Simple Summary
Hepatocellular carcinoma (HCC) is the fourth most common cause of cancer-related deaths worldwide. HCC mostly results from liver cirrhosis and its genetic predisposition is believed to be rare. A liver transplantation is considered a curative therapy for HCC; however, de novo tumor development is a feared complication in immunosuppressed transplant recipients. Having analyzed the prevalence of pathogenic/likely pathogenic germline variants in cancer-predisposition genes in 334 HCC patients considered for liver transplantation, we found only 7/334 (2.1%) carriers of pathogenic variants in established cancer-predisposition genes (PMS2, 4×NBN, FH or RET). Interestingly, two MRN complex genes (NBN and RAD50) were significantly more frequent among patients over controls. Therefore, we conclude that the genetic predisposition to HCC is rare and HCC does not meet the criteria for routine germline genetic testing; however, germline testing could be considered in liver transplant recipients as the variant carriers may benefit from tailored follow-up or targeted therapy.
Abstract
Hepatocellular carcinoma (HCC) mainly stems from liver cirrhosis and its genetic predisposition is believed to be rare. However, two recent studies describe pathogenic/likely pathogenic germline variants (PV) in cancer-predisposition genes (CPG). As the risk of de novo tumors might be increased in PV carriers, especially in immunosuppressed patients after a liver transplantation, we analyzed the prevalence of germline CPG variants in HCC patients considered for liver transplantation. Using the panel NGS targeting 226 CPGs, we analyzed germline DNA from 334 Czech HCC patients and 1662 population-matched controls. We identified 48 PVs in 35 genes in 47/334 patients (14.1%). However, only 7/334 (2.1%) patients carried a PV in an established CPG (PMS2, 4×NBN, FH or RET). Only the PV carriers in two MRN complex genes (NBN and RAD50) were significantly more frequent among patients over controls. We found no differences in clinicopathological characteristics between carriers and non-carriers. Our study indicated that the genetic component of HCC is rare. The HCC diagnosis itself does not meet criteria for routine germline CPG genetic testing. However, a low proportion of PV carriers may benefit from a tailored follow-up or targeted therapy and germline testing could be considered in liver transplant recipients.
1. Introduction
Hepatocellular carcinoma (HCC) is the fourth most frequent cause of cancer-related deaths and the fifth most frequent malignancy globally []. With 854,000 new cases and 810,000 deaths annually, HCC represents 7% of all malignancies. Diagnosis of HCC is responsible for 90% of primary liver tumors []. Its incidence increases with age and peaks at the age of 70; however, the age at diagnosis is significantly lower in Chinese and black African populations. Males are affected 2–2.5× more often than females. The incidence also varies geographically, with the highest incidence reported in low- and middle-resource countries from Southeastern Asia and Sub-Saharan Africa, accounting for more than 85% of the new global cases of HCC. In Europe, the incidence is significantly lower except for Southern Europe [].
Approximately 90% of HCC cases occur in cirrhotic liver patients associated with chronic hepatitis B or C; alcoholic or metabolic liver disease, including non-alcoholic steatohepatitis (NASH); hereditary hemochromatosis or alpha-1-antitrypsin deficiency []. One-third of patients with liver cirrhosis develop HCC. The annual risk of HCC development in cirrhotic patients is estimated to be 1–8%, depending on the liver disease severity [,]. Liver transplantation represents the curative therapy with the best long-term results []. The 1-year and 5-year survival rates of liver transplant recipients are 90% and 70%, respectively, with de novo malignancies being the most frequent cause of late mortality in immunosuppressed liver transplant recipients []. To reduce mortality, guidelines for prevention and management of de novo tumors have been published recently [].
In contrast to other cancer types, the hereditary component of HCC is considered rare []. However, recently published studies revealed that 11.4–12.6% of HCC patients carried pathogenic/likely pathogenic germline variants (PV) in some cancer-predisposition genes (CPG), including established high-penetrant genes causing hereditary breast/ovarian cancer (BRCA1, BRCA2, PALB2) or Lynch syndrome (MLH1, MSH2, MSH6) [,].
We hypothesized that the risk of de novo malignancies after liver transplantation might be increased in immunosuppressed PV carriers in CPGs. To this end, we aimed to identify the prevalence of PV in a retrospective, precisely clinicopathologically characterized single-center cohort of 334 consecutive liver transplant candidates with HCC in this study.
2. Materials and Methods
2.1. Patients
The study group consisted of 334 HCC patients (258 males and 76 females) referred to the Institute for Clinical and Experimental Medicine in Prague as liver transplantation candidates between August 2002 and September 2021. In the majority of patients (329/334, 98.5%), liver cirrhosis was diagnosed in accordance with the diagnostic guidelines before HCC onset []. There was no evidence of liver cirrhosis found for only five HCC patients. The etiology of the liver cirrhosis was based on patients’ medical history and laboratory data (Table 1).

Table 1.
Clinicopathological characteristics in all 334 HCC patients.
Regarding the treatment modalities, 299 patients underwent liver transplantation, 34 patients were referred to palliative oncological therapy or best supportive care, and a single patient underwent liver resection. The median follow-up was 4.2 years (range 0.1–22.2 years). Demographic, laboratory and histopathological data were extracted from the hospital electronic information system. All but two patients were Caucasian of Czech origin, gave written informed consent to storing of their blood samples, and agreed to use of the blood samples for future research, including genetic testing.
2.2. Controls
Data from two population-matched control groups were used for germline variant evaluation. For variant prioritization, we used a group of “super-controls” consisting of 791 healthy, non-cancer, older individuals aged >60 years (92 males and 697 females), without personal and first-degree family member cancer history. For case-control analyses, we used an independent control group consisting of 1662 unselected population-matched controls (PMC) provided by the National Center for Medical Genomics (http://ncmg.cz, accessed on 1 April 2022), in details described previously [].
2.3. Library Preparation, Sequencing and Bioinformatics
Patients’ genomic DNA was isolated from peripheral blood using the Qiagen QIAamp DNA blood kit (Qiagen, Hilden, Germany). One hundred ng of gDNA was processed for the NGS library preparation using a KAPA HyperPlus Kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions. Briefly, gDNA was enzymatically fragmented for 12.5 min at 37 °C, targeting 200 bp DNA fragments. The preparation of libraries, including the use of in-house-designed adapters and dual index primers used in a six-cycle, ligation-mediated polymerase chain reaction (LM-PCR), as well as the primers subsequently used in post-capture PCR, have been described in Soukupova et al. []. The prepared pre-library was eluted to a final volume of 30 µL, checked with a High Sensitivity DNA kit using a Bioanalyzer 2100 (both from Agilent Technologies, Santa Clara, CA, USA) and quantified with dsDNA High Sensitivity Assay Kits (Qubit assays) using a Qubit Flex Fluorimeter (both from Thermo Fisher Scientific, Waltham, MA, USA). Seventy-two barcoded samples were equimolarly pooled yielding a 1.5 µg DNA pool in 45 µL volume, concentrated if necessary. Pooled samples were then hybridized at 55 °C for 16–20 h using a KAPA HyperCapture Reagent kit from Roche and a Roche-made custom-designed CZECANCA (CZEch CAncer paNel for Clinical Application) panel capturing 226 established and candidate CPG [] (Supplementary Table S2). The post-hybridization clean-up and amplification (in 11 cycles) were performed according to the manufacturer’s instructions for <40 Mbp capture target size with minor workflow modifications, including the in-house-designed post-capture PCR primers, as described previously []. The final library concentration was measured with the Qubit dsDNA HS Assay Kit and the targeted gene enrichment was checked using qPCR with in-house designed primers (available upon request). Finally, two libraries (each consisting of 72 samples) were proportionally pooled together at a final 1.5 pM concentration and prepared for NextSeq sequencing by adding 0.03 pM PhiX. Sequencing was performed on an Illumina NextSeq 500 instrument using the NextSeq 500/550 Mid Output Kit v2.5 for 150 cycles (Illumina, San Diego, CA, USA).
The sequencing data stored as FASTQ files were generated from NextSeq using an Illumina BaseSpace Sequence Hub and processed as described in Soukupova et al. [] with minor upgrades. Briefly, the FASTQ files were mapped to a reference genome hg19 using Novoalign v2.08.03 (http://www.novocraft.com/products/novoalign/, accessed on 1 April 2022), providing the corresponding SAM and afterward BAM files using Picard tools v1.129 (https://broadinstitute.github.io/picard/, accessed on 1 April 2022). The BAM files served for identification of single nucleotide variants (SNV), medium-size insertions and deletions (indels), as well as copy number variants (CNV). For SNV analyses, VCF files were generated from the BAM files (following the exclusion of PCR duplicates) using GATK toolkit v3.8.1 (https://gatk.broadinstitute.org/, accessed on 1 April 2022) [] and annotated with SnpEff v4.3 (https://pcingola.github.io/SnpEff/, accessed on 1 April 2022) []. Medium-sized indels were characterized using Pindel (http://gmt.genome.wustl.edu/packages/pindel/, accessed on 1 April 2022) [] and CNV were identified with CNVkit (https://pypi.org/project/CNVkit/, accessed on 1 April 2022), as described previously [].
2.4. Variant and Gene Prioritization
The variant prioritization aimed to identify clinically significant PVs. From the raw called variants, we sequentially filtered out variants (i) with low sequencing quality (<150); (ii) localized in repetitive and low-complexity DNA sequences (using RepeatMasker []); and (iii) non-coding (3’/5’UTR, downstream/upstream/intergenic/intragenic/deep intronic) variants and in-frame indels. Further, we filtered out variants with low clinical impact including those present (iv) in the group of super-controls with minor allele frequency (MAF) > 0.4%; (v) in the general population (gnomAD, 1000 Genomes Project, NHLBI GO ESP, ExAC databases [,,,]) with MAF > 0.4%; and (vi) interpreted as benign/likely benign (B/LB) by ClinVar []. Additionally, we excluded variants (vii) in last exons; (viii) in introns out of conserved splice site (>2 bp from an exon boundary); (ix) synonymous variants and (x) sequencing errors except for known PV. Finally, we filtered out variants without ClinVar interpretation as pathogenic/likely pathogenic, unless they caused premature termination, frameshift or aberrant splicing (1–2 bp from an exon).
Identified PV were confirmed using Sanger sequencing and/or MLPA and divided as (i) variants in established high-to-moderate CPGs (N = 48; including genes with germline variants of probable prognostic or predictive potential) or (ii) candidate CPGs (N = 178; including genes with uncertain prognostic or predictive effects of their germline variants; Figure 1 and Supplementary Table S2).
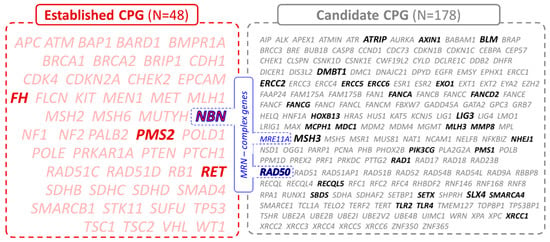
Figure 1.
List of 226 cancer-predisposition genes divided into established (N = 48, in red) and candidate (N = 178, in grey) CPGs based on their clinical significance. The genes of the MRN complex are highlighted in blue. The PVs in CPGs highlighted in bold were found in this study.
2.5. Statistical Analyses
Student’s t-test or the non-parametric Mann–Whitney and Kruskal–Wallis tests were used for continuous data, and categorical data were analyzed using the chi-square test. The survival rates were assessed with Kaplan–Meier analysis and the log-rank test was used to compare survival rates between individual groups. All statistical analyses were two-sided and a p-value of <0.05 was considered statistically significant. Statistical analysis was performed using the GraphPad Prism 9.3.1 software (GraphPad). Risk scores for PV carriers in HCC patients vs. PMC were calculated as odds ratio (OR) and 95% confidence interval (95% CI).
3. Results
3.1. Germline Variants in Established and Candidate CPG
Altogether, 334 patients’ DNA samples were sequenced with a mean coverage of 119× enabling reliable copy number variant (CNV) calling. Identified variants were prioritized as described in the Methods section, yielding 48 PV in 35 genes found in 47/334 (14.1%) patients (Table 2). However, only 7/334 (2.1%) patients harbored a PV in 4/48 established high-to-moderate CPGs, including PMS2, NBN, FH and RET (Table 2). The most frequent was a frameshift variant c.657del5 (c.657_661delACAAA) in NBN found in four patients. The remaining PVs included a novel 8907 bp deletion affecting exons 11–12 in PMS2 (Figure 2) and missense PVs in FH and RET. In 1662 PMCs, we detected four carriers of PMS2 and NBN variants, respectively, two carriers of PVs in RET and none in FH. However, a statistically significant difference in frequency was observed only for NBN (p = 0.012; Table 3).
Table 2.
Characterization of PV carriers with HCC in established high-to-moderate (A) and candidate (B) cancer predisposition genes. The PVs were present in the heterozygous state in all carriers.
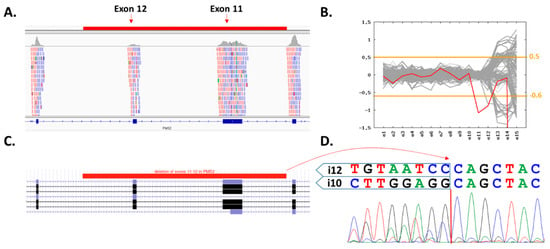
Figure 2.
Characterization of exons 11–12 (8907 bp) PMS2 deletion. (A) Integrated Genome Viewer (IGV) visualization of the coverage decrease suggesting a presence of heterozygous two-exon deletion (highlighted region in red). (B) Visualization of CNV analyses generated using the CNVkit (patient’s sample in red). The inconsistent region covering exons 12–15 contains numerous PMS2 pseudogenes. (C) Schematic region of deletion in UCSC genome browser. (D) Characterization of the deletion breakpoint with Sanger sequencing.
Table 3.
Germline PV identified in A. established high-to-moderate and B. candidate cancer predisposition genes. Statistically significant differences between variant frequencies of patients and controls are highlighted in bold.
In addition, we detected PVs in 31/178 candidate CPGs in 40/334 (12.0%) patients (including a patient harboring simultaneous germline variants in ATRIP and RAD50; Table 2). Overall 104/1662 (6.3%) individuals among PMC carried germline variants in these 31 genes (Table 3). Germline variants in HCC patients were recurrently found in only eight candidate genes including DMBT1, RAD50, ATRIP, BLM, ERCC2, LIG3, MSH3 and SLX4; however, only DMBT1, RAD50 and LIG3 germline variants showed a significant difference in HCC patients compared to PMC (Table 3).
Notably, PVs in seven HCC patients affected the genes coding for proteins of the MRN (MRE11-RAD50-NBN) complex, including four carriers of c.657del5 in NBN and three carriers of different variants in RAD50 (Table 2). PVs in NBN and RAD50 were significantly enriched in analyzed HCC patients over PMC (7/334; 2.1% vs. 7/1662; 0.4%; p = 0.001).
3.2. Clinical Characterization of PV Carriers
Patients with PVs in established CPGs, candidate CPGs, or in MRN complex genes differed from the variant non-carriers neither in demographic characteristics (age, cause of cirrhosis or occurrence of HCC in non-cirrhotic liver, diabetes or obesity), nor in tumor characteristics (angioinvasion, cholangiogenic differentiation, recurrence after liver transplantation). Moreover, the variant carriers did not present an increased frequency of multiple primary tumors (either before or after the liver transplantation) or a higher rate of primary malignancies in their first-degree relatives (Supplementary Table S3). The survival of patients was comparable between non-carriers and carriers of established CPG, candidate CPG and MRN genes (Supplementary Figure S1).
4. Discussion
In this single-center study, we performed germline genetic testing on 334 patients with HCC indicated for liver transplants. PVs in the analyzed genes were found in 47/334 (14.1%) patients; however, only 7/334 patients (2.1%) carried a PV in established high-to-moderate CPGs. Of these genes, only variants in FH can be considered as high-penetrant and were previously described in HCC patients []. Moreover, NBN was the most frequently altered gene (Table 3) with four identified carriers of a recurrent Slavic c.657del5 variant [] that moderately increases the risk of various cancer types in our population [,]. The NBN gene encodes for a protein stabilizing the MRN complex that regulates double-stranded DNA break repair []. Interestingly, we also identified three HCC patients who carried a PV in RAD50 encoding another MRN complex protein. Thus, in total, seven (2.1%) HCC patients carried a PV in MRN complex genes compared to only 7/1662 (0.4%) controls (p = 0.001). While PV carriers in NBN and RAD50 were observed also in previous HCC studies (Table 4), none was found in MRE11, the third gene of the MRN complex; however, its germline variants are rare []. Interestingly, germline variants in NBN were linked to HCC susceptibility in cirrhotic patients with chronic HBV infection previously [,]. In animal models, an increased formation of liver tumors was observed in mice hemizygous for the Nbn gene []. These findings suggest the possible involvement of the MRN complex in HCC development; however, further research, including mechanistic studies of HCC pathogenesis and large analyses in HCC patients are required.
Table 4.
Comparison of germline panel studies in HCC patients. The table describes only genes that were analyzed in at least two cohorts and where a carrier of heterozygous PV was found. Established high-to-moderate cancer predisposition genes are highlighted in bold letters.
The overall frequency of PV carriers in our HCC patients (14.1%) corresponds to the results published previously (Table 4) by Mezina et al. [], who identified 25/217 (11.5%) carriers in prospective and 30/219 (13.7%) in retrospective cohorts of HCC patients. Another small study by Uson Junior et al. identified seven (15.9%) PVs in a set of 44 HCC patients []. However, the panel of genes analyzed in these studies varied, with ours being the largest (Table 4). The proportion of deleterious variants declined when only PVs in high-to-moderate CPGs were considered (Table 4). However, unlike ours, Mezina et al.’s retrospective study identified nine patients with germline BRCA1/BRCA2 variants (entirely absent in our cohort) and four patients with germline alterations in Lynch syndrome genes.
The varying frequencies of PV carriers (2.1–11.4%) in high-to-moderate CPGs in the abovementioned studies reflect different enrollment criteria and diverse characteristics of the HCC cohorts. While HCC patients in three studies (this report, the prospective arm of Mezina et al.’s study, andUson Junior et al.’s study) were first enrolled and germline genetic testing was performed subsequently, individuals with the HCC diagnosis were selected retroactively from a large dataset of patients (analyzed in the commercial laboratory; Invitae) in the retrospective arm of the study by Mezina et al. Prospective studies were characterized by a low frequency of PVs in the genes conferring high overall cancer risk (APC, BRCA1, BRCA2, PALB2, Lynch syndrome genes) that are routinely tested for hereditary cancer syndromes (Table 4). In contrast, carriers of PVs in such genes were enriched in the retrospective (Invitae) cohort in Mezina et al.’s study []. We speculate that the HCC diagnosis among carriers from this retrospective cohort may represent a confounding event in individuals with HCC risk factors (alcohol abuse, HBV/HCV infections, etc.). Additionally, compared to our data (Supplementary Table S3), the HCC patients in Mezina et al.’s study are characterized by a high frequency of individuals with second primary tumors (17.1 vs. 38.4% in the prospective study) and a high frequency of analyzed patients with positive family cancer history (39.2 vs. over 80% in both prospective and retrospective studies). Also, the retrospective study of Mezina and colleagues included an unusual proportion of female patients compared to their prospective study and our report (56.2 vs. 16.8 and 22.6%, respectively). Moreover, it is possible that the proportion of PV carriers in highly penetrant genes in our study is artificially lower due to the potential early onset of their first cancer before HCC (median age of our cohort is 63 years). Thus, these PV carriers would develop HCC as their second tumor and, hence, they would not be eligible for liver transplantation, referred to the specialized tertiary care center and included in our study.
For additional evidence, we looked for HCC patients from the Czech CZECANCA consortium database []. Among 10,480 cancer patients, we identified 20 individuals with HCC diagnosis of which two were PV carriers in established CPGs (BRCA1 and CHEK2; Supplementary Table S4). These findings resemble results from Mezina et al.’s retrospective study [] indicating that PVs in HCC patients are likely found incidentally and can hardly be considered a genetic cause of HCC. It is of note that the risk for HCC development has not been estimated (or even documented) for any of the CPGs mentioned in this report. The results of our study support previous assumptions expecting a low hereditary component of HCC.
Mezina et al. also suggested germline variants in FANCA and BRIP1 as candidates for HCC susceptibility. While the frequency of FANCA variants was comparable among our HCC patients and PMC, BRIP1 variants were not detected in our study. Moreover, we found rare germline variants in PMS1 [], and other DNA damage response (DDR) genes ERCC2 and XRCC1 (associated with an increased risk of liver cirrhosis and its potential transformation into HCC in HBV-positive patients) [,], but we failed to identify PVs in other CPGs (including BAP1, DICER1, HNF1A, MET, TERT and VHL) associated with HCC in other studies [,,,,,].
Concerning the clinicopathological characteristics, only 5/334 individuals in our cohort developed HCC in the non-cirrhotic liver, corresponding to an expected causal effect of cirrhosis on HCC development. None of the non-cirrhotic patients carried a PV in the analyzed genes. Due to the low overall frequency of variant carriers, we did not notice any considerable differences in the carriers’ clinicopathological or tumor characteristics compared to the non-carriers.
Despite the low frequency of germline variants, germline genetic testing of HCC patients could be a prospect for precision medicine or targeted therapy. The PV carriers in Lynch syndrome genes and BRCA1/BRCA2 could benefit from treatment with immune checkpoint (PD-1/PD-L1 inhibitors) and PARP (PARPi) inhibitors, respectively [,]. Moreover, a widening PARPi indication could include the PV carriers in the MRN complex and/or other DDR genes []. Genetic testing might be of particular importance in a subgroup of HCC patients indicated for liver transplantation. The high lifetime cancer risk in PV carriers in CPGs could strongly accelerate the development of de novo tumors in immunosuppressed transplant recipients. Several such cases have been reported in individuals with various organ transplantation episodically [,,,,], but a systematic study in liver transplantation recipients is still missing. Our study indicates that a larger cohort of HCC patients indicated for liver transplantation will be required to perform such analysis due to the low frequency of PV carriers in CPGs among the patients. However, it must be stressed that genetic testing results must not influence liver transplantation eligibility.
The strength of this study includes the rigorous enrollment of well-characterized HCC patients unbiased from the recruitment of patients indicated for germline genetic testing. The study was limited by the predominance of younger liver transplant candidates with less advanced HCC, complying with the criteria for liver transplantation. The germline genetic testing was limited to the 226 cancer-predisposition genes included in the CZECANCA panel, which was designed for the analysis of cancer predisposition but does not cover some of the known cirrhosis-predisposing genes (i.e., APOB, HFE, PNPLA3, SERPINA1) [,,,,].
5. Conclusions
We conclude that the low overall prevalence of PV carriers makes germline genetic testing in HCC diagnosis rather unnecessary unless the patients fulfil other criteria for germline genetic testing (including the presence of indicative second primary tumors or positive family cancer history). However, germline genetic testing might be considered for liver transplant recipients to reduce late mortality from de novo malignancies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers15010201/s1, Figure S1: Survival of variant carriers in (A) established genes, (B) candidate genes, (C) MRN genes compared to non-carriers, Table S1: List of CZECANCA consortium members; Table S2: List of 226 genes included in the CZECANCA panel (and used transcription variants), divided into two groups based on their clinical significance; Table S3: Clinicopathological characteristics in 334 HCC patients’ subgroups of carriers of PVs in cancer-predisposition genes (any CPG) and non-carriers (None); Table S4: Characterization of HCC patients from retrospective CZECANCA consortium database.
Author Contributions
K.H.: Investigation; Formal analysis and data curation; Writing—Original Draft. S.F.: Resources; Investigation; Formal analysis and data curation; Writing—Original Draft. P.Z.: Formal analysis and data curation; Software; Writing—Review & Editing. P.N.: Formal analysis and data curation; Software. M.C.: Investigation. M.N.: Resources. B.O.: Investigation. J.K.: Investigation. M.H.: Investigation. V.S.: Resources; Investigation. T.Z.: Resources; Funding acquisition. M.S.: Resources. M.K.: Resources. C.C.: Resources; Investigation. J.N.: Investigation; Writing—Review & Editing. J.S. (Jan Sperl): Conceptualization and methodology; Resources; Funding acquisition; Writing—Review & Editing. M.B.: Investigation; Writing—Review & Editing. S.J.: Investigation. M.V.: Writing—Review & Editing. M.J. (Marketa Janatova): Investigation. P.K.: Conceptualization and methodology; Investigation; Formal analysis and data curation; Funding acquisition; Writing—Review & Editing. Z.K.: Conceptualization and methodology; Resources; Investigation; Formal analysis and data curation; Funding acquisition; Writing—Original Draft. M.J. (Milan Jirsa): Conceptualization and methodology; Resources; Funding acquisition; Writing—Review & Editing. J.S. (Jana Soukupova): Investigation; Formal analysis and data curation; Writing—Review & Editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been supported by Ministry of Health of the Czech Republic: NU20-03-00285; Ministry of Health of the Czech Republic: NU20-03-00283; Ministry of Health of the Czech Republic: RVO-VFN 00064165; Ministry of Health of the Czech Republic: RVO-IKEM 00023001; Charles University: COOPERATIO; Charles University: SVV260516; Ministry of Education Youth and Sports of the Czech Republic: LX22NPO05102.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of Thomayer’s Hospital and the Institute for Clinical and Experimental Medicine, Prague, Czech Republic (protocol code G14-08-51; date of approval 13 August 2014).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data generated and analyzed in this study are included in this publication. Identified variants were submitted to ClinVars under accession no. SCV002569169–SCV002569175, or are available from the corresponding author upon reasonable request.
Acknowledgments
We thank Lucie Budisova for her excellent technical assistance and the National Center for Medical Genomics (http://ncmg.cz, accessed on 1 April 2022) for their provision of PMC sequencing data.
Conflicts of Interest
Z.K. declares research funding from Roche, unrelated to the project. No other potential conflicts of interest were reported. We declare that the results summarized in this manuscript have not been published previously and have not been submitted for consideration to any other journal.
Abbreviations
| CI | confidence interval |
| CNV | copy number variants |
| CPG | cancer-predisposition genes |
| DDR | DNA damage repair |
| HBV | hepatitis B virus |
| HCC | hepatocellular carcinoma |
| HCV | hepatitis C virus |
| MAF | minor allele frequency |
| MRN | MRE11-RAD50-NBN |
| NASH | non-alcoholic steatohepatitis |
| OR | odds ratio |
| PARPi | PARP1 inhibitors |
| PV | pathogenic/likely pathogenic germline variant |
| PMC | population-matched controls |
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; Artaman, A.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies from 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar]
- European Association For The Study of The Liver; European Organisation for Research and Treatment of Cancer. EASL-EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [Google Scholar] [CrossRef]
- Sangiovanni, A.; Prati, G.M.; Fasani, P.; Ronchi, G.; Romeo, R.; Manini, M.; Del Ninno, E.; Morabito, A.; Colombo, M. The natural history of compensated cirrhosis due to hepatitis C virus: A 17-year cohort study of 214 patients. Hepatology 2006, 43, 1303–1310. [Google Scholar] [CrossRef]
- van der Meer, A.J.; Veldt, B.J.; Feld, J.J.; Wedemeyer, H.; Dufour, J.F.; Lammert, F.; Duarte-Rojo, A.; Heathcote, E.J.; Manns, M.P.; Kuske, L.; et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA 2012, 308, 2584–2593. [Google Scholar] [CrossRef]
- Colmenero, J.; Tabrizian, P.; Bhangui, P.; Pinato, D.J.; Rodriguez-Peralvarez, M.L.; Sapisochin, G.; Bhoori, S.; Pascual, S.; Senzolo, M.; Al-Adra, D.; et al. De Novo Malignancy after Liver Transplantation: Risk Assessment, Prevention, and Management-Guidelines From the ILTS-SETH Consensus Conference. Transplantation 2022, 106, e30–e45. [Google Scholar] [CrossRef]
- Daniel, K.E.; Eickhoff, J.; Lucey, M.R. Why do patients die after a liver transplantation? Clin. Transpl. 2017, 31, e12906. [Google Scholar] [CrossRef]
- Ozturk, M. Genetic aspects of hepatocellular carcinogenesis. Semin. Liver Dis. 1999, 19, 235–242. [Google Scholar] [CrossRef]
- Uson Junior, P.L.; Kunze, K.L.; Golafshar, M.A.; Riegert-Johnson, D.; Boardman, L.; Borad, M.J.; Ahn, D.; Sonbol, M.B.; Faigel, D.O.; Fukami, N.; et al. Germline Cancer Susceptibility Gene Testing in Unselected Patients with Hepatobiliary Cancers: A Multi-Center Prospective Study. Cancer Prev. Res. 2022, 15, 121–128. [Google Scholar] [CrossRef]
- Mezina, A.; Philips, N.; Bogus, Z.; Erez, N.; Xiao, R.; Fan, R.; Olthoff, K.M.; Reddy, K.R.; Samadder, N.J.; Nielsen, S.M.; et al. Multigene Panel Testing in Individuals With Hepatocellular Carcinoma Identifies Pathogenic Germline Variants. JCO Precis. Oncol. 2021, 5, 988–1000. [Google Scholar] [CrossRef]
- Lhotova, K.; Stolarova, L.; Zemankova, P.; Vocka, M.; Janatova, M.; Borecka, M.; Cerna, M.; Jelinkova, S.; Kral, J.; Volkova, Z.; et al. Multigene Panel Germline Testing of 1333 Czech Patients with Ovarian Cancer. Cancers 2020, 12, 956. [Google Scholar] [CrossRef] [PubMed]
- Soukupova, J.; Zemankova, P.; Lhotova, K.; Janatova, M.; Borecka, M.; Stolarova, L.; Lhota, F.; Foretova, L.; Machackova, E.; Stranecky, V.; et al. Validation of CZECANCA (CZEch CAncer paNel for Clinical Application) for targeted NGS-based analysis of hereditary cancer syndromes. PLoS ONE 2018, 13, e0195761. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang Le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org (accessed on 29 November 2022).
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Exome Variant Server. Available online: https://evs.gs.washington.edu/EVS/ (accessed on 1 April 2022).
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Varon, R.; Seemanova, E.; Chrzanowska, K.; Hnateyko, O.; Piekutowska-Abramczuk, D.; Krajewska-Walasek, M.; Sykut-Cegielska, J.; Sperling, K.; Reis, A. Clinical ascertainment of Nijmegen breakage syndrome (NBS) and prevalence of the major mutation, 657del5, in three Slav populations. Eur. J. Hum. Genet. 2000, 8, 900–902. [Google Scholar] [CrossRef]
- Wieme, G.; Kral, J.; Rosseel, T.; Zemankova, P.; Parton, B.; Vocka, M.; Van Heetvelde, M.; Kleiblova, P.; Blaumeiser, B.; Soukupova, J.; et al. Prevalence of Germline Pathogenic Variants in Cancer Predisposing Genes in Czech and Belgian Pancreatic Cancer Patients. Cancers 2021, 13, 4430. [Google Scholar] [CrossRef] [PubMed]
- Elkholi, I.E.; Foulkes, W.D.; Rivera, B. MRN Complex and Cancer Risk: Old Bottles, New Wine. Clin. Cancer Res. 2021, 27, 5465–5471. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Rybicka, M.; Woziwodzka, A.; Sznarkowska, A.; Romanowski, T.; Stalke, P.; Dreczewski, M.; Verrier, E.R.; Baumert, T.F.; Bielawski, K.P. Liver Cirrhosis in Chronic Hepatitis B Patients Is Associated with Genetic Variations in DNA Repair Pathway Genes. Cancers 2020, 12, 3295. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Xiao, R.; Chen, X.; Yuan, C.; Sun, Y.; Li, J. A non-synonymous polymorphism in NBS1 is associated with progression from chronic hepatitis B virus infection to hepatocellular carcinoma in a Chinese population. Onco Targets Ther. 2018, 11, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Dumon Jones, V.; Frappart, P.-O.; Tong, W.-M.; Sajithlal, G.; Hulla, W.; Schmid, G.; Herceg, Z.; Digweed, M.; Wang, Z.-Q. Nbn Heterozygosity Renders Mice Susceptible to Tumor Formation and Ionizing Radiation-Induced Tumorigenesis. Cancer Res. 2003, 63, 7263–7269. [Google Scholar]
- Soukupova, J.; Zemankova, P.; Kleiblova, P.; Janatova, M.; Kleibl, Z. CZECANCA: CZEch CAncer paNel for Clinical Application—Design and Optimization of the Targeted Sequencing Panel for the Identification of Cancer Susceptibility in High-risk Individuals from the Czech Republic. Klin. Onkol. 2016, 29 (Suppl. 1), S46–S54. [Google Scholar] [CrossRef]
- Li, X.; Wu, Y.; Suo, P.; Liu, G.; Li, L.; Zhang, X.; Chen, S.; Xu, M.; Song, L. Identification of a novel germline frameshift mutation p.D300fs of PMS1 in a patient with hepatocellular carcinoma: A case report and literature review. Medicine 2020, 99, e19076. [Google Scholar] [CrossRef]
- Chau, C.; van Doorn, R.; van Poppelen, N.M.; van der Stoep, N.; Mensenkamp, A.R.; Sijmons, R.H.; van Paassen, B.W.; van den Ouweland, A.M.W.; Naus, N.C.; van der Hout, A.H.; et al. Families with BAP1-Tumor Predisposition Syndrome in The Netherlands: Path to Identification and a Proposal for Genetic Screening Guidelines. Cancers 2019, 11, 1114. [Google Scholar] [CrossRef]
- Caruso, S.; Calderaro, J.; Letouze, E.; Nault, J.C.; Couchy, G.; Boulai, A.; Luciani, A.; Zafrani, E.S.; Bioulac-Sage, P.; Seror, O.; et al. Germline and somatic DICER1 mutations in familial and sporadic liver tumors. J. Hepatol. 2017, 66, 734–742. [Google Scholar] [CrossRef]
- Fu, J.; Wang, T.; Zhai, X.; Xiao, X. Primary hepatocellular adenoma due to biallelic HNF1A mutations and its co-occurrence with MODY 3: Case-report and review of the literature. Endocrine 2020, 67, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Tovar, E.A.; Graveel, C.R. MET in human cancer: Germline and somatic mutations. Ann. Transl. Med. 2017, 5, 205. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Pietrelli, A.; Pingitore, P.; Dongiovanni, P.; Caddeo, A.; Walker, L.; Baselli, G.; Pelusi, S.; Rosso, C.; Vanni, E.; et al. Telomerase reverse transcriptase germline mutations and hepatocellular carcinoma in patients with nonalcoholic fatty liver disease. Cancer Med. 2017, 6, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, J.J.; Frier, Q.J.; Sumarriva, D.; Oberley, M.; Bolton, D.; Deveras, R.A. Germline VHL Mutation Discovered in Association with EGFR-Positive Lung Cancer and Metachronous Hepatocellular Carcinoma: A Case Report. Case Rep. Oncol. 2021, 14, 1392–1398. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.M.; Mahan, K.; Mettler, T.; Dunitz, J.M.; Khoruts, A. Case report of synchronous post-lung transplant colon cancers in the era of colorectal cancer screening recommendations in cystic fibrosis: Screening “too early” before it’s too late. BMC Gastroenterol. 2019, 19, 137. [Google Scholar] [CrossRef]
- Gozdowska, J.; Bieniasz, M.; Wszoła, M.; Kieszek, R.; Domagała, P.; Drozdowski, J.; Tomaszek, A.; Kwiatkowski, A.; Chmura, A.; Durlik, M. Determining eligibility for and preparation to kidney transplantation of a patient with Lynch syndrome—A case report and literature review. Ann. Transplant. 2014, 19, 124–128. [Google Scholar]
- Qudaih, A.T.; Al Ashour, B.H.; Naim, A.K.; Joudeh, A.A. Kidney Transplant Recipient With Multiple Contemporaneous Malignancies Secondary to Muir-Torre Syndrome. Cureus 2021, 13, e16642. [Google Scholar] [CrossRef]
- Wassano, N.S.; Sergi, F.; Ferro, G.; Genzini, T.; D’Alpino Peixoto, R. Rapid Disease Progression of Liver Metastases following Resection in a Liver-Transplanted Patient with Probable Lynch Syndrome—A Case Report and Review of the Literature. Case Rep. Oncol. 2017, 10, 244–251. [Google Scholar] [CrossRef]
- Yang, R.L.; Kurian, A.W.; Winton, L.M.; Weill, D.; Patel, K.; Kingham, K.; Wapnir, I.L. Addressing inherited predisposition for breast cancer in transplant recipients. J. Surg. Oncol. 2016, 113, 605–608. [Google Scholar] [CrossRef]
- Trepo, E.; Nahon, P.; Bontempi, G.; Valenti, L.; Falleti, E.; Nischalke, H.D.; Hamza, S.; Corradini, S.G.; Burza, M.A.; Guyot, E.; et al. Association between the PNPLA3 (rs738409 C>G) variant and hepatocellular carcinoma: Evidence from a meta-analysis of individual participant data. Hepatology 2014, 59, 2170–2177. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; Manjunath, H.; Yopp, A.C.; Beg, M.S.; Marrero, J.A.; Gopal, P.; Waljee, A.K. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Pelusi, S.; Baselli, G.; Pietrelli, A.; Dongiovanni, P.; Donati, B.; McCain, M.V.; Meroni, M.; Fracanzani, A.L.; Romagnoli, R.; Petta, S.; et al. Rare Pathogenic Variants Predispose to Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 3682. [Google Scholar] [CrossRef] [PubMed]
- Pirisi, M.; Toniutto, P.; Uzzau, A.; Fabris, C.; Avellini, C.; Scott, C.; Apollonio, L.; Beltrami, C.A.; Beltrami, C.; Bresadola, F. Carriage of HFE mutations and outcome of surgical resection for hepatocellular carcinoma in cirrhotic patients. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2000, 89, 297–302. [Google Scholar]
- Strnad, P.; Buch, S.; Hamesch, K.; Fischer, J.; Rosendahl, J.; Schmelz, R.; Brueckner, S.; Brosch, M.; Heimes, C.V.; Woditsch, V.; et al. Heterozygous carriage of the alpha1-antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut 2019, 68, 1099. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).