
HIV-1 Protease as DNA Immunogen against Drug Resistance in HIV-1 Infection: DNA Immunization with Drug Resistant HIV-1 Protease Protects Mice from Challenge with Protease-Expressing Cells

 , , , ,
, , , ,  , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Design and Generation of Consensus FSU-A Protease Genes
2.2. Preparation of Lentiviral Vectors Encoding PR Variants
2.3. Recombinant HIV-1 Proteases and Protease-Specific Antibodies
2.4. Peptides Representing PR Epitopes and In Silico Assessment of the Effects of DR Mutations on Immunogenicity
2.5. Eukaryotic Cell Transfection
2.6. Analysis of Protease Expression through Western Blotting
2.7. Evaluation of Protease Activity
2.8. Lentiviral Transduction of 4T1luc2 Cells and Isolation of PR_A-Expressing Clones
2.9. Experiments in Laboratory Mice
2.10. DNA Immunization of Mice
3. Humoral Immune Responses Assessed by ELISA
4. Assessment of T-Cell Immune Response by Fluorospot and Flow Cytometry
4.1. Immunotoxicity Tests
4.2. Tumor Implantation and Follow-Up of Tumor Growth in Naïve and Immunized Mice
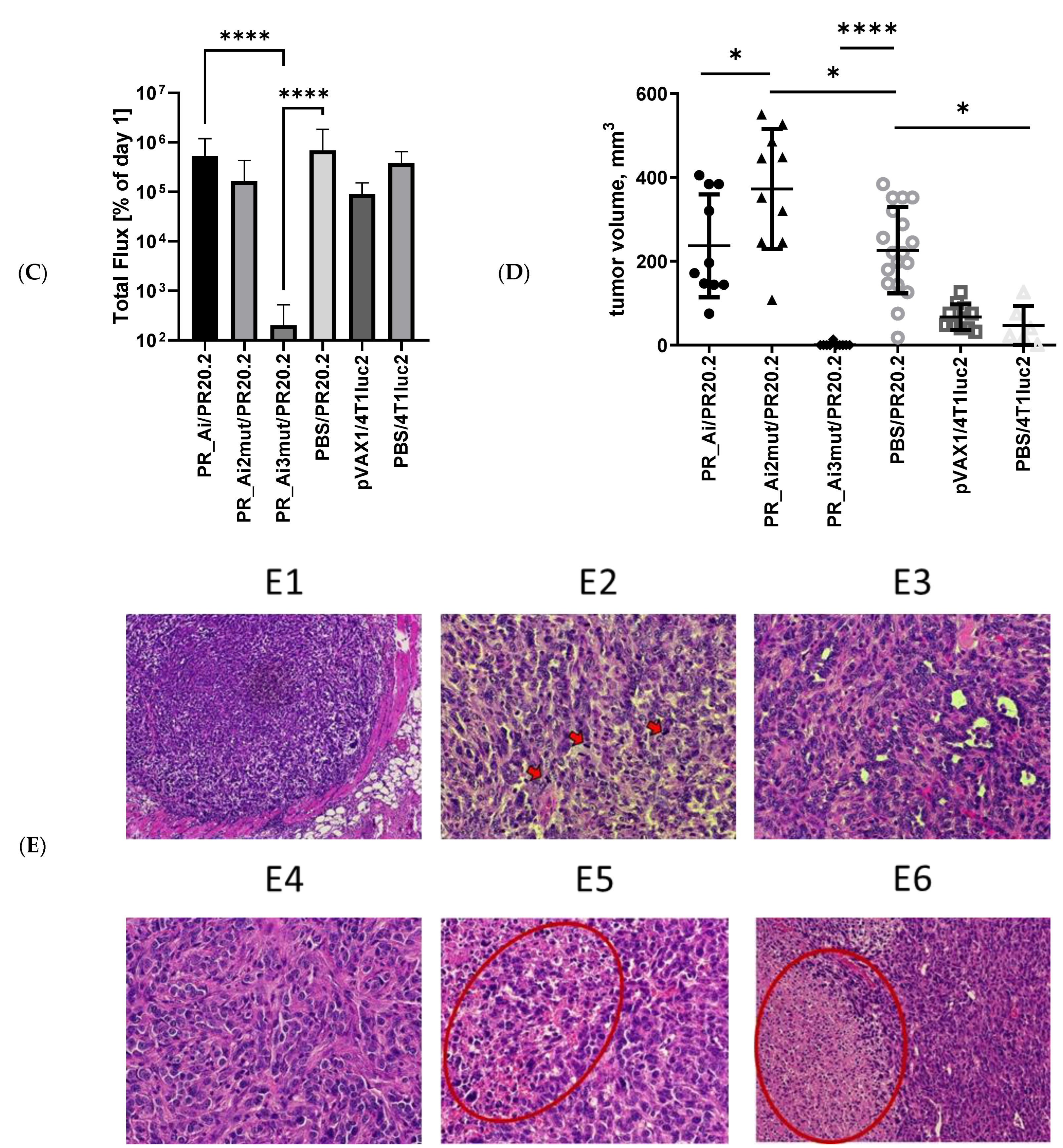
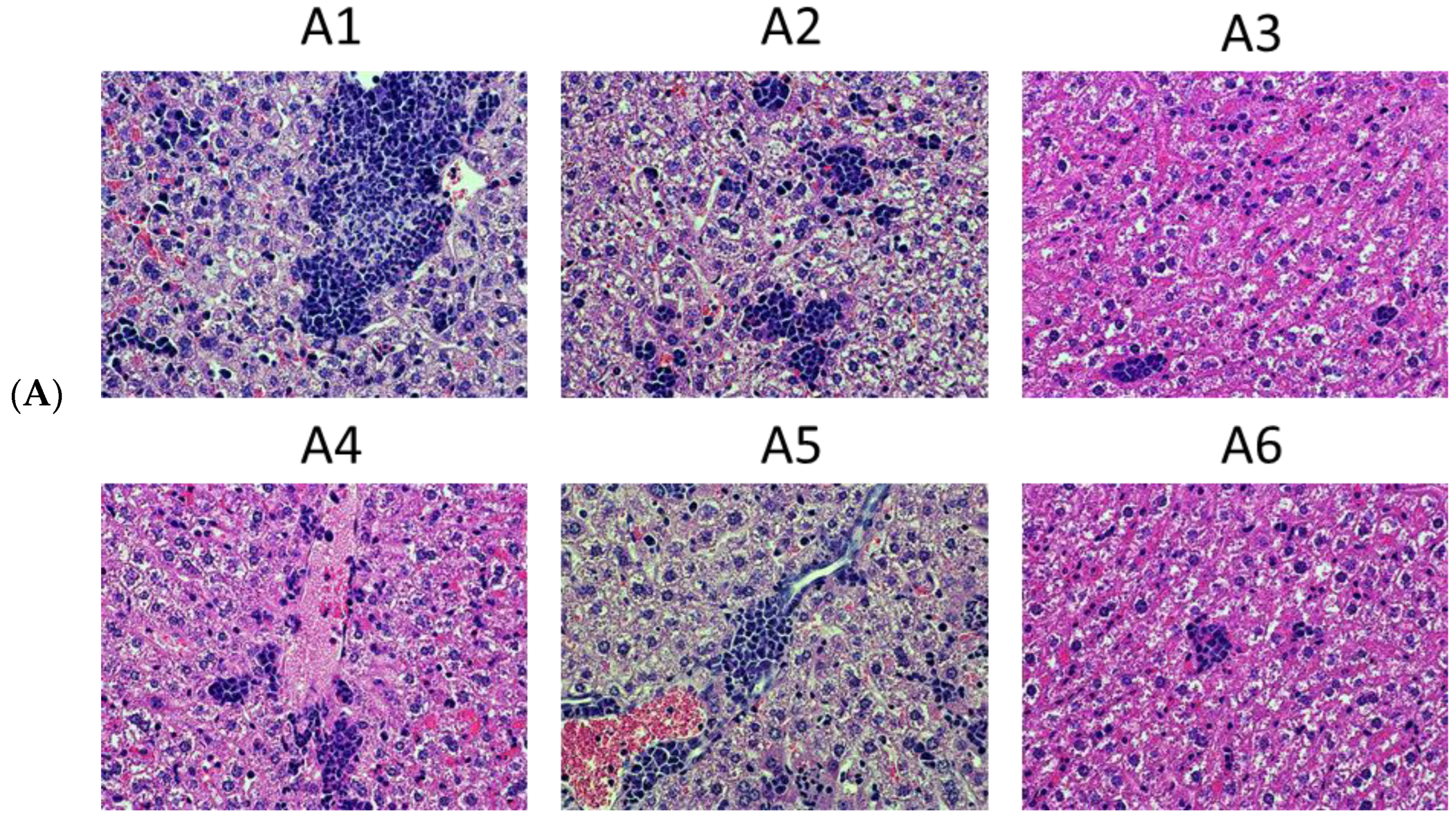
4.3. Tumor Histology and Histological Assessment of the Metastases
4.4. Statistical Analysis
5. Results
5.1. Design of the Consensus Protease of HIV-1 FSU_A Strain and Its Variants with Drug-Resistance-Conferring Mutations
5.2. Synthetic Genes’ Direct Expression of Enzymatically Active HIV-1 Proteases in Bacteria
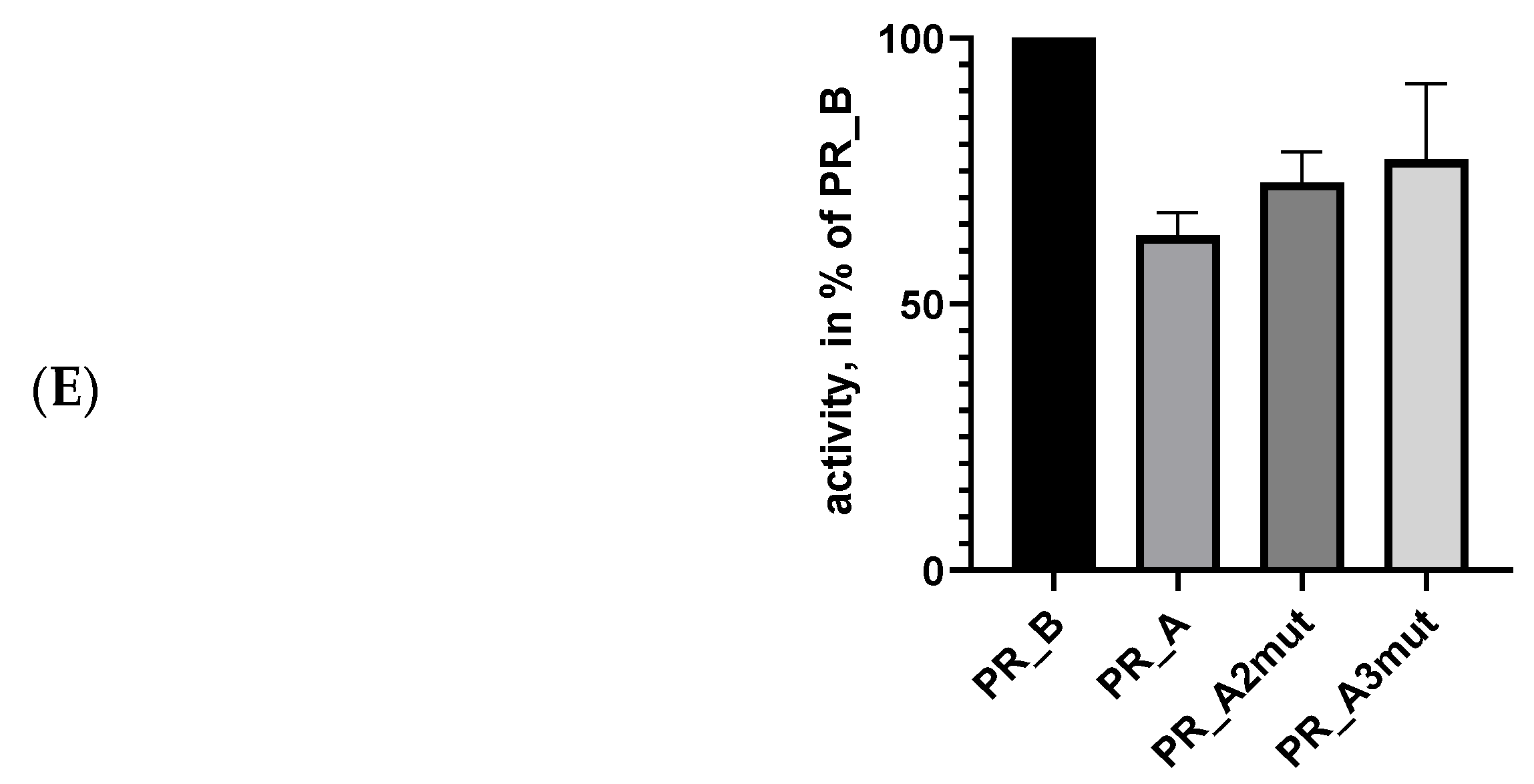
5.3. Suppression of Protease Activity Increases Protease Content in the Expressing Cells
5.4. Immunogenic Performance of Plasmids Encoding PR_A Variants in Mice
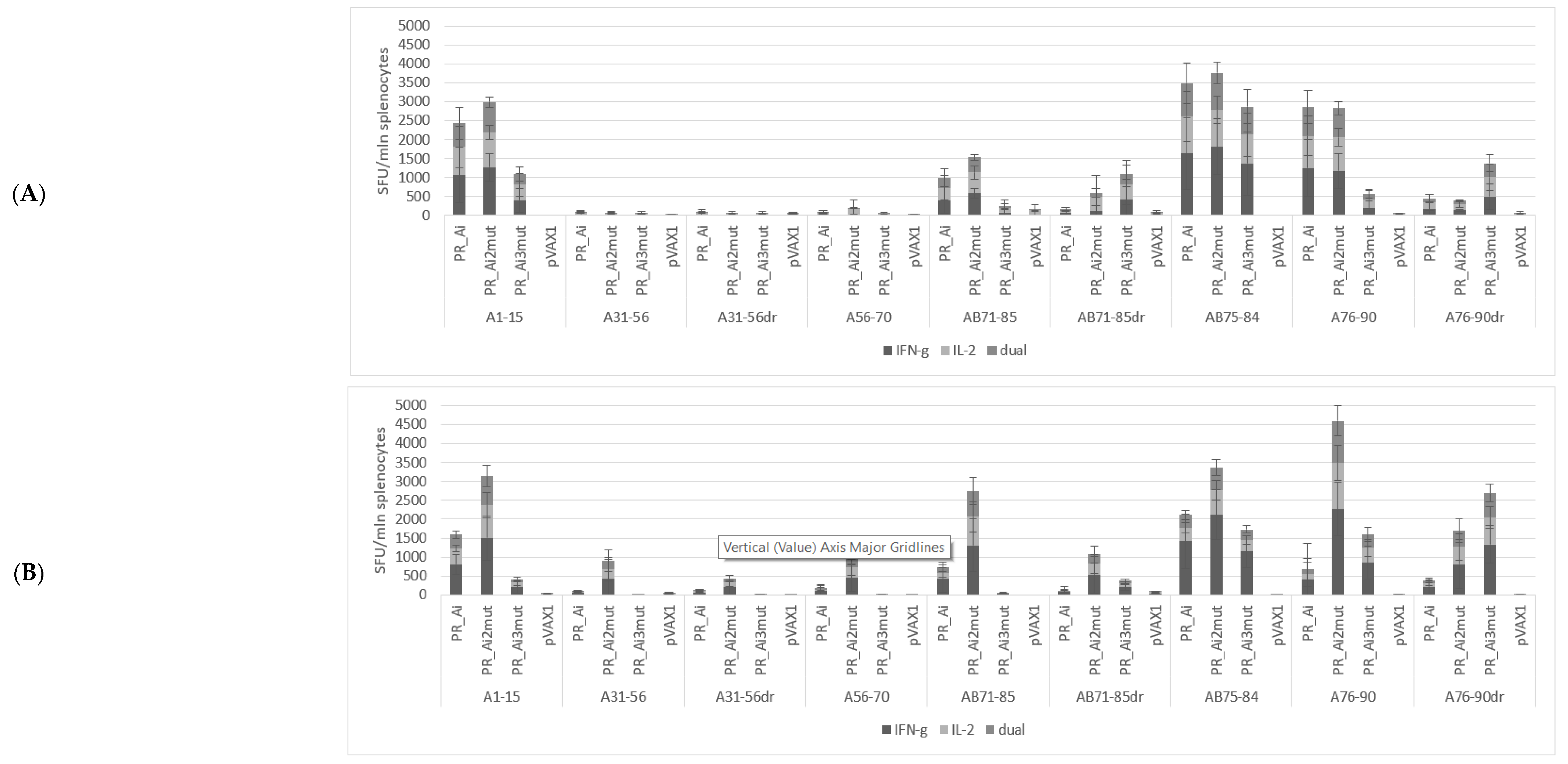
5.4.1. PR_A and PR_Ai Variants Are Strongly Immunogenic after Single DNA Immunization
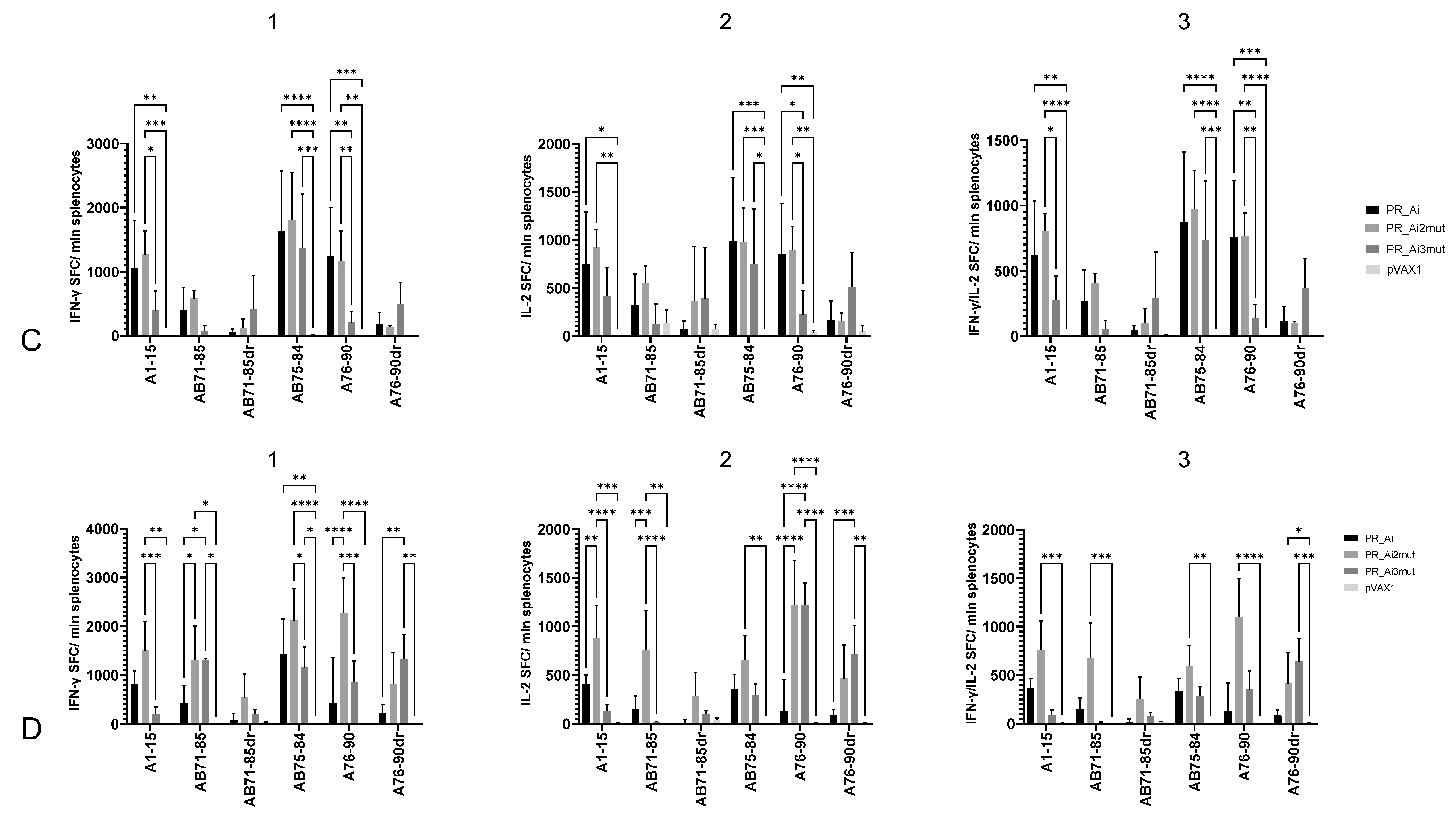
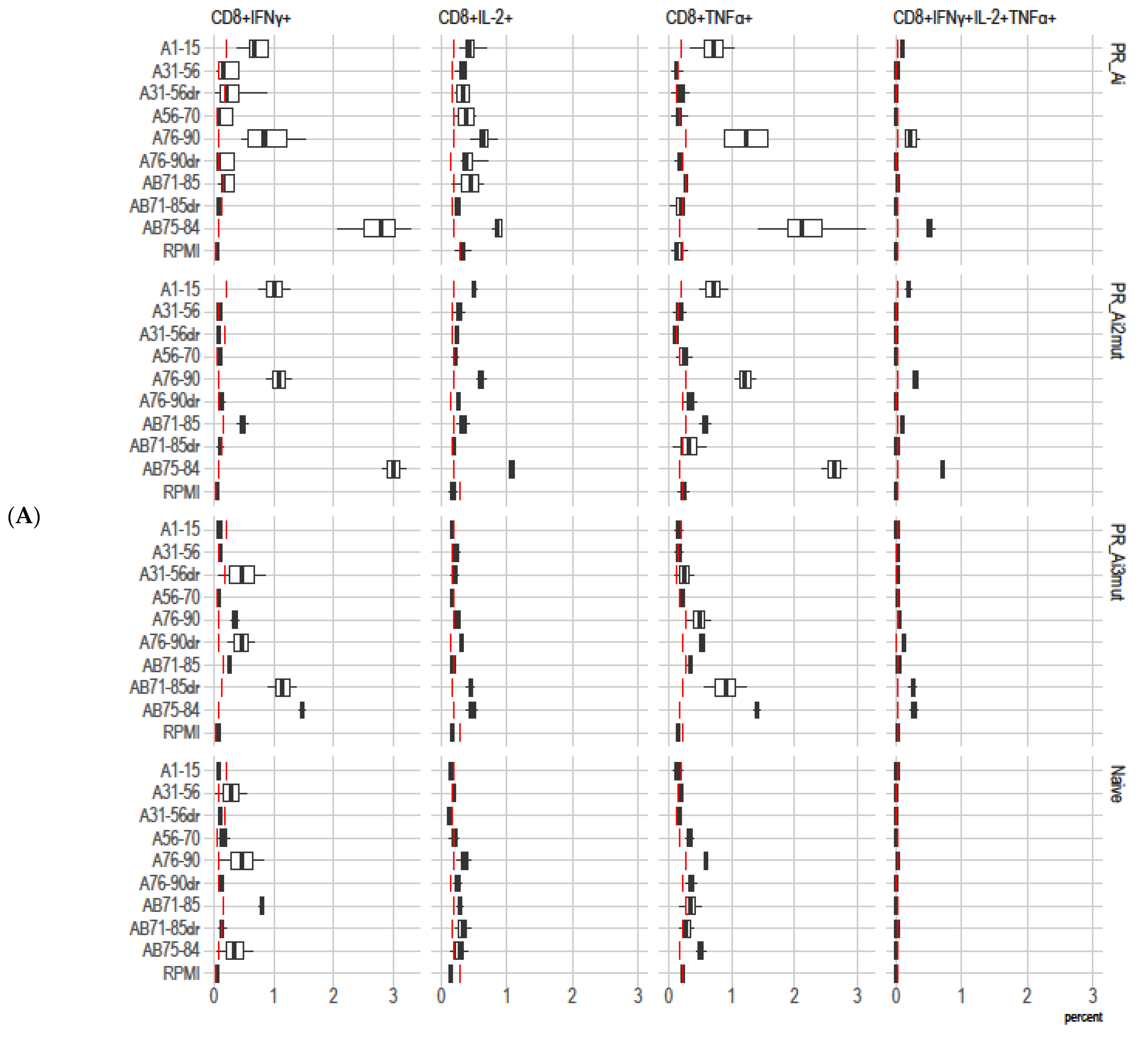
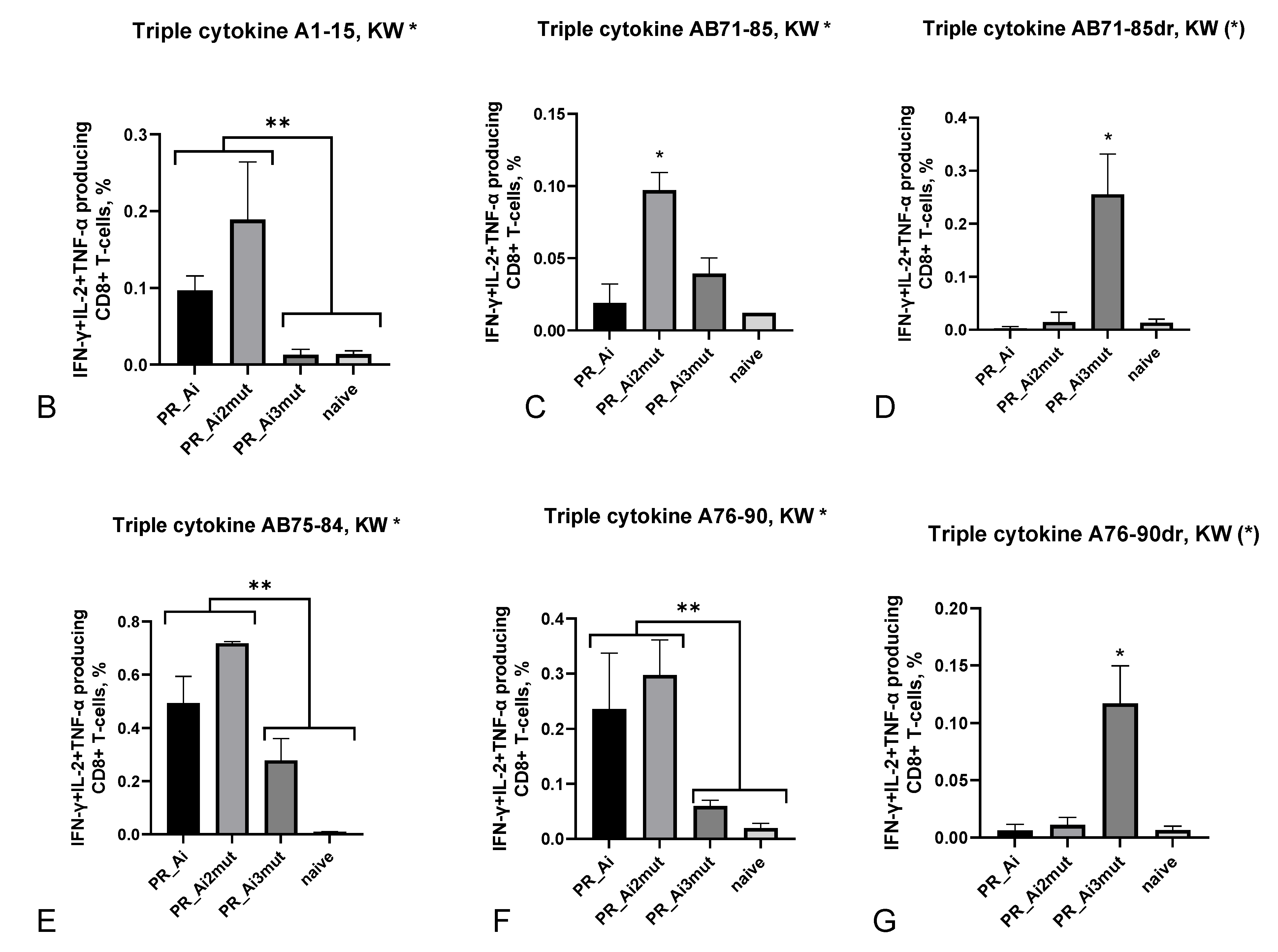
5.4.2. Homologous Prime–Boost Immunization with PR_A Gene Variants Induces a Cellular Response against Peptides Bearing Drug Resistance Mutations
5.4.3. Protease A Variants Exert No Toxic Reactions/No Immunotoxicity in DNA-Immunized Mice
5.4.4. Assessment of Protective Capacity of PR Immunization: Growth of Tumor Cells Expressing PR Can Be Prevented Only by Immunization with PR Gene Identical to That Expressed by Tumor Cells
6. Discussion
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cohen, J. Tracing the regional rise of HIV. Science 2010, 329, 161. [Google Scholar] [CrossRef] [PubMed]
- Hamers, F.F.; Downs, A.M. HIV in central and eastern Europe. Lancet 2003, 361, 1035–1044. [Google Scholar] [CrossRef]
- The Lancet. Russia’s burgeoning HIV epidemic. Lancet 2019, 393, 612. [Google Scholar] [CrossRef] [PubMed]
- Karamov, E.; Epremyan, K.; Siniavin, A.; Zhernov, Y.; Cuevas, M.T.; Delgado, E.; Sanchez-Martinez, M.; Carrera, C.; Kornilaeva, G.; Turgiev, A.; et al. HIV-1 Genetic Diversity in Recently Diagnosed Infections in Moscow: Predominance of AFSU, Frequent Branching in Clusters, and Circulation of the Iberian Subtype G Variant. AIDS Res. Hum. Retrovir. 2018, 34, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Beloukas, A.; Psarris, A.; Giannelou, P.; Kostaki, E.; Hatzakis, A.; Paraskevis, D. Molecular epidemiology of HIV-1 infection in Europe: An overview. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 46, 180–189. [Google Scholar] [CrossRef]
- Bobkov, A.; Cheingsong-Popov, R.; Selimova, L.; Ladnaya, N.; Kazennova, E.; Kravchenko, A.; Fedotov, E.; Saukhat, S.; Zverev, S.; Pokrovsky, V.; et al. An HIV type 1 epidemic among injecting drug users in the former Soviet Union caused by a homogeneous subtype A strain. AIDS Res. Hum. Retrovir. 1997, 13, 1195–1201. [Google Scholar] [CrossRef]
- Holt, E. Disputed figures do not hide Russia’s HIV epidemic. Lancet HIV 2019, 6, e216–e217. [Google Scholar] [CrossRef]
- Mansky, L.M.; Temin, H.M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [Google Scholar] [CrossRef]
- Gupta, R.K.; Gregson, J.; Parkin, N.; Haile-Selassie, H.; Tanuri, A.; Andrade Forero, L.; Kaleebu, P.; Watera, C.; Aghokeng, A.; Mutenda, N.; et al. HIV-1 drug resistance before initiation or re-initiation of first-line antiretroviral therapy in low-income and middle-income countries: A systematic review and meta-regression analysis. Lancet Infect. Dis. 2018, 18, 346–355. [Google Scholar] [CrossRef]
- Hamers, R.L.; Rinke de Wit, T.F.; Holmes, C.B. HIV drug resistance in low-income and middle-income countries. Lancet HIV 2018, 5, e588–e596. [Google Scholar] [CrossRef]
- Bobkova, M. Current status of HIV-1 diversity and drug resistance monitoring in the former USSR. AIDS Rev. 2013, 15, 204–212. [Google Scholar]
- Chavda, V.P.; Hossain, M.K.; Beladiya, J.; Apostolopoulos, V.J.B. Nucleic acid vaccines for COVID-19: A paradigm shift in the vaccine development arena. Biologics 2021, 1, 337–356. [Google Scholar] [CrossRef]
- Qin, F.; Xia, F.; Chen, H.; Cui, B.; Feng, Y.; Zhang, P.; Chen, J.; Luo, M. A Guide to Nucleic Acid Vaccines in the Prevention and Treatment of Infectious Diseases and Cancers: From Basic Principles to Current Applications. Front. Cell Dev. Biol. 2021, 9, 830. [Google Scholar] [CrossRef]
- McBrien, J.B.; Kumar, N.A.; Silvestri, G. Mechanisms of CD8(+) T cell-mediated suppression of HIV/SIV replication. Eur. J. Immunol. 2018, 48, 898–914. [Google Scholar] [CrossRef]
- Owen, R.E.; Heitman, J.W.; Hirschkorn, D.F.; Lanteri, M.C.; Biswas, H.H.; Martin, J.N.; Krone, M.R.; Deeks, S.G.; Norris, P.J. Immunology, N.C.f.H.A.V. HIV+ elite controllers have low HIV-specific T-cell activation yet maintain strong, polyfunctional T-cell responses. AIDS 2010, 24, 1095–1105. [Google Scholar] [CrossRef]
- Masopust, D. Developing an HIV cytotoxic T-lymphocyte vaccine: Issues of CD8 T-cell quantity, quality and location. J. Intern. Med. 2009, 265, 125–137. [Google Scholar] [CrossRef]
- Martins, M.A.; Tully, D.C.; Pedreno-Lopez, N.; von Bredow, B.; Pauthner, M.G.; Shin, Y.C.; Yuan, M.; Lima, N.S.; Bean, D.J.; Gonzalez-Nieto, L.; et al. Mamu-B*17(+) Rhesus Macaques Vaccinated with env, vif, and nef Manifest Early Control of SIVmac239 Replication. J. Virol. 2018, 92, e00690-18. [Google Scholar] [CrossRef]
- Hutnick, N.A.; Myles, D.J.; Hirao, L.; Scott, V.L.; Ferraro, B.; Khan, A.S.; Lewis, M.G.; Miller, C.J.; Bett, A.J.; Casimiro, D.; et al. An optimized SIV DNA vaccine can serve as a boost for Ad5 and provide partial protection from a high-dose SIVmac251 challenge. Vaccine 2012, 30, 3202–3208. [Google Scholar] [CrossRef]
- Pallikkuth, S.; Bolivar, H.; Fletcher, M.A.; Babic, D.Z.; De Armas, L.R.; Gupta, S.; Termini, J.M.; Arheart, K.L.; Stevenson, M.; Tung, F.Y.; et al. A therapeutic HIV-1 vaccine reduces markers of systemic immune activation and latent infection in patients under highly active antiretroviral therapy. Vaccine 2020, 38, 4336–4345. [Google Scholar] [CrossRef]
- Tung, F.Y.; Tung, J.K.; Pallikkuth, S.; Pahwa, S.; Fischl, M.A. A therapeutic HIV-1 vaccine enhances anti-HIV-1 immune responses in patients under highly active antiretroviral therapy. Vaccine 2016, 34, 2225–2232. [Google Scholar] [CrossRef]
- Rosenberg, E.S.; Graham, B.S.; Chan, E.S.; Bosch, R.J.; Stocker, V.; Maenza, J.; Markowitz, M.; Little, S.; Sax, P.E.; Collier, A.C.; et al. Safety and immunogenicity of therapeutic DNA vaccination in individuals treated with antiretroviral therapy during acute/early HIV-1 infection. PLoS ONE 2010, 5, e10555. [Google Scholar] [CrossRef] [PubMed]
- Palma, P.; Gudmundsdotter, L.; Finocchi, A.; Eriksson, L.E.; Mora, N.; Santilli, V.; Aquilani, A.; Manno, E.C.; Zangari, P.; Romiti, M.L.; et al. Immunotherapy with an HIV-DNA Vaccine in Children and Adults. Vaccines 2014, 2, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Gotch, F.M.; Imami, N.; Hardy, G. Candidate vaccines for immunotherapy in HIV. HIV Med. 2001, 2, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Tunggal, H.C.; Munson, P.V.; O’Connor, M.A.; Hajari, N.; Dross, S.E.; Bratt, D.; Fuller, J.T.; Bagley, K.; Fuller, D.H. Effects of therapeutic vaccination on the control of SIV in rhesus macaques with variable responsiveness to antiretroviral drugs. PLoS ONE 2021, 16, e0253265. [Google Scholar] [CrossRef] [PubMed]
- Smidt, W. Potential elucidation of a novel CTL epitope in HIV-1 protease by the protease inhibitor resistance mutation L90M. PLoS ONE 2013, 8, e71888. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, A.C.; Chapman, J.M.; Heiken, B.D.; Hoh, R.; Kallas, E.G.; Martin, J.N.; Hecht, F.M.; Deeks, S.G.; Nixon, D.F. Antiretroviral drug therapy alters the profile of human immunodeficiency virus type 1-specific T-cell responses and shifts the immunodominant cytotoxic T-lymphocyte response from Gag to Pol. J. Virol. 2007, 81, 11543–11548. [Google Scholar] [CrossRef]
- Mason, R.D.; Bowmer, M.I.; Howley, C.M.; Gallant, M.; Myers, J.C.; Grant, M.D. Antiretroviral drug resistance mutations sustain or enhance CTL recognition of common HIV-1 Pol epitopes. J. Immunol. 2004, 172, 7212–7219. [Google Scholar] [CrossRef]
- Laughlin, C.; Schleif, A.; Heilman, C.A. Addressing viral resistance through vaccines. Future Virol. 2015, 10, 1011–1022. [Google Scholar] [CrossRef]
- Boberg, A.; Isaguliants, M. Vaccination against drug resistance in HIV infection. Expert Rev. Vaccines 2008, 7, 131–145. [Google Scholar] [CrossRef]
- Kohl, N.E.; Emini, E.A.; Schleif, W.A.; Davis, L.J.; Heimbach, J.C.; Dixon, R.; Scolnick, E.M.; Sigal, I.S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA 1988, 85, 4686–4690. [Google Scholar] [CrossRef]
- Brik, A.; Wong, C.H. HIV-1 protease: Mechanism and drug discovery. Org. Biomol. Chem. 2003, 1, 5–14. [Google Scholar] [CrossRef]
- Louis, J.M.; Ishima, R.; Torchia, D.A.; Weber, I.T. HIV-1 protease: Structure, dynamics, and inhibition. Adv. Pharmacol. 2007, 55, 261–298. [Google Scholar] [CrossRef]
- Ryom, L.; Boesecke, C.; Bracchi, M.; Ambrosioni, J.; Pozniak, A.; Arribas, J.; Behrens, G.; Mallon, P.; Puoti, M.; Rauch, A.; et al. Highlights of the 2017 European AIDS Clinical Society (EACS) Guidelines for the treatment of adult HIV-positive persons version 9.0. HIV Med. 2018, 19, 309–315. [Google Scholar] [CrossRef]
- Musana, H.; Ssensamba, J.T.; Nakafeero, M.; Mugerwa, H.; Kiweewa, F.M.; Serwadda, D.; Ssali, F. Predictors of failure on second-line antiretroviral therapy with protease inhibitor mutations in Uganda. AIDS Res. Ther. 2021, 18, 1–10. [Google Scholar] [CrossRef]
- Obasa, A.E.; Ambikan, A.T.; Gupta, S.; Neogi, U.; Jacobs, G.B. Increased acquired protease inhibitor drug resistance mutations in minor HIV-1 quasispecies from infected patients suspected of failing on national second-line therapy in South Africa. BMC Infect. Dis. 2021, 21, 214. [Google Scholar] [CrossRef]
- Frontieres, M.S. Untangling the Web of Antiretroviral Price Reductions, 18th ed.; Médecins Sans Frontières: Geneva, Switzerland, 2016. [Google Scholar]
- WHO. Antiretroviral Therapy for HIV Infection in Adults and Adolescents: Recommendations for a Public Health Approach: 2010 Revision; WHO: Geneva, Switzerland, 2010.
- Eholie, S.P.; Moh, R.; Benalycherif, A.; Gabillard, D.; Ello, F.; Messou, E.; Zoungrana, J.; Diallo, I.; Diallo, M.; Bado, G.; et al. Implementation of an intensive adherence intervention in patients with second-line antiretroviral therapy failure in four west African countries with little access to genotypic resistance testing: A prospective cohort study. Lancet HIV 2019, 6, e750–e759. [Google Scholar] [CrossRef]
- Cesar, C.; Shepherd, B.E.; Jenkins, C.A.; Ghidinelli, M.; Castro, J.L.; Veloso, V.G.; Cortes, C.P.; Padgett, D.; Crabtree-Ramirez, B.; Gotuzzo, E.; et al. Use of third line antiretroviral therapy in Latin America. PLoS ONE 2014, 9, e106887. [Google Scholar] [CrossRef]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef]
- Hallengard, D.; Haller, B.K.; Petersson, S.; Boberg, A.; Maltais, A.K.; Isaguliants, M.; Wahren, B.; Brave, A. Increased expression and immunogenicity of HIV-1 protease following inactivation of the enzymatic activity. Vaccine 2011, 29, 839–848. [Google Scholar] [CrossRef]
- Giry-Laterriere, M.; Verhoeyen, E.; Salmon, P. Lentiviral vectors. Methods Mol. Biol. 2011, 737, 183–209. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Isaguliants, M.G.; Gudima, S.O.; Ivanova, O.V.; Levi, M.; Hinkula, J.; Garaev, M.M.; Kochetkov, S.N.; Wahren, B. Immunogenic properties of reverse transcriptase of HIV type 1 assessed by DNA and protein immunization of rabbits. AIDS Res. Hum. Retrovir. 2000, 16, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Latanova, A.A.; Petkov, S.; Kilpelainen, A.; Jansons, J.; Latyshev, O.E.; Kuzmenko, Y.V.; Hinkula, J.; Abakumov, M.A.; Valuev-Elliston, V.T.; Gomelsky, M.; et al. Codon optimization and improved delivery/immunization regimen enhance the immune response against wild-type and drug-resistant HIV-1 reverse transcriptase, preserving its Th2-polarity. Sci. Rep. 2018, 8, 8078. [Google Scholar] [CrossRef] [PubMed]
- Gazagne, A.; Claret, E.; Wijdenes, J.; Yssel, H.; Bousquet, F.; Levy, E.; Vielh, P.; Scotte, F.; Goupil, T.L.; Fridman, W.H.; et al. A Fluorospot assay to detect single T lymphocytes simultaneously producing multiple cytokines. J. Immunol. Methods 2003, 283, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Finak, G.; Frelinger, J.; Jiang, W.; Newell, E.W.; Ramey, J.; Davis, M.M.; Kalams, S.A.; De Rosa, S.C.; Gottardo, R. OpenCyto: An open source infrastructure for scalable, robust, reproducible, and automated, end-to-end flow cytometry data analysis. PLoS Comput. Biol. 2014, 10, e1003806. [Google Scholar] [CrossRef] [PubMed]
- Theil, K.S. 22 Bone Marrow Processing and Normal Morphology. Lab. Hematol. Pract. 2012, 281. [Google Scholar]
- Jansons, J.; Skrastina, D.; Kurlanda, A.; Petkov, S.; Avdoshina, D.; Kuzmenko, Y.; Krotova, O.; Trofimova, O.; Gordeychuk, I.; Sominskaya, I.; et al. Reciprocal Inhibition of Immunogenic Performance in Mice of Two Potent DNA Immunogens Targeting HCV-Related Liver Cancer. Microorganisms 2021, 9, 1073. [Google Scholar] [CrossRef]
- Bayurova, E.; Jansons, J.; Skrastina, D.; Smirnova, O.; Mezale, D.; Kostyusheva, A.; Kostyushev, D.; Petkov, S.; Podschwadt, P.; Valuev-Elliston, V.; et al. HIV-1 Reverse Transcriptase Promotes Tumor Growth and Metastasis Formation via ROS-Dependent Upregulation of Twist. Oxidative Med. Cell. Longev. 2019, 2019, 6016278. [Google Scholar] [CrossRef]
- Pulaski, B.A.; Ostrand-Rosenberg, S. Mouse 4T1 breast tumor model. Curr. Protoc. Immunol. 2001, 39, 20.2.1–20.2.16. [Google Scholar] [CrossRef]
- Baklaushev, V.P.; Kilpelainen, A.; Petkov, S.; Abakumov, M.A.; Grinenko, N.F.; Yusubalieva, G.M.; Latanova, A.A.; Gubskiy, I.L.; Zabozlaev, F.G.; Starodubova, E.S.; et al. Luciferase Expression Allows Bioluminescence Imaging But Imposes Limitations on the Orthotopic Mouse (4T1) Model of Breast Cancer. Sci. Rep. 2017, 7, 7715. [Google Scholar] [CrossRef]
- Elston, C.W.; Ellis, I.O. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: Experience from a large study with long-term follow-up. Histopathology 1991, 19, 403–410. [Google Scholar] [CrossRef]
- Shafer, R.W.; Schapiro, J.M. HIV-1 drug resistance mutations: An updated framework for the second decade of HAART. AIDS Rev. 2008, 10, 67. [Google Scholar]
- Rezacova, P.; Brynda, J.; Fabry, M.; Horejsi, M.; Stouracova, R.; Lescar, J.; Chitarra, V.; Riottot, M.M.; Sedlacek, J.; Bentley, G.A. Inhibition of HIV protease by monoclonal antibodies. J. Mol. Recognit. JMR 2002, 15, 272–276. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Miyazaki, M. Refolding techniques for recovering biologically active recombinant proteins from inclusion bodies. Biomolecules 2014, 4, 235–251. [Google Scholar] [CrossRef]
- Prabu-Jeyabalan, M.; Nalivaika, E.A.; King, N.M.; Schiffer, C.A. Viability of a drug-resistant human immunodeficiency virus type 1 protease variant: Structural insights for better antiviral therapy. J. Virol. 2003, 77, 1306–1315. [Google Scholar] [CrossRef]
- Pazhanisamy, S.; Stuver, C.M.; Cullinan, A.B.; Margolin, N.; Rao, B.G.; Livingston, D.J. Kinetic characterization of human immunodeficiency virus type-1 protease-resistant variants. J. Biol. Chem. 1996, 271, 17979–17985. [Google Scholar] [CrossRef]
- Schwartz, O.; Marechal, V.; Friguet, B.; Arenzana-Seisdedos, F.; Heard, J.M. Antiviral activity of the proteasome on incoming human immunodeficiency virus type 1. J. Virol. 1998, 72, 3845–3850. [Google Scholar] [CrossRef]
- Schubert, U.; Ott, D.E.; Chertova, E.N.; Welker, R.; Tessmer, U.; Princiotta, M.F.; Bennink, J.R.; Krausslich, H.G.; Yewdell, J.W. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 2000, 97, 13057–13062. [Google Scholar] [CrossRef]
- Sawant, S.; Tucker, B.; Senanayake, P.; Waters, D.D.; Patel, S.; Rye, K.-A.; Ong, K.L.; Cochran, B.J. The association between lipid levels and leukocyte count: A cross-sectional and longitudinal analysis of three large cohorts. Am. Heart J. Plus Cardiol. Res. Pract. 2021, 4, 100024. [Google Scholar] [CrossRef]
- Topçiu-Shufta, V.; Haxhibeqiri, V. Lipid Disorders in Uremia. In Cellular Metabolism and Related Disorders; IntechOpen: London, UK, 2019. [Google Scholar]
- Cox, R.A.; García-Palmieri, M.R. Cholesterol, Triglycerides, and Associated Lipoproteins, 3rd ed; Butterworths: Boston, MA, USA, 2011. [Google Scholar]
- Khan, M.A.; Anjum, F. Thymic Hyperplasia; StatPearls Publishing: Tampa, FL, USA, 2020. [Google Scholar]
- Chen, C.; Su, X.; Hu, Z. Immune promotive effect of bioactive peptides may be mediated by regulating the expression of SOCS1/miR-155. Exp. Ther. Med. 2019, 18, 1850–1862. [Google Scholar] [CrossRef]
- Aliyu, M.; Zohora, F.; Saboor-Yaraghi, A.A. Spleen in innate and adaptive immunity regulation. AIMS Allergy Immunol. 2021, 5, 1–17. [Google Scholar] [CrossRef]
- Santra, S.; Korber, B.T.; Muldoon, M.; Barouch, D.H.; Nabel, G.J.; Gao, F.; Hahn, B.H.; Haynes, B.F.; Letvin, N.L. A centralized gene-based HIV-1 vaccine elicits broad cross-clade cellular immune responses in rhesus monkeys. Proc. Natl. Acad. Sci. USA 2008, 105, 10489–10494. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Corbitt, N.; Pankhong, P.; Shin, T.; Khan, A.; Sardesai, N.Y.; Weiner, D.B. Immunogenicity of a novel engineered HIV-1 clade C synthetic consensus-based envelope DNA vaccine. Vaccine 2011, 29, 7173–7181. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Harro, C.D.; Robertson, M.N.; Lally, M.A.; O’Neill, L.D.; Edupuganti, S.; Goepfert, P.A.; Mulligan, M.J.; Priddy, F.H.; Dubey, S.A.; Kierstead, L.S.; et al. Safety and immunogenicity of adenovirus-vectored near-consensus HIV type 1 clade B gag vaccines in healthy adults. AIDS Res. Hum. Retrovir. 2009, 25, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Korber, B. HIV-1 vaccine development after STEP. Annu. Rev. Med. 2010, 61, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Diez-Fuertes, F.; Cabello, M.; Thomson, M.M. Bayesian phylogeographic analyses clarify the origin of the HIV-1 subtype A variant circulating in former Soviet Union’s countries. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2015, 33, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Meek, T.D.; Dayton, B.D.; Metcalf, B.W.; Dreyer, G.B.; Strickler, J.E.; Gorniak, J.G.; Rosenberg, M.; Moore, M.L.; Magaard, V.W.; Debouck, C. Human immunodeficiency virus 1 protease expressed in Escherichia coli behaves as a dimeric aspartic protease. Proc. Natl. Acad. Sci. USA 1989, 86, 1841–1845. [Google Scholar] [CrossRef]
- Miller, M.; Jaskolski, M.; Rao, J.K.; Leis, J.; Wlodawer, A. Crystal structure of a retroviral protease proves relationship to aspartic protease family. Nature 1989, 337, 576–579. [Google Scholar] [CrossRef]
- Rose, J.R.; Salto, R.; Craik, C.S. Regulation of autoproteolysis of the HIV-1 and HIV-2 proteases with engineered amino acid substitutions. J. Biol. Chem. 1993, 268, 11939–11945. [Google Scholar] [CrossRef]
- Blanco, R.; Carrasco, L.; Ventoso, I. Cell killing by HIV-1 protease. J. Biol. Chem. 2003, 278, 1086–1093. [Google Scholar] [CrossRef]
- Rumlova, M.; Krizova, I.; Keprova, A.; Hadravova, R.; Dolezal, M.; Strohalmova, K.; Pichova, I.; Hajek, M.; Ruml, T. HIV-1 protease-induced apoptosis. Retrovirology 2014, 11, 1–15. [Google Scholar] [CrossRef]
- Mildner, A.M.; Rothrock, D.J.; Leone, J.W.; Bannow, C.A.; Lull, J.M.; Reardon, I.M.; Sarcich, J.L.; Howe, W.J.; Tomich, C.S.; Smith, C.W.; et al. The HIV-1 protease as enzyme and substrate: Mutagenesis of autolysis sites and generation of a stable mutant with retained kinetic properties. Biochemistry 1994, 33, 9405–9413. [Google Scholar] [CrossRef]
- Nijhuis, M.; Schuurman, R.; de Jong, D.; Erickson, J.; Gustchina, E.; Albert, J.; Schipper, P.; Gulnik, S.; Boucher, C.A. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. Aids 1999, 13, 2349–2359. [Google Scholar] [CrossRef]
- Chang, M.W.; Torbett, B.E. Accessory mutations maintain stability in drug-resistant HIV-1 protease. J. Mol. Biol. 2011, 410, 756–760. [Google Scholar] [CrossRef]
- Muzammil, S.; Ross, P.; Freire, E. A major role for a set of non-active site mutations in the development of HIV-1 protease drug resistance. Biochemistry 2003, 42, 631–638. [Google Scholar] [CrossRef]
- Ahmad, S.; Kumar, V.; Ramanand, K.B.; Rao, N.M. Probing protein stability and proteolytic resistance by loop scanning: A comprehensive mutational analysis. Protein Sci. A Publ. Protein Soc. 2012, 21, 433–446. [Google Scholar] [CrossRef]
- Mueller, S.M.; Spriewald, B.M.; Bergmann, S.; Eismann, K.; Leykauf, M.; Korn, K.; Walter, H.; Schmidt, B.; Arnold, M.L.; Harrer, E.G.; et al. Influence of major HIV-1 protease inhibitor resistance mutations on CTL recognition. J. Acquir. Immune Defic. Syndr. 2011, 56, 109–117. [Google Scholar] [CrossRef]
- Karlsson, A.C.; Deeks, S.G.; Barbour, J.D.; Heiken, B.D.; Younger, S.R.; Hoh, R.; Lane, M.; Sallberg, M.; Ortiz, G.M.; Demarest, J.F.; et al. Dual pressure from antiretroviral therapy and cell-mediated immune response on the human immunodeficiency virus type 1 protease gene. J. Virol. 2003, 77, 6743–6752. [Google Scholar] [CrossRef][Green Version]
- Lemieux, J.; Jobin, C.; Simard, C.; Neron, S. A global look into human T cell subsets before and after cryopreservation using multiparametric flow cytometry and two-dimensional visualization analysis. J. Immunol. Methods 2016, 434, 73–82. [Google Scholar] [CrossRef]
- Yu, P.; Lee, Y.; Liu, W.; Krausz, T.; Chong, A.; Schreiber, H.; Fu, Y.X. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J. Exp. Med. 2005, 201, 779–791. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene. Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]
- Gatanaga, H.; Murakoshi, H.; Hachiya, A.; Hayashida, T.; Chikata, T.; Ode, H.; Tsuchiya, K.; Sugiura, W.; Takiguchi, M.; Oka, S. Naturally selected rilpivirine-resistant HIV-1 variants by host cellular immunity. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2013, 57, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Thorball, C.W.; Fellay, J.; Boni, J.; Yerly, S.; Perreau, M.; Hirsch, H.H.; Kusejko, K.; Thurnheer, M.C.; Battegay, M.; et al. Systematic screening of viral and human genetic variation identifies antiretroviral resistance and immune escape link. eLife 2021, 10, e67388. [Google Scholar] [CrossRef] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Series | Group | Nn Mice | DNA Immunogen | Immunization | Tumor Challenge | Immune Response Tests | Immunotoxicity Tests | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Prime, Dose | Boost, Dose | (Cell Line) | T-Cell Response | Antibody Response | Blood Formula | Blood Biochemistry | Other Tests | ||||
| I | I-I | 5 | PR_A | 20 ug × 2 sites | none | no | IFN-γ/IL-2 Fluorospot | yes | no | no | no |
| I-2 | 5 | PR_Ai | |||||||||
| I-3 | 5 | PR_B | |||||||||
| I-4 | 5 | PR_Bi | |||||||||
| I-5 | 5 | pVAX1 | |||||||||
| II | II-1 | 9 | PR_Ai | 20 ug × 2 sites | 20 ug × 2 sites | no | IFN-γ/IL-2 FluorospotFACS | yes | no | no | no |
| II-2 | 9 | PR_Ai2mut | |||||||||
| II-3 | 9 | PR_Ai3mut | |||||||||
| II-4 | 9 | pVAX1 | |||||||||
| III | III-1 | 6 | PR_Ai | 20 ug × 3 sites | 20 ug × 3 sites | no | no | no | Supplementary Table S6 | Supplementary Table S8 | Supplementary Tables S9 and S10 |
| III-2 | 6 | PR_Ai2mut + PR_Ai3mut (1:1) | |||||||||
| III-3 | 6 | pVAX1 | |||||||||
| III-4 | 6 | PBS | |||||||||
| IV | IV-1 | 5 | PR_Ai | 20 ug × 2 sites | 20 ug × 2 sites | 4T1luc2_PRAi3mut (PR20.2) | IFN-γ/IL-2 Fluorospot | no | no | no | no |
| IV-2 | 5 | PR_Ai2mut | |||||||||
| IV-3 | 5 | PR_Ai3mut | |||||||||
| IV-4 | 9 | PBS | |||||||||
| IV-5 | 5 | pVAX1 | 4T1luc2 | ||||||||
| IV-6 | 3 | PBS | |||||||||
| Immunogen | PR_Ai * | PR DR mix * | PR_Ai ** | PR DR Mix ** | pVAX1 ** | PBS | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day of Assessment Post-Boost | Day 1 | Day 12 | Day 1 | Day 12 | Day 1 | Day 12 | Day 1 | Day 12 | Day 1 | Day 12 | Day 1 | Day 12 |
| Thymus % (AVE ± stdv) | 0.13 ± 0.03 | 0.17 ± 0.03 | 0.14 ± 0.04 | 0.12 ± 0.05 | 0.13 ± 0.03 | 0.17 ± 0.03 | 0.14 ± 0.04 | 0.12 ± 0.05 | 0.11 ± 0.03 | 0.12 ± 0.02 | 0.16 ± 0.03 | 0.10 ± 0.04 |
| Stats, Thymus % *** | ns * | p = 0.017 | ns | ns | ns | p = 0.004 | ns | ns | p = 0.03 | ns | N/A | N/A |
| Spleen % (AVE ± stdv) | 0.49 ± 0.09 | 0.44 ± 0.03 | 0.39 ± 0.06 | 0.38 ± 0.04 | 0.49 ± 0.09 | 0.44 ± 0.03 | 0.39 ± 0.06 | 0.38 ± 0.04 | 0.39 ± 0.03 | 0.48 ± 0.13 | 0.43 ± 0.03 | 0.44 ± 0.02 |
| Stats, Spleen % *** | ns | ns | ns | ns | ns | ns | ns | p = 0.008 | ns | ns | N/A | N/A |
| Axil LN (AVE ± stdv) | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.02 ± 0.01 | 0.04 ± 0.02 | 0.02 ± 0.01 | 0.04 ± 0.02 | 0.02 ± 0.01 |
| Stats, Axil LN *** | ns | ns | p = 0.009 | ns | ns | ns | ns | ns | ns | ns | N/A | N/A |
| WBC (AVE ± stdv) | 16.22 ± 3.41 | 12.70 ± 2.44 | 9.42 ± 3.95 | 7.38 ± 2.56 | 16.22 ± 3.41 | 12.70 ± 2.44 | 9.42 ± 3.95 | 7.38 ± 2.56 | 10.80 ± 3.54 | 11.90 ± 1.62 | 8.97 ± 1.09 | 9.68 ± 3.13 |
| Stats, WBC *** | p = 0.026 | ns | ns | p = 0.009 | p = 0.002 | ns | ns | ns | ns | ns | N/A | N/A |
| LYM (AVE ± stdv) | 63.97 ± 15.96 | 70.60 ± 2.33 | 70.92 ± 7.75 | 74.50 ± 1.33 | 63.97 ± 15.96 | 70.60 ± 2.33 | 70.92 ± 7.75 | 74.50 ± 1.33 | 71.53 ± 2.91 | 71.50 ± 4.24 | 63.27 ± 5.02 | 66.82 ± 7.02 |
| Stats, LYM *** | ns | ns | ns | ns | ns | ns | p = 0.04 | p = 0.008 | p = 0.01 | ns | N/A | N/A |
| GRAN (AVE ± stdv) | 33.57 ± 15.90 | 25.52 ± 3.00 | 26.72 ± 15.89 | 22.80 ± 1.13 | 33.57 ± 15.90 | 25.52 ± 3.00 | 26.72 ± 15.89 | 22.80 ± 1.13 | 25.75 ± 2.68 | 26.04 ± 4.09 | 43.02 ± 4.92 | 30.45 ± 6.39 |
| Stats, GRAN *** | ns | ns | ns | ns | ns | ns | p = 0.04 | p = 0.004 | p = 0.01 | ns | N/A | N/A |
| RBC (AVE ± stdv) | 8.90 ± 0.23 | 10.00 ± 0.20 | 9.20 ± 1.11 | 9.74 ± 0.53 | 8.90 ± 0.23 | 10.00 ± 0.20 | 9.20 ± 1.11 | 9.74 ± 0.53 | 9.66 ± 0.20 | 10.10 ± 0.32 | 8.90 ± 0.09 | 9.86 ± 0.30 |
| Stats, RBC *** | p = 0.002 | ns | ns | ns | ns | ns | ns | ns | p = 0.004 | ns | N/A | N/A |
| PLT (AVE ± stdv) | 918.17 ± 47.72 | 807.50 ± 180.74 | 950.17 ± 63.10 | 806.17 ± 239.41 | 918.17 ± 47.72 | 807.50 ± 180.74 | 950.17 ± 63.10 | 806.17 ± 239.41 | 1066.83 ± 63.55 | 712.20 ± 177.18 | 899.17 ± 128.71 | 874.83 ± 158.26 |
| Stats, PLT *** | p = 0.004 | ns | p = 0.016 | ns | ns | ns | ns | ns | p = 0.016 | ns | N/A | N/A |
| Metamyelocytes (AVE ± stdv) | 5.00 ± 1.41 | 4.00 ± 0.63 | 5.17 ± 0.41 | 4.00 ± 0.63 | 5.00 ± 1.41 | 4.00 ± 0.63 | 5.17 ± 0.41 | 4.00 ± 0.63 | 5.50 ± 0.55 | 3.40 ± 0.55 | 5.67 ± 0.52 | 3.83 ± 1.17 |
| Stats, Metamyelocytes *** | p = 0.015 | ns | ns | ns | ns | ns | ns | ns | ns | ns | N/A | N/A |
| Immunogen | PR_Ai * | PR DR mix * | PR_Ai ** | PR DR Mix ** | pVAX1 ** | PBS | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Assessment Day | Day 1 | Day 12 | Day 1 | Day 12 | Day 1 | Day 12 | Day 1 | Day 12 | Day 1 | Day 12 | Day 1 | Day 12 |
| HCT (AVE ± stdv) | 40.50 ± 0.55 | 46.17 ± 0.94 | 42.33 ± 5.61 | 45.23 ± 1.77 | 40.50 ± 0.55 | 46.17 ± 0.94 | 42.33 ± 5.61 | 45.23 ± 1.77 | 44.50 ± 1.05 | 44.98 ± 2.57 | 40.83 ± 0.75 | 45.77 ± 1.47 |
| HCT | p = 0.002 | ns | ns | ns | ns | ns | ns | ns | p = 0.04 | ns | N/A | N/A |
| HGB (AVE ± stdv) | 128.00 ± 2.19 | 145.17 ± 2.64 | 133.17 ± 17.14 | 142.00 ± 7.54 | 128.00 ± 2.19 | 145.17 ± 2.64 | 133.17 ± 17.14 | 142.00 ± 7.54 | 144.20 ± 8.04 | 139.83 ± 2.99 | 129.67 ± 2.25 | 147.67 ± 5.50 |
| HGB | p = 0.002 | ns | ns | ns | ns | ns | ns | ns | p = 0.004 | ns | N/A | N/A |
| Total protein (AVE ± stdv) | 55.76 ± 17.62 | 67.72 ± 10.10 | 67.88 ± 3.06 | 65.44 ± 16.41 | 55.76 ± 17.62 | 67.72 ± 10.10 | 67.88 ± 3.06 | 65.44 ± 16.41 | 81.00 ± 12.72 | 56.54 ± 7.76 | 64.28 ± 12.75 | 63.00 ± 12.81 |
| Total protein | ns | ns | p = 0.016 | ns | ns | ns | ns | ns | ns | ns | N/A | N/A |
| ALT (AVE ± stdv) | 51.66 ± 22.64 | 35.16 ± 4.27 | 35.22 ± 16.53 | 29.04 ± 5.30 | 51.66 ± 22.64 | 35.16 ± 4.27 | 35.22 ± 16.53 | 29.04 ± 5.30 | 31.98 ± 7.51 | 46.20 ± 13.06 | 40.38 ± 8.78 | 41.70 ± 14.70 |
| ALT | ns | ns | ns | p = 0.03 | ns | ns | ns | ns | ns | ns | N/A | N/A |
| Urea (AVE ± stdv) | 4.51 ± 0.44 | 7.71 ± 1.57 | 4.76 ± 1.83 | 5.32 ± 0.96 | 4.51 ± 0.44 | 7.71 ± 1.57 | 4.76 ± 1.83 | 5.32 ± 0.96 | 4.56 ± 0.87 | 8.93 ± 2.57 | 5.65 ± 1.20 | 8.93 ± 2.57 |
| Urea | ns | ns | ns | p = 0.03 | ns | ns | ns | p = 0.03 | p = 0.009 | ns | N/A | N/A |
| TGC (AVE ± stdv) | 0.98 ± 0.23 | 1.39 ± 0.23 | 0.94 ± 0.11 | 1.73 ± 0.24 | 0.98 ± 0.23 | 1.39 ± 0.23 | 0.94 ± 0.11 | 1.73 ± 0.24 | 0.74 ± 0.05 | 1.45 ± 0.28 | 1.26 ± 0.21 | 1.26 ± 0.24 |
| TGC | ns | ns | p = 0.008 | ns | ns | ns | p = 0.028 | p = 0.03 | p = 0.046 | ns | N/A | N/A |
| Cholesterol (AVE ± stdv) | 1.82 ± 0.61 | 2.39 ± 0.21 | 2.36 ± 0.29 | 2.20 ± 0.43 | 1.82 ± 0.61 | 2.39 ± 0.21 | 2.36 ± 0.29 | 2.20 ± 0.43 | 2.58 ± 0.27 | 2.16 ± 0.56 | 2.09 ± 0.35 | 2.13 ± 0.31 |
| Cholesterol | p = 0.03 | ns | ns | ns | ns | ns | ns | ns | p = 0.016 | ns | N/A | N/A |
| Tumor Volume Correlated with | ||||||
|---|---|---|---|---|---|---|
| A1–15 | A76–90 | |||||
| IFN-γ | IL-2 | IFN-γ/IL-2 | IFN-γ | IL-2 | IFN-γ/IL-2 | |
| Spearman r | −0.4202 | −0.3659 | −0.6293 | −0.4147 | −0.4229 | −0.6121 |
| 95% confidence interval | −0.7106 to −0.007482 | −0.6773 to 0.05673 | −0.8277 to −0.2911 | −0.7073 to −0.0008411 | −0.7122 to −0.01072 | −0.8187 to −0.2654 |
| p (two-tailed) | 0.0409 | 0.0787 | 0.0010 | 0.0439 | 0.0395 | 0.0015 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petkov, S.; Kilpeläinen, A.; Bayurova, E.; Latanova, A.; Mezale, D.; Fridrihsone, I.; Starodubova, E.; Jansons, J.; Dudorova, A.; Gordeychuk, I.; et al. HIV-1 Protease as DNA Immunogen against Drug Resistance in HIV-1 Infection: DNA Immunization with Drug Resistant HIV-1 Protease Protects Mice from Challenge with Protease-Expressing Cells. Cancers 2023, 15, 238. https://doi.org/10.3390/cancers15010238
Petkov S, Kilpeläinen A, Bayurova E, Latanova A, Mezale D, Fridrihsone I, Starodubova E, Jansons J, Dudorova A, Gordeychuk I, et al. HIV-1 Protease as DNA Immunogen against Drug Resistance in HIV-1 Infection: DNA Immunization with Drug Resistant HIV-1 Protease Protects Mice from Challenge with Protease-Expressing Cells. Cancers. 2023; 15(1):238. https://doi.org/10.3390/cancers15010238
Chicago/Turabian StylePetkov, Stefan, Athina Kilpeläinen, Ekaterina Bayurova, Anastasia Latanova, Dzeina Mezale, Ilse Fridrihsone, Elizaveta Starodubova, Juris Jansons, Alesja Dudorova, Ilya Gordeychuk, and et al. 2023. "HIV-1 Protease as DNA Immunogen against Drug Resistance in HIV-1 Infection: DNA Immunization with Drug Resistant HIV-1 Protease Protects Mice from Challenge with Protease-Expressing Cells" Cancers 15, no. 1: 238. https://doi.org/10.3390/cancers15010238
APA StylePetkov, S., Kilpeläinen, A., Bayurova, E., Latanova, A., Mezale, D., Fridrihsone, I., Starodubova, E., Jansons, J., Dudorova, A., Gordeychuk, I., Wahren, B., & Isaguliants, M. (2023). HIV-1 Protease as DNA Immunogen against Drug Resistance in HIV-1 Infection: DNA Immunization with Drug Resistant HIV-1 Protease Protects Mice from Challenge with Protease-Expressing Cells. Cancers, 15(1), 238. https://doi.org/10.3390/cancers15010238