar] [CrossRef]
- McCusker, M.E.; Coté, T.R.; Clegg, L.X.; Sobin, L.H. Primary malignant neoplasms of the appendix. Cancer 2002, 94, 3307–3312. [Google Scholar] [CrossRef] [PubMed]
- McGory, M.L.; Maggard, M.A.; Kang, H.; O’Connell, J.B.; Ko, C.Y. Malignancies of the Appendix: Beyond Case Series Reports. Dis. Colon Rectum 2005, 48, 2264–2271. [Google Scholar] [CrossRef] [PubMed]
- Benedix, F.; Reimer, A.; Gastinger, I.; Mroczkowski, P.; Lippert, H.; Kube, R. Primary appendiceal carcinoma–Epidemiology, surgery and survival: Results of a German multi-center study. Eur. J. Surg. Oncol. (EJSO) 2010, 36, 763–771. [Google Scholar] [CrossRef]
- Hrabe, J. Neuroendocrine Tumors of the Appendix, Colon, and Rectum. Surg. Oncol. Clin. North. Am. 2020, 29, 267–279. [Google Scholar] [CrossRef]
- Amr, B.; Froghi, F.; Edmond, M.; Haq, K.; Kochupapy, R.T. Management and outcomes of appendicular neuroendocrine tumours: Retrospective review with 5-year follow-up. Eur. J. Surg. Oncol. (EJSO) 2015, 41, 1243–1246. [Google Scholar] [CrossRef]
- Unver, N.; Coban, G.; Arıcı, D.S.; Buyukpınarbasılı, N.; Gucin, Z.; Malya, F.; Onaran, O.I.; Topalan, K. Unusual Histopathological Findings in Appendectomy Specimens: A Retrospective Analysis of 2047 Cases. Int. J. Surg. Pathol. 2018, 27, 142–146. [Google Scholar] [CrossRef]
- Coşkun, H.; Bostanci, O.; Dilege, M.E.; Mihmanli, M.; Yilmaz, B.; Akgün, I.; Yildirim, S. Carcinoid Tumors of Ap-pendix: Treatment and Outcome. Ulus. Travma. Acil. Cerrahi. Derg. 2006, 12, 150–154. [Google Scholar]
- Alexandraki, K.I.; Kaltsas, G.A.; Grozinsky-Glasberg, S.; Chatzellis, E.; Grossman, A.B. Appendiceal Neuroendo-crine Neoplasms:Diagnosis and Management. Endocr. Relat. Cancer 2016, 23, R27–R41. [Google Scholar] [CrossRef]
- Goede, A.C.; Caplin, M.E.; Winslet, M.C. Carcinoid tumour of the appendix. Br. J. Surg. 2003, 90, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Modlin, I.M.; Lye, K.D.; Kidd, M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer 2003, 97, 934–959. [Google Scholar] [CrossRef] [PubMed]
- Stinner, B.; Rothmund, M. Neuroendocrine tumours (carcinoids) of the appendix. Best Pr. Res. Clin. Gastroenterol. 2005, 19, 729–738. [Google Scholar] [CrossRef]
- Maggard, M.A.; O’Connell, J.B.; Ko, C.Y. Updated Population-Based Review of Carcinoid Tumors. Ann. Surg. 2004, 240, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Hassan, M.M.; Phan, A.T.; Dagohoy, C.G.; Leary, C.C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.-N.; Rashid, A.; et al. One hundred years after "carcinoid": Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J. Clin. Oncol. 2008, 26, 3063–3072. [Google Scholar] [CrossRef]
- Sadot, E.; Keidar, A.; Shapiro, R.; Wasserberg, N. Laparoscopic accuracy in prediction of appendiceal pathology: Oncologic and inflammatory aspects. Am. J. Surg. 2013, 206, 805–809. [Google Scholar] [CrossRef]
- Rindi, G. Nomenclature and Classification of Neuroendocrine Neoplasms of the Digestive System. In WHO Classification of Tumours of the Digestive System; Bosman, T.F., Hruban, R.H., Theise, N.D., Eds.; International Agency for Research on Cancer (IARC): Lion, France, 2010. [Google Scholar]
- Volante, M.; Grillo, F.; Massa, F.; Maletta, F.; Mastracci, L.; Campora, M.; Ferro, J.; Vanoli, A.; Papotti, M. Neuroendocrine neoplasms of the appendix, colon and rectum. Pathologica 2021, 113, 19–27. [Google Scholar] [CrossRef]
- Rindi, G.; Mete, O.; Uccella, S.; Basturk, O.; La Rosa, S.; Brosens, L.A.A.; Ezzat, S.; de Herder, W.W.; Klimstra, D.S.; Papotti, M.; et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr. Pathol. 2022, 33, 115–154. [Google Scholar] [CrossRef]
- Hsu, C.; Rashid, A.; Xing, Y.; Chiang, Y.-J.; Chagpar, R.B.; Fournier, K.F.; Chang, G.J.; You, Y.N.; Feig, B.W.; Cormier, J.N. Varying malignant potential of appendiceal neuroendocrine tumors: Importance of histologic subtype. J. Surg. Oncol. 2012, 107, 136–143. [Google Scholar] [CrossRef]
- Cai, W.; Tan, Y.; Ge, W.; Ding, K.; Hu, H. Pattern and risk factors for distant metastases in gastrointestinal neuroendocrine neoplasms: A population-based study. Cancer Med. 2018, 7, 2699–2709. [Google Scholar] [CrossRef]
- Smith, J.D.; Reidy, D.L.; Goodman, K.A.; Shia, J.; Nash, G.M. A Retrospective Review of 126 High-Grade Neuroendocrine Carcinomas of the Colon and Rectum. Ann. Surg. Oncol. 2014, 21, 2956–2962. [Google Scholar] [CrossRef] [PubMed]
- Maru, D.M.; Khurana, H.; Rashid, A.; Correa, A.M.; Anandasabapathy, S.; Krishnan, S.; Komaki, R.; Ajani, J.A.; Swisher, S.G.; Hofstetter, W.L. Retrospective Study of Clinicopathologic Features and Prognosis of High-grade Neuroendocrine Carcinoma of the Esophagus. Am. J. Surg. Pathol. 2008, 32, 1404–1411. [Google Scholar] [CrossRef] [PubMed]
- Brathwaite, S.; Rock, J.; Yearsley, M.M.; Bekaii-Saab, T.; Wei, L.; Frankel, W.L.; Hays, J.; Wu, C.; Abdel-Misih, S. Mixed Adeno-neuroendocrine Carcinoma: An Aggressive Clinical Entity. Ann. Surg. Oncol. 2016, 23, 2281–2286. [Google Scholar] [CrossRef] [PubMed]
- Abreu, R.P.N.D.S. Appendiceal neuroendocrine tumors: Approach and treatment. J. Coloproctology 2018, 38, 337–342. [Google Scholar] [CrossRef]
- Mullen, J.T.; Savarese, D.M. Carcinoid tumors of the appendix: A population-based study. J. Surg. Oncol. 2011, 104, 41–44. [Google Scholar] [CrossRef]
- Sandor, A.; Modlin, I.M. A Retrospective Analysis of 1570 Appendiceal Carcinoids. Am. J. Gastroenterol. 1998, 93, 422–428. [Google Scholar] [CrossRef]
- Bayhan, Z.; Yildiz, Y.A.; Akdeniz, Y.; Gonullu, E.; Altintoprak, F.; Mantoglu, B.; Capoglu, R.; Akkaya, Z.K. Appendix Neuroendocrine Tumor: Retrospective Analysis of 4026 Appendectomy Patients in a Single Center. Emerg. Med. Int. 2020, 2020, 1–6. [Google Scholar] [CrossRef]
- Shapiro, R.; Eldar, S.; Sadot, E.; Venturero, M.; Papa, M.Z.; Zippel, D.B. The significance of occult carcinoids in the era of laparoscopic appendectomies. Surg. Endosc. 2010, 24, 2197–2199. [Google Scholar] [CrossRef]
- Connor, S.J.; Hanna, G.B.; Frizelle, F.A. Appendiceal tumors. Dis. Colon Rectum 1998, 41, 75–80. [Google Scholar] [CrossRef]
- Rault-Petit, B.; Cao, C.D.; Guyétant, S.; Guimbaud, R.; Rohmer, V.; Julié, C.; Baudin, E.; Goichot, B.; Coriat, R.; Tabarin, A.; et al. Current Management and Predictive Factors of Lymph Node Metastasis of Appendix Neuroendocrine Tumors. Ann. Surg. 2019, 270, 165–171. [Google Scholar] [CrossRef]
- Roggo, A.; Wood, W.C.; Ottinger, L.W. Carcinoid Tumors of the Appendix. Ann. Surg. 1993, 217, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, A.; Riva, N.; Ricci, M.; Liverani, C.; La Manna, F.; De Vita, A.; Ibrahim, T.; Foca, F.; Mercatali, L.; Amadori, D.; et al. First-line chemotherapy in patients with metastatic gastroenteropancreatic neuroendocrine carcinoma. OncoTargets Ther. 2015, 8, 3613–3619. [Google Scholar] [CrossRef] [PubMed]
- Turaga, K.K.; Pappas, S.G.; Gamblin, T.C. Importance of Histologic Subtype in the Staging of Appendiceal Tumors. Ann. Surg. Oncol. 2012, 19, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- De Mestier, L.; Cros, J.; Neuzillet, C.; Hentic, O.; Egal, A.; Muller, N.; Bouché, O.; Cadiot, G.; Ruszniewski, P.; Couvelard, A.; et al. Digestive System Mixed Neuroendocrine-Non-Neuroendocrine Neoplasms. Neuroendocrinology 2017, 105, 412–425. [Google Scholar] [CrossRef]
- Ludmir, E.B.; McCall, S.J.; Cardona, D.M.; Perkinson, K.R.; Guy, C.D.; Zhang, X. Mixed Adenoneuroendocrine Carcinoma, Amphicrine Type, of the Small Bowel. Am. J. Clin. Pathol. 2016, 145, 703–709. [Google Scholar] [CrossRef]
- Ozemir, I.A.; Baysal, H.; Zemheri, E.; Bilgic, C.; Yigitbasi, R.; Alimoglu, O. Goblet cell carcinoid of the appendix accompanied by adenomatous polyp with high-grade dysplasia at the cecum. Turk. J. Surg. 2018, 34, 234–236. [Google Scholar] [CrossRef]
- Guzman, C.; Boddhula, S.; Panneerselvam, N.; Dodhia, C.; Hellenthal, N.J.; Monie, D.; Monzon, J.R.; Kaufman, T. Appendiceal Carcinoid Tumors: Is There a Survival Advantage to Colectomy over Appendectomy? J. Gastrointest. Surg. 2019, 24, 1149–1157. [Google Scholar] [CrossRef]
- Iwafuchi, M.; Watanabe, H.; Kijima, H.; Ajioka, Y.; Shimoda, T.; Ito, S. ARGYROPHIL, NON-ARGENTAFFIN CARCINOIDS OF THE APPENDIX VERMIFORMIS Immunohistochemical and Ultrastructural Studies. Pathol. Int. 1987, 37, 1237–1247. [Google Scholar] [CrossRef]
- Carr, N.J.; Sobin, L.H. Neuroendocrine tumors of the appendix. Semin. Diagn. Pathol. 2004, 21, 108–119. [Google Scholar] [CrossRef]
- Sohn, J.H.; Cho, M.-Y.; Park, Y.; Kim, H.; Kim, W.H.; Kim, J.M.; Jung, E.S.; Kim, K.-M.; Lee, J.H.; Chan, H.K.; et al. Prognostic Significance of Defining L-Cell Type on the Biologic Behavior of Rectal Neuroendocrine Tumors in Relation with Pathological Parameters. Cancer Res. Treat. 2015, 47, 813–822. [Google Scholar] [CrossRef]
- Cloyd, J.M.; Ejaz, A.; Konda, B.; Makary, M.S.; Pawlik, T.M. Neuroendocrine liver metastases: A contemporary review of treatment strategies. HepatoBiliary Surg. Nutr. 2020, 9, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Warner, R.R.; Aronson, A.; Wisnivesky, J.P.; Kim, M.K. Lymph Node Metastasis in the Prognosis of Gastroenteropancreatic Neuroendocrine Tumors. Pancreas 2017, 46, 1214–1218. [Google Scholar] [CrossRef]
- Sansone, A.; Lauretta, R.; Vottari, S.; Chiefari, A.; Barnabei, A.; Romanelli, F.; Appetecchia, M. Specific and Non-Specific Biomarkers in Neuroendocrine Gastroenteropancreatic Tumors. Cancers 2019, 11, 1113. [Google Scholar] [CrossRef] [PubMed]
- Marotta, V.; Zatelli, M.C.; Sciammarella, C.; Ambrosio, M.R.; Bondanelli, M.; Colao, A.; Faggiano, A. Chromogranin A as circulating marker for diagnosis and management of neuroendocrine neoplasms: More flaws than fame. Endocrine-Related Cancer 2018, 25, R11–R29. [Google Scholar] [CrossRef] [PubMed]
- Mahata, S.K.; Corti, A. Chromogranin A and its fragments in cardiovascular, immunometabolic, and cancer regulation. Ann. New York Acad. Sci. 2019, 1455, 34–58. [Google Scholar] [CrossRef]
- Yao, J.C.; Pavel, M.; Lombard-Bohas, C.; Van Cutsem, E.; Voi, M.; Brandt, U.; He, W.; Chen, D.; Capdevila, J.; De Vries, E.G.E.; et al. Everolimus for the Treatment of Advanced Pancreatic Neuroendocrine Tumors: Overall Survival and Circulating Biomarkers From the Randomized, Phase III RADIANT-3 Study. J. Clin. Oncol. 2016, 34, 3906–3913. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Pavel, M.; Phan, A.T.; Kulke, M.H.; Hoosen, S.; Peter, J.S.; Cherfi, A.; Öberg, K.E. Chromogranin A and Neuron-Specific Enolase as Prognostic Markers in Patients with Advanced pNET Treated with Everolimus. J. Clin. Endocrinol. Metab. 2011, 96, 3741–3749. [Google Scholar] [CrossRef]
- Chou, W.-C.; Chen, J.-S.; Hung, Y.-S.; Hsu, J.-T.; Chen, T.-C.; Sun, C.-F.; Lu, C.-H.; Hwang, T.-L. Plasma Chro-mogranin A Levels Predict Survival and Tumor Response in Patients with Advanced Gastroenteropancreatic Neuroendocrine Tumors. Anticancer Res. 2014, 34, 5661–5669. [Google Scholar] [PubMed]
- Chou, W.-C.; Hung, Y.-S.; Hsu, J.-T.; Chen, J.-S.; Lu, C.-H.; Hwang, T.-L.; Rau, K.-M.; Yeh, K.-Y.; Chen, T.-C.; Sun, C.-F. Chromogranin A Is a Reliable Biomarker for Gastroenteropancreatic Neuroendocrine Tumors in an Asian Population of Patients. Neuroendocrinology 2012, 95, 344–350. [Google Scholar] [CrossRef]
- Hendy, G.N.; Bevan, S.; Mattei, M.G.; Mouland, A.J. Chromogranin A. Clin. Invest. Med. 1995, 18, 47–65. [Google Scholar]
- Gut, P.; Czarnywojtek, A.; Fischbach, J.; Bączyk, M.; Ziemnicka, K.; Wrotkowska, E.; Gryczyńska, M.; Ruchała, M. Chromogranin A–unspecific neuroendocrine marker. Clinical utility and potential diagnostic pitfalls. Arch. Med Sci. 2016, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kaltsas, G.; Caplin, M.; Davies, P.; Ferone, D.; Garcia-Carbonero, R.; Grozinsky-Glasberg, S.; Hörsch, D.; Janson, E.T.; Kianmanesh, R.; Kos-Kudla, B.; et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: Pre- and Perioperative Therapy in Patients with Neuroendocrine Tumors. Neuroendocrinology 2017, 105, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Halperin, D.M.; Shen, C.; Dasari, A.; Xu, Y.; Chu, Y.; Zhou, S.; Shih, Y.-C.T.; Yao, J.C. Frequency of carcinoid syndrome at neuroendocrine tumour diagnosis: A population-based study. Lancet Oncol. 2017, 18, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Ewang-Emukowhate, M.; Nair, D.; Caplin, M. The role of 5-hydroxyindoleacetic acid in neuroendocrine tumors: The journey so far. Int. J. Endocr. Oncol. 2019, 6, IJE17. [Google Scholar] [CrossRef]
- Griniatsos, J.; Michail, O. Appendiceal neuroendocrine tumors: Recent insights and clinical implications. World J. Gastrointest. Oncol. 2010, 2, 192–196. [Google Scholar] [CrossRef]
- Murray, S.E.; Lloyd, R.V.; Sippel, R.S.; Chen, H.; Oltmann, S.C. Postoperative surveillance of small appendiceal carcinoid tumors. Am. J. Surg. 2013, 207, 342–345. [Google Scholar] [CrossRef]
- Reubi, J.C.; Waser, B.; Cescato, R.; Gloor, B.; Stettler, C.; Christ, E. Internalized Somatostatin Receptor Subtype 2 in Neuroendocrine Tumors of Octreotide-Treated Patients. J. Clin. Endocrinol. Metab. 2010, 95, 2343–2350. [Google Scholar] [CrossRef]
- van Adrichem, R.C.; Kamp, K.; van Deurzen, C.H.; Biermann, K.; Feelders, R.A.; Franssen, G.J.; Kwekkeboom, D.J.; Hofland, L.J.; de Herder, W.W. Is There an Additional Value of Using Somatostatin Receptor Subtype 2a Immunohistochemistry Compared to Somatostatin Receptor Scintigraphy Uptake in Predicting Gastroenteropancreatic Neuroendocrine Tumor Response? Neuroendocrinology 2015, 103, 560–566. [Google Scholar] [CrossRef]
- Hope, T.A.; Bergsland, E.K.; Bozkurt, M.F.; Graham, M.; Heaney, A.P.; Herrmann, K.; Howe, J.; Kulke, M.H.; Kunz, P.L.; Mailman, J.; et al. Appropriate Use Criteria for Somatostatin Receptor PET Imaging in Neuroendocrine Tumors. J. Nucl. Med. 2017, 59, 66–74. [Google Scholar] [CrossRef]
- Weber, M.; Kessler, L.; Schaarschmidt, B.M.; Fendler, W.P.; Lahner, H.; Antoch, G.; Umutlu, L.; Herrmann, K.; Rischpler, C. Treatment-related changes in neuroendocrine tumors as assessed by textural features derived from 68Ga-DOTATOC PET/MRI with simultaneous acquisition of apparent diffusion coefficient. BMC Cancer 2020, 20, 1–12. [Google Scholar] [CrossRef]
- Sadowski, S.M.; Neychev, V.; Millo, C.; Shih, J.; Nilubol, N.; Herscovitch, P.; Pacak, K.; Marx, S.J.; Kebebew, E. Prospective Study of 68Ga-DOTATATE Positron Emission Tomography/Computed Tomography for Detecting Gastro-Entero-Pancreatic Neuroendocrine Tumors and Unknown Primary Sites. J. Clin. Oncol. 2016, 34, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.H.; Adsay, V.; Schuster, D.; Russell, M.C.; Cardona, K.; Delman, K.A.; Winer, J.H.; Altinel, D.; Sarmiento, J.M.; El-Rayes, B.; et al. Octreoscan Versus FDG-PET for Neuroendocrine Tumor Staging: A Biological Approach. Ann. Surg. Oncol. 2015, 22, 2295–2301. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Y.; Tuli, A.; Muhleman, M.; Sen, U.; Gavane, S.; Heiba, S.; Kostakoglu, L. Impact and Potential Pitfalls of Ga-68 DOTATATE PET/CT. J. Nucl. Med. 2018, 59, 1221. [Google Scholar]
- Mojtahedi, A.; Thamake, S.; Tworowska, I.; Ranganathan, D.; Delpassand, E.S. The Value of (68)Ga-DOTATATE PET/CT in Diagnosis and Management of Neuroendocrine Tumors Compared to Current FDA Approved Imag-ing Modalities: A Review of Literature. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 426–434. [Google Scholar] [PubMed]
- Venkitaraman, B.; Karunanithi, S.; Kumar, A.; Khilnani, G.C.; Kumar, R. Role of 68Ga-DOTATOC PET/CT in initial evaluation of patients with suspected bronchopulmonary carcinoid. Eur. J. Nucl. Med. 2014, 41, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, A.; Kebebew, E. The utility of 68Ga-DOTATATE positron-emission tomography/computed tomography in the diagnosis, management, follow-up and prognosis of neuroendocrine tumors. Futur. Oncol. 2018, 14, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Menda, Y.; O’Dorisio, T.M.; Howe, J.R.; Schultz, M.; Dillon, J.S.; Dick, D.; Watkins, G.L.; Ginader, T.; Bushnell, D.L.; Sunderland, J.J.; et al. Localization of Unknown Primary Site with 68Ga-DOTATOC PET/CT in Patients with Metastatic Neuroendocrine Tumor. J. Nucl. Med. 2017, 58, 1054–1057. [Google Scholar] [CrossRef]
- Sadowski, S.M.; Millo, C.; Cottle-Delisle, C.; Merkel, R.; Yang, L.A.; Herscovitch, P.; Pacak, K.; Simonds, W.F.; Marx, S.J.; Kebebew, E. Results of 68Gallium-DOTATATE PET/CT Scanning in Patients with Multiple Endocrine Neoplasia Type 1. J. Am. Coll. Surg. 2015, 221, 509–517. [Google Scholar] [CrossRef]
- Holmager, P.; Willemoe, G.L.; Nielsen, K.; Grøndahl, V.; Klose, M.; Andreassen, M.; Langer, S.W.; Hansen, C.P.; Kjær, A.; Federspiel, B.H.; et al. Neuroendocrine neoplasms of the appendix: Characterization of 335 patients referred to the Copenhagen NET Center of Excellence. Eur. J. Surg. Oncol. (EJSO) 2021, 47, 1357–1363. [Google Scholar] [CrossRef]
- Pawa, N.; Clift, A.K.; Osmani, H.; Drymousis, P.; Cichocki, A.; Flora, R.; Goldin, R.; Patsouras, D.; Baird, A.; Malczewska, A.; et al. Surgical Management of Patients with Neuroendocrine Neoplasms of the Appendix: Appendectomy or More. Neuroendocrinology 2017, 106, 242–251. [Google Scholar] [CrossRef]
- Mosquera, C.; Fitzgerald, T.L.; Vora, H.; Grzybowski, M. Novel nomogram combining depth of invasion and size can accurately predict the risk for regional nodal metastases for appendiceal neuroendocrine tumors (A-NET). J. Surg. Oncol. 2017, 116, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Rorstad, O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J. Surg. Oncol. 2005, 89, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, S.C.; Gaertner, W.; Stewart, D.; Davids, J.; Alavi, K.; Paquette, I.M.; Steele, S.R.; Feingold, D.L. The Amer-ican Society of Colon and Rectal Surgeons, Clinical Practice Guidelines for the Management of Appendiceal Ne-oplasms. Dis. Colon. Rectum. 2019, 62, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Anthony, L.B.; Strosberg, J.R.; Klimstra, D.S.; Maples, W.J.; O’dorisio, T.M.; Warner, R.R.P.; Wiseman, G.A.; Al, L.; Benson, B.; Pommier, R.F. The NANETS Consensus Guidelines for the Diagnosis and Management of Gastrointestinal Neuroendocrine Tumors (NETs) Well-Differentiated NETs of the Distal Colon and Rectum. Pancreas 2010, 39, 767–774. [Google Scholar] [CrossRef]
- Boudreaux, J.P.; Klimstra, D.S.; Hassan, M.M.; Woltering, E.A.; Jensen, R.T.; Goldsmith, S.J.; Nutting, C.; Bushnell, D.L.; Caplin, M.E.; Yao, J.C. The NANETS Consensus Guideline for the Diagnosis and Management of Neuroendocrine Tumors Well-Differentiated Neuroendocrine Tumors of the Jejunum, Ileum, Appendix, and Cecum. Pancreas 2010, 39, 753–766. [Google Scholar] [CrossRef]
- Landry, J.P.; Mattison, Y.B.; Ramirez, R.A.; Boudreaux, J.P.; Woltering, E.A.; Maluccio, M.A.; Thiagarajan, R. Management of Appendiceal Neuroendocrine Tumors: Beyond Tumor Size. J. Cancer Immunol. 2020, 2, 194–198. [Google Scholar] [CrossRef]
- Landry, J.P.; Ms, B.A.V.; Ramirez, R.A.; Boudreaux, J.P.; Woltering, E.A.; Thiagarajan, R. Management of Appendiceal Neuroendocrine Tumors: Metastatic Potential of Small Tumors. Ann. Surg. Oncol. 2020, 28, 751–757. [Google Scholar] [CrossRef]
- Kleiman, D.A.; Finnerty, B.; Beninato, T.; Zarnegar, R.; Nandakumar, G.; Fahey, T.J.; Lee, S.W. Features Associated With Metastases Among Well-Differentiated Neuroendocrine (Carcinoid) Tumors of the Appendix. Dis. Colon Rectum 2015, 58, 1137–1143. [Google Scholar] [CrossRef]
- Raoof, M.; Dumitra, S.; O’Leary, M.P.; Singh, G.; Fong, Y.; Lee, B. Mesenteric Lymphadenectomy in Well-Differentiated Appendiceal Neuroendocrine Tumors. Dis. Colon Rectum 2017, 60, 674–681. [Google Scholar] [CrossRef]
- Nussbaum, D.P.; Speicher, P.J.; Gulack, B.C.; Keenan, J.E.; Ganapathi, A.M.; Englum, B.R.; Tyler, D.S.; Blazer, D.G. Management of 1- to 2-cm Carcinoid Tumors of the Appendix: Using the National Cancer Data Base to Address Controversies in General Surgery. J. Am. Coll. Surg. 2015, 220, 894–903. [Google Scholar] [CrossRef]
- Pape, U.-F.; Niederle, B.; Costa, F.; Gross, D.; Kelestimur, F.; Kianmanesh, R.; Knigge, U.; Öberg, K.; Pavel, M.; Perren, A.; et al. ENETS Consensus Guidelines for Neuroendocrine Neoplasms of the Appendix (Excluding Goblet Cell Carcinomas). Neuroendocrinology 2016, 103, 144–152. [Google Scholar] [CrossRef]
- Plöckinger, U.; Couvelard, A.; Falconi, M.; Sundin, A.; Salazar, R.; Christ, E.; de Herder, W.W.; Gross, D.; Knapp, W.H.; Knigge, U.P.; et al. Consensus Guidelines for the Management of Patients with Digestive Neuroendocrine Tumours: Well-Differentiated Tumour/Carcinoma of the Appendix and Goblet Cell Carcinoma. Neuroendocrinology 2007, 87, 20–30. [Google Scholar] [CrossRef]
- Sommer, C.; Pause, F.G.; Diezi, M.; Rougemont, A.-L.; Wildhaber, B.E. A National Long-Term Study of Neuroendocrine Tumors of the Appendix in Children: Are We Too Aggressive? Eur. J. Pediatr. Surg. 2018, 29, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Dockerty, M.B.; Judd, E.S. Carcinoid Tumors of the Vermiform Appendix. Cancer 1968, 21, 270–278. [Google Scholar] [CrossRef]
- Sarshekeh, A.M.; Advani, S.; Halperin, D.M.; Conrad, C.; Shen, C.; Yao, J.C.; Dasari, A. Regional lymph node involvement and outcomes in appendiceal neuroendocrine tumors: A SEER database analysis. Oncotarget 2017, 8, 99541–99551. [Google Scholar] [CrossRef] [PubMed]
- Groth, S.S.; Virnig, B.A.; Al-Refaie, W.B.; Jarosek, S.L.; Jensen, E.; Tuttle, T.M. Appendiceal carcinoid tumors: Predictors of lymph node metastasis and the impact of right hemicolectomy on survival. J. Surg. Oncol. 2010, 103, 39–45. [Google Scholar] [CrossRef]
- Daskalakis, K.; Alexandraki, K.; Kassi, E.; Tsoli, M.; Angelousi, A.; Ragkousi, A.; Kaltsas, G. The risk of lymph node metastases and their impact on survival in patients with appendiceal neuroendocrine neoplasms: A systematic review and meta-analysis of adult and paediatric patients. Endocrine 2019, 67, 20–34. [Google Scholar] [CrossRef]
- Hesketh1, K.T. The Management of Primary Adenocarcinoma of the Vermiform Appendix. Gut 1963, 4, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Bednarczuk, T.; Bolanowski, M.; Zemczak, A.; Bałdys-Waligórska, A.; Blicharz-Dorniak, J.; Boratyn-Nowicka, A.; Borowska, M.; Cichocki, A.; Ćwikła, J.B.; Falconi, M.; et al. Nowotwory neuroendokrynne jelita cienkiego i wyrostka robaczkowego—zasady postępowania (rekomendowane przez Polską Sieć Guzów Neuroendokrynnych). Endokrynol. Polska 2017, 68, 223–236. [Google Scholar] [CrossRef]
- Pavel, M.; Baudin, E.; Couvelard, A.; Krenning, E.; Öberg, K.; Steinmüller, T.; Anlauf, M.; Wiedenmann, B.; Salazar, R. ENETS Consensus Guidelines for the Management of Patients with Liver and Other Distant Metastases from Neuroendocrine Neoplasms of Foregut, Midgut, Hindgut, and Unknown Primary. Neuroendocrinology 2012, 95, 157–176. [Google Scholar] [CrossRef]
- Limani, P.; Tschuor, C.; Gort, L.; Balmer, B.; Gu, A.; Ceresa, C.; Raptis, D.A.; Lesurtel, M.; Puhan, M.; Breitenstein, S.; et al. Nonsurgical Strategies in Patients With NET Liver Metastases: A Protocol of Four Systematic Reviews. JMIR Res. Protoc. 2014, 3, e9. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Yoshida, N.; Iwagami, S.; Iwatsuki, M.; Baba, H. Surgical treatment for gastrointestinal neuroendocrine tumors. Ann. Gastroenterol. Surg. 2020, 4, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.T.; Howe, J. Management of Small Bowel Neuroendocrine Tumors. J. Oncol. Pr. 2018, 14, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Howe, J.R.; Cardona, K.; Fraker, D.L.; Kebebew, E.; Untch, B.R.; Wang, Y.-Z.; Law, C.H.; Liu, E.H.; Kim, M.K.; Menda, Y.; et al. The Surgical Management of Small Bowel Neuroendocrine Tumors. Pancreas 2017, 46, 715–731. [Google Scholar] [CrossRef]
- Machairas, N. Currently available treatment options for neuroendocrine liver metastases. Ann. Gastroenterol. 2021, 34, 130–141. [Google Scholar] [CrossRef]
- Howe, J.R. It May Not Be Too Little or Too Late: Resecting Primary Small Bowel Neuroendocrine Tumors in the Presence of Metastatic Disease. Ann. Surg. Oncol. 2020, 27, 2583–2585. [Google Scholar] [CrossRef]
- La Salvia, A.; Partelli, S.; Tampellini, M.; Tamburrino, D.; Falconi, M.; Scagliotti, G.V.; Brizzi, M.P. Management of hepatic metastases of well/moderately differentiated neuroendocrine tumors of the digestive tract. J. Cancer Metastasis Treat. 2016, 2, 294. [Google Scholar] [CrossRef]
- Frilling, A.; Modlin, I.M.; Kidd, M.; Russell, C.; Breitenstein, S.; Salem, R.; Kwekkeboom, D.; Lau, W.-Y.; Klersy, C.; Vilgrain, V.; et al. Recommendations for management of patients with neuroendocrine liver metastases. Lancet Oncol. 2014, 15, e8–e21. [Google Scholar] [CrossRef]
- Ejaz, A.; Reames, B.N.; Maithel, S.; Poultsides, G.A.; Bauer, T.W.; Fields, R.C.; Weiss, M.J.; Marques, H.P.; Aldrighetti, L.; Pawlik, T.M. Cytoreductive debulking surgery among patients with neuroendocrine liver metastasis: A multi-institutional analysis. Hpb 2017, 20, 277–284. [Google Scholar] [CrossRef]
- Clift, A.K.; Frilling, A. Management of patients with hepatic metastases from neuroendocrine tumors. Ann. Saudi Med. 2014, 34, 279–290. [Google Scholar] [CrossRef]
- Graff-Baker, A.N.; Sauer, D.A.; Pommier, S.J.; Pommier, R.F. Expanded criteria for carcinoid liver debulking: Maintaining survival and increasing the number of eligible patients. Surgery 2014, 156, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.E.; Pommier, S.J.; Pommier, R.F. Expanded criteria for debulking of liver metastasis also apply to pancreatic neuroendocrine tumors. Surgery 2018, 163, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.E.; Sherman, S.K.; O’Dorisio, T.M.; Bellizzi, A.M.; Howe, J.R. Liver-directed surgery of neuroendocrine metastases: What is the optimal strategy? Surgery 2015, 159, 320–335. [Google Scholar] [CrossRef] [PubMed]
- Lehrman, E.D.; Fidelman, N. Liver-Directed Therapy for Neuroendocrine Tumor Liver Metastases in the Era of Peptide Receptor Radionuclide Therapy. Semin. Interv. Radiol. 2020, 37, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Gonulal, B.; Bilgic, Y.; Akbulut, S.; Karabulut, E.; Samdanci, E.T. Management and Survival Analysis of Gastrointestinal Neuroendocrine Tumors by Different Tumor Characteristics: Tertiary Center Experience. J. Gastrointest. Cancer 2021, 1–6. [Google Scholar] [CrossRef]
- Zheng, Z.; Chen, C.; Jiang, L.; Zhou, X.; Dai, X.; Song, Y.; Li, Y. Incidence and risk factors of gastrointestinal neuroendocrine neoplasm metastasis in liver, lung, bone, and brain: A population-based study. Cancer Med. 2019, 8, 7288–7298. [Google Scholar] [CrossRef]
- Kessler, J.; Singh, G.; Ituarte, P.H.; Allen, R.; Chang, S.; Li, D. A Comparison of Liver-Directed Therapy and Systemic Therapy for the Treatment of Liver Metastases in Patients with Gastrointestinal Neuroendocrine Tumors: Analysis of the California Cancer Registry. J. Vasc. Interv. Radiol. 2020, 32, 393–402. [Google Scholar] [CrossRef]
- Duan, H.; Ferri, V.; Fisher, G.A.; Shaheen, S.; Davidzon, G.A.; Iagaru, A.; Aparici, C.M. Evaluation of Liver and Renal Toxicity in Peptide Receptor Radionuclide Therapy for Somatostatin Receptor Expressing Tumors: A 2-Year Follow-Up. Oncol. 2022, 27, 447–452. [Google Scholar] [CrossRef]
- Gonzalez-Aguirre, A.; Ziv, E. Liver-Directed Therapy for Gastroenteropancreatic NETs in the Era of Peptide Receptor Radionuclide Therapy. Dig. Dis. Interv. 2020, 4, 282–290. [Google Scholar] [CrossRef]
- Spina, J.C.; Hume, I.; Pelaez, A.; Peralta, O.; Quadrelli, M.; Monaco, R.G. Expected and Unexpected Imaging Findings after 90Y Transarterial Radioembolization for Liver Tumors. Radiographics 2019, 39, 578–595. [Google Scholar] [CrossRef]
- Stueven, A.K.; Kayser, A.; Wetz, C.; Amthauer, H.; Wree, A.; Tacke, F.; Wiedenmann, B.; Roderburg, C.; Jann, H. Somatostatin Analogues in the Treatment of Neuroendocrine Tumors: Past, Present and Future. Int. J. Mol. Sci. 2019, 20, 3049. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, R.; Barnabei, A.; Rizza, L.; Isidori, A.; Rota, F.; Di Giacinto, P.; Paoloni, A.; Torino, F.; Corsello, S.M.; Lenzi, A.; et al. Somatostatin Analogs Therapy in Gastroenteropancreatic Neuroendocrine Tumors: Current Aspects and New Perspectives. Front. Endocrinol. 2014, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Porras, M.; Cárdenas-Salas, J.; Álvarez-Escolá, C. Somatostatin Analogs in Clinical Practice: A Review. Int. J. Mol. Sci. 2020, 21, 1682. [Google Scholar] [CrossRef]
- Ruszniewski, P.; Ducreux, M.; Chayvialle, J.; Blumberg, J.; Cloarec, D.; Michel, H.; Raymond, J.M.; Dupas, J.L.; Gouerou, H.; Jian, R.; et al. Treatment of the carcinoid syndrome with the longacting somatostatin analogue lanreotide: A prospective study in 39 patients. Gut 1996, 39, 279–283. [Google Scholar] [CrossRef]
- Kos-Kudła, B. Treatment of neuroendocrine tumors: New recommendations based on the CLARINET study. Contemp. Oncol. 2015, 19, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Rinke, A.; Müller, H.-H.; Schade-Brittinger, C.; Klose, K.-J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.-F.; Bläker, M.; et al. Placebo-Controlled, Double-Blind, Prospective, Randomized Study on the Effect of Octreotide LAR in the Control of Tumor Growth in Patients With Metastatic Neuroendocrine Midgut Tumors: A Report From the PROMID Study Group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef] [PubMed]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in Metastatic Enteropancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Ferolla, P.; Faggiano, A.; Grimaldi, F.; Ferone, D.; Scarpelli, G.; Ramundo, V.; Severino, R.; Bellucci, M.C.; Camera, L.M.; Lombardi, G.; et al. Shortened interval of long-acting octreotide administration is effective in patients with well-differentiated neuroendocrine carcinomas in progression on standard doses. J. Endocrinol. Investig. 2012, 35, 326–331. [Google Scholar] [CrossRef]
- Gariani, K.; Meyer, P.; Philippe, J.; Clinician, A. Implications of Somatostatin Analogues in the Treatment of Acromegaly. Eur. Endocrinol. 2010, 9, 132–135. [Google Scholar] [CrossRef]
- Pauwels, E.; Cleeren, F.; Bormans, G.; Deroose, C.M. Somatostatin receptor PET ligands—The next generation for clinical practice. Am. J. Nucl. Med. Mol. Imaging 2018, 8, 311–331. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30510849%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC6261874 (accessed on 20 November 2022).
- Sollini, M.; Erba, P.A.; Fraternali, A.; Casali, M.; Di Paolo, M.L.; Froio, A.; Frasoldati, A.; Versari, A. PET and PET/CT with68Gallium-Labeled Somatostatin Analogues in Non GEP-NETs Tumors. Sci. World J. 2014, 2014, 1–19. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Nesti, C.; Spanjol, M.; Kollár, A.; Bütikofer, L.; Gloy, V.L.; Dumont, R.; Seiler, C.; Christ, E.R.; Radojewski, P.; et al. Treatment for gastrointestinal and pancreatic neuroendocrine tumours: A network meta-analysis. Cochrane Database Syst. Rev. 2021, 2021, CD013700. [Google Scholar]
- Zanini, S.; Renzi, S.; Giovinazzo, F.; Bermano, G. mTOR Pathway in Gastroenteropancreatic Neuroendocrine Tumor (GEP-NETs). Front. Endocrinol. 2020, 11, 562505. [Google Scholar] [CrossRef]
- Gajate, P.; Martínez-Sáez, O.; Alonso-Gordoa, T.; Grande, E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manag. Res. 2017, 9, 215–224. [Google Scholar] [CrossRef]
- Chan, D.L.; Segelov, E.; Singh, S. Everolimus in the management of metastatic neuroendocrine tumours. Ther. Adv. Gastroenterol. 2016, 10, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.E.; Hainsworth, J.D.; Baudin, E.; Peeters, M.; Höersch, D.; Winkler, R.E.; Klimovsky, J.; Lebwohl, D.; Jehl, V.; Wolin, E.M.; et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): A randomised, placebo-controlled, phase 3 study. Lancet 2011, 378, 2005–2012. [Google Scholar] [CrossRef]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M.; et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet 2015, 387, 968–977. [Google Scholar] [CrossRef]
- Paplomata, E.; Zelnak, A.; O’Regan, R. Everolimus: Side effect profile and management of toxicities in breast cancer. Breast Cancer Res. Treat. 2013, 140, 453–462. [Google Scholar] [CrossRef]
- Arena, C.; Bizzoca, M.E.; Caponio, V.C.A.; Troiano, G.; Zhurakivska, K.; Leuci, S.; Muzio, L.L. Everolimus therapy and side-effects: A systematic review and meta-analysis. Int. J. Oncol. 2021, 59, 1–9. [Google Scholar] [CrossRef]
- Vitale, G.; Cozzolino, A.; Malandrino, P.; Minotta, R.; Puliani, G.; Saronni, D.; Faggiano, A.; Colao, A. Role of FGF System in Neuroendocrine Neoplasms: Potential Therapeutic Applications. Front. Endocrinol. 2021, 12, 665631. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, E.; Ignazzi, A.; De Michele, F.; Caruso, M.L. PDGFRα expression as a novel therapeutic marker in well-differentiated neuroendocrine tumors. Cancer Biol. Ther. 2018, 20, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, M.; Von Arx, C.; De Divitiis, C.; Ottaiano, A.; Tatangelo, F.; Romano, G.M.; Tafuto, S. Antiangiogenic Therapy in Pancreatic Neuroendocrine Tumors. Anticancer. Res. 2016, 36, 5025–5030. [Google Scholar] [CrossRef] [PubMed]
- Spada, F.; Antonuzzo, L.; Marconcini, R.; Radice, D.; Antonuzzo, A.; Ricci, S.; Di Costanzo, F.; Fontana, A.; Gelsomino, F.; Luppi, G.; et al. Oxaliplatin-Based Chemotherapy in Advanced Neuroendocrine Tumors: Clinical Outcomes and Preliminary Correlation with Biological Factors. Neuroendocrinology 2016, 103, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Kunz, P.L.; Balise, R.R.; Fehrenbacher, L.; Pan, M.; Venook, A.P.; Fisher, G.A.; Tempero, M.A.; Ko, A.H.; Korn, W.M.; Hwang, J.; et al. Oxaliplatin–Fluoropyrimidine Chemotherapy Plus Bevacizumab in Advanced Neuroendocrine Tumors. Pancreas 2016, 45, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shen, L.; Zhou, Z.; Li, J.; Bai, C.; Chi, Y.; Li, Z.; Xu, N.; Li, E.; Liu, T.; et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET-ep): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, J.; Fazio, N.; Lopez, C.; Teulé, A.; Valle, J.W.; Tafuto, S.; Custodio, A.; Reed, N.; Raderer, M.; Grande, E.; et al. Lenvatinib in Patients With Advanced Grade 1/2 Pancreatic and Gastrointestinal Neuroendocrine Tumors: Results of the Phase II TALENT Trial (GETNE1509). J. Clin. Oncol. 2021, 39, 2304–2312. [Google Scholar] [CrossRef]
- Bongiovanni, A.; Liverani, C.; Recine, F.; Fausti, V.; Mercatali, L.; Vagheggini, A.; Spadazzi, C.; Miserocchi, G.; Cocchi, C.; Di Menna, G.; et al. Phase-II Trials of Pazopanib in Metastatic Neuroendocrine Neoplasia (mNEN): A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Chan, J.A.; Faris, J.E.; Murphy, J.E.; Blaszkowsky, L.S.; Kwak, E.L.; McCleary, N.J.; Fuchs, C.S.; Meyerhardt, J.A.; Ng, K.; Zhu, A.X.; et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J. Clin. Oncol. 2017, 35, 228. [Google Scholar] [CrossRef]
- Fine, R.L.; Gulati, A.P.; Krantz, B.; Moss, R.; Schreibman, S.; Tsushima, D.A.; Mowatt, K.B.; Dinnen, R.D.; Mao, Y.; Stevens, P.D.; et al. Capecitabine and temozolomide (CAPTEM) for metastatic, well-differentiated neuroendocrine cancers: The Pancreas Center at Columbia University experience. Cancer Chemother. Pharmacol. 2013, 71, 663–670. [Google Scholar] [CrossRef]
- Sahu, A.; Jefford, M.; Lai-Kwon, J.; Thai, A.; Hicks, R.J.; Michael, M. CAPTEM in Metastatic Well-Differentiated Intermediate to High Grade Neuroendocrine Tumors: A Single Centre Experience. J. Oncol. 2019, 2019, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fine, R.L.; Gulati, A.P.; Tsushima, D.; Mowatt, K.B.; Oprescu, A.; Bruce, J.N.; Chabot, J.A. Prospective phase II study of capecitabine and temozolomide (CAPTEM) for progressive, moderately, and well-differentiated metastatic neuroendocrine tumors. J. Clin. Oncol. 2014, 32, 179. [Google Scholar] [CrossRef]
- Faure, M.; Niccoli, P.; Autret, A.; Cavaglione, G.; Mineur, L.; Raoul, J.-L. Systemic chemotherapy with FOLFOX in metastatic grade 1/2 neuroendocrine cancer. Mol. Clin. Oncol. 2016, 6, 44–48. [Google Scholar] [CrossRef] [PubMed]
- McNamara, M.G.; Scoazec, J.Y.; Walter, T. Extrapulmonary poorly differentiated NECs, including molecular and immune aspects. Endocrine-Related Cancer 2020, 27, R219–R238. [Google Scholar] [CrossRef] [PubMed]
- Casas, F.; Ferrer, F.; Farr s, B.; Casals, J.; Biete, A. Primary Small Cell Carcinoma of the Esophagus. Cancer 1997, 80, 1366–1372. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Sorbye, H.; Baudin, E.; Raymond, E.; Wiedenmann, B.; Niederle, B.; Sedlackova, E.; Toumpanakis, C.; Anlauf, M.; Cwikla, J.M.; et al. Vienna Consensus Conference participants. ENETS Consensus Guidelines for High-Grade Gastroenteropancreatic Neuroendocrine Tumors and Neuroendocrine Carcinomas. Neuroendocrinology 2016, 103, 186–194. [Google Scholar] [CrossRef]
- Rinke, A.; Gress, T.M. Neuroendocrine Cancer, Therapeutic Strategies in G3 Cancers. Digestion 2017, 95, 109–114. [Google Scholar] [CrossRef]
- Sorbye, H.; Welin, S.; Langer, S.W.; Vestermark, L.W.; Holt, N.; Osterlund, P.; Dueland, S.; Hofsli, E.; Guren, M.G.; Ohrling, K.; et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): The NORDIC NEC study. Ann. Oncol. 2012, 24, 152–160. [Google Scholar] [CrossRef]
- Walter, T.; Tougeron, D.; Baudin, E.; Le Malicot, K.; Lecomte, T.; Malka, D.; Hentic, O.; Manfredi, S.; Bonnet, I.; Guimbaud, R.; et al. Poorly differentiated gastro-entero-pancreatic neuroendocrine carcinomas: Are they really heterogeneous? Insights from the FFCD-GTE national cohort. Eur. J. Cancer 2017, 79, 158–165. [Google Scholar] [CrossRef]
- Brandi, G.; Paragona, M.; Campana, D.; Brighi, N.; Bondi, A.; Pantaleo, M.A.; Corbelli, J.; Barbera, M.A.; Biasco, G. Good performance of platinum-based chemotherapy for high-grade gastroenteropancreatic and unknown primary neuroendocrine neoplasms. J. Chemother. 2017, 30, 53–58. [Google Scholar] [CrossRef]
- Lamarca, A.; Frizziero, M.; Barriuso, J.; McNamara, M.G.; Hubner, R.; Valle, J.W. Platinum-etoposide chemotherapy for extra-pulmonary high grade neuroendocrine carcinoma (EP-G3-NEC): A survey of clinical practice. Endocr. Abstr. 2017, 52. [Google Scholar] [CrossRef]
- Mollazadegan, K.; Welin, S.; Crona, J. Systemic Treatment of Gastroenteropancreatic Neuroendocrine Carcinoma. Curr. Treat. Options Oncol. 2021, 22, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Morizane, C.; Machida, N.; Honma, Y.; Okusaka, T.; Boku, N.; Kato, K.; Nomura, S.; Hiraoka, N.; Sekine, S.; Taniguchi, H.; et al. Randomized phase III study of etoposide plus cisplatin versus irinotecan plus cisplatin in advanced neuroendocrine carcinoma of the digestive system: A Japan Clinical Oncology Group study (JCOG1213). J. Clin. Oncol. 2022, 40, 501. [Google Scholar] [CrossRef]
- Hentic, O.; Hammel, P.; Couvelard, A.; Rebours, V.; Zappa, M.; Palazzo, M.; Maire, F.; Goujon, G.; Gillet, A.; Levy, P.; et al. FOLFIRI regimen: An effective second-line chemotherapy after failure of etoposide–platinum combination in patients with neuroendocrine carcinomas grade 3. Endocrine-Related Cancer 2012, 19, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Malka, D.; Planchard, D.; Scoazec, J.Y.; Caramella, C.; Guigay, J.; Boige, V.; Leboulleux, S.; Burtin, P.; Berdelou, A.; et al. Post-first-line FOLFOX chemotherapy for grade 3 neuroendocrine carcinoma. Endocrine-Related Cancer 2015, 22, 289–298. [Google Scholar] [CrossRef]
- Patel, S.P.; Othus, M.; Chae, Y.K.; Giles, F.J.; Hansel, D.E.; Singh, P.P.; Fontaine, A.; Shah, M.H.; Kasi, A.; Al Baghdadi, T.; et al. A Phase II Basket Trial of Dual Anti–CTLA-4 and Anti–PD-1 Blockade in Rare Tumors (DART SWOG 1609) in Patients with Nonpancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2020, 26, 2290–2296. [Google Scholar] [CrossRef]
- Klein, O.; Kee, D.; Markman, B.; Michael, M.; Underhill, C.R.; Carlino, M.S.; Jackett, L.; Lum, C.; Scott, C.L.; Nagrial, A.; et al. Immunotherapy of Ipilimumab and Nivolumab in Patients with Advanced Neuroendocrine Tumors: A Subgroup Analysis of the CA209-538 Clinical Trial for Rare Cancers. Clin. Cancer Res. 2020, 26, 4454–4459. [Google Scholar] [CrossRef]
- Apostolidis, L.; Bergmann, F.; Haag, G.M.; Jaeger, D.; Winkler, E.C. Treatment outcomes of patients with mixed neuroendocrine-nonneuroendocrine neoplasms (MiNEN). J. Clin. Oncol. 2018, 36, e16163. [Google Scholar] [CrossRef]
- Ahmed, M. Gastrointestinal Neuroendocrine Tumors in 2020. World J. Gastrointest. Oncol. 2020, 12, 791–807. [Google Scholar] [CrossRef]
- Thomas, K.E.; Voros, B.A.; Boudreaux, J.P.; Thiagarajan, R.; Woltering, E.A.; Ramirez, R.A. Current Treatment Options in Gastroenteropancreatic Neuroendocrine Carcinoma. Oncol. 2019, 24, 1076–1088. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).