
Extracellular Vesicle Membrane Protein Profiling and Targeted Mass Spectrometry Unveil CD59 and Tetraspanin 9 as Novel Plasma Biomarkers for Detection of Colorectal Cancer

 , , , , , ,
, , , , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Specimens
2.2. Cell Cultures
2.3. EV Isolation from CRC Cell Lines
2.4. EV Isolation from Human Plasma Samples
2.5. Transmission Electron Microscopy
2.6. Nanoparticles Tracking Analysis (NTA)
2.7. Flow Cytometry Analysis of EV-Bound Beads
2.8. Western Blot Analysis
2.9. In-Solution Digestion of EVs for 2D-LC-MS/MS
2.10. Two-Dimensional LC-MS/MS Analysis (2D-LC-MS/MS)
2.11. Data Processing
2.12. Gene Ontology Annotation and Topological Analysis of Target Proteins
2.13. Selection of Signature Peptides and Accurate Inclusion Mass Screening (AIMS)
2.14. EV Digestion and iTRAQ Labelling of EV Derived Peptides
2.15. 2D LC-MS/MS Analysis Coupled with iTRAQ
2.16. Sequence Database Search and Quantitative Data Analysis for iTRAQ
2.17. Synthesis of Surrogate Peptides
2.18. Preparation of EV Samples for MS-Based Targeted Protein Quantification
2.19. LC-PIS-MS, MS Data Processing and Generation of Response Curves
2.20. Statistical Analysis
3. Results
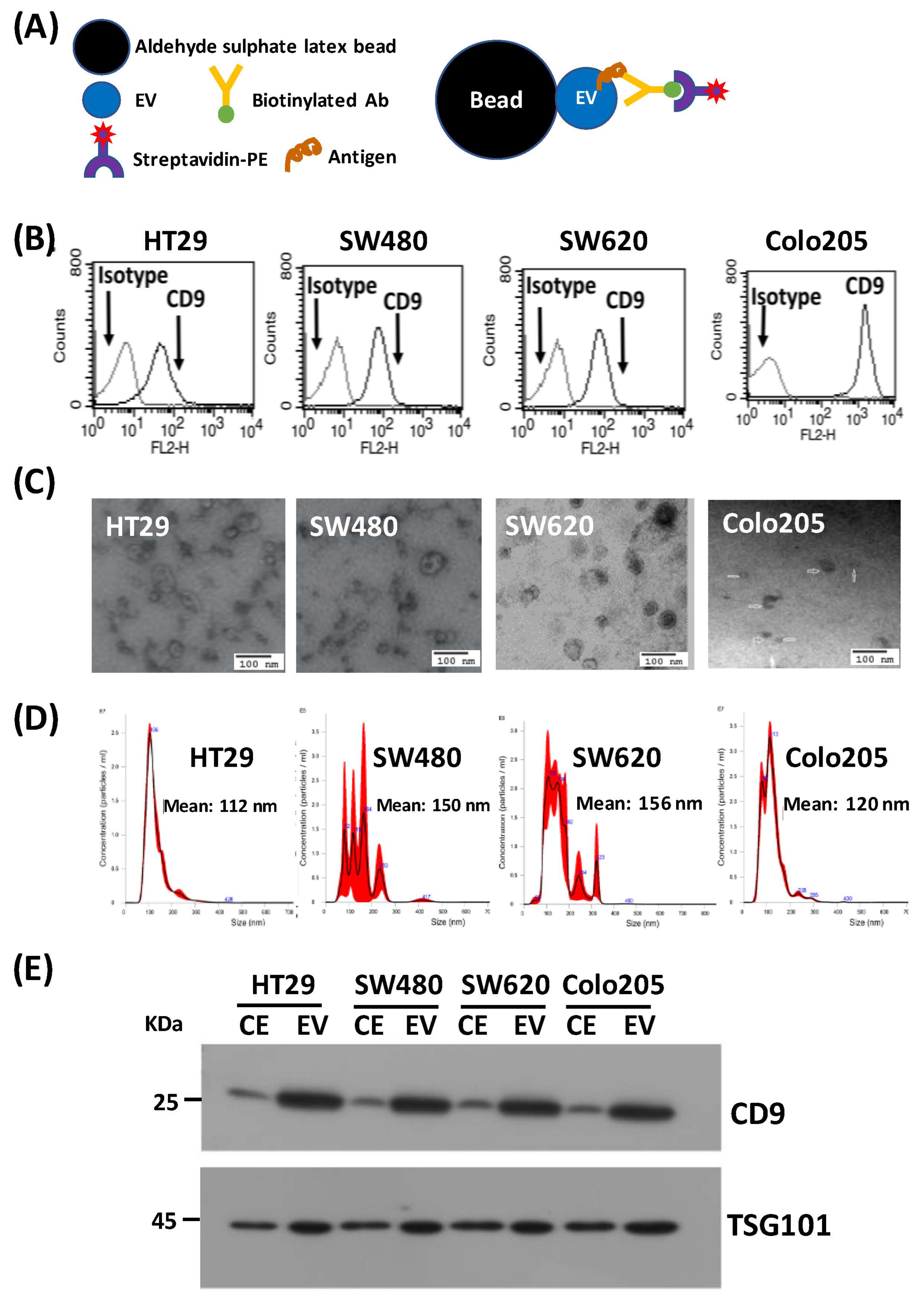
3.1. Study Design and Characterization of EVs
3.2. Identification of EV Proteins by 2D-LC-MS/MS and Annotation of Identified Proteins
3.3. Prioritization of Target Proteins and Confirmation of Their Presence in Plasma-Derived EVs from HCs and CRC Patients
3.4. Quantitative Proteome Profiling of EV Samples from CRC Patients and HCs
3.5. Selection of Candidates for MS-Based Targeted Protein Quantification
3.6. Quantification of Selected Targets in Individual Plasma-Derived EVs by Targeted MS
3.7. Generation of Candidate Plasma EV Protein Biomarker Panels
3.8. Association of Plasma-Derived EV Levels of ADAM10, CD59 and TSPAN9 and Plasma CEA Levels with Clinicopathological Characteristics of CRC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Doubeni, C.A.; Gabler, N.B.; Wheeler, C.M.; McCarthy, A.M.; Castle, P.E.; Halm, E.A.; Schnall, M.D.; Skinner, C.S.; Tosteson, A.N.A.; Weaver, D.L.; et al. Timely follow-up of positive cancer screening results: A systematic review and recommendations from the PROSPR Consortium. CA Cancer J. Clin. 2018, 68, 199–216. [Google Scholar] [CrossRef] [PubMed]
- van der Stok, E.P.; Spaander, M.C.W.; Grünhagen, D.J.; Verhoef, C.; Kuipers, E.J. Surveillance after curative treatment for colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Rusiecki, J.; Cifu, A.S. Colonoscopy surveillance after colorectal cancer resection. JAMA 2017, 318, 2346–2347. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Matsuda, T.; Saito, Y. Surveillance after endoscopic and surgical resection of colorectal cancer. Best Pract. Res. Clin. Gastroenterol. 2016, 30, 959–970. [Google Scholar] [CrossRef]
- Kahi, C.J.; Boland, C.R.; Dominitz, J.A.; Giardiello, F.M.; Johnson, D.A.; Kaltenbach, T.; Lieberman, D.; Levin, T.R.; Robertson, D.J.; Rex, D.K. United States Multi-Society Task Force on Colorectal Cancer. Colonoscopy Surveillance after Colorectal Cancer Resection: Recommendations of the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2016, 150, 758–768.e11. [Google Scholar] [CrossRef]
- Vacante, M.; Ciuni, R.; Basile, F.; Biondi, A. The liquid biopsy in the management of colorectal cancer: An overview. Biomedicines 2020, 8, 308. [Google Scholar] [CrossRef]
- Ding, Y.; Li, W.; Wang, K.; Xu, C.; Hao, M.; Ding, L. Perspectives of the Application of Liquid Biopsy in Colorectal Cancer. Biomed. Res. Int. 2020, 2020, 6843180. [Google Scholar] [CrossRef]
- Poulet, G.; Massias, J.; Taly, V. Liquid Biopsy: General Concepts. Acta Cytol. 2019, 63, 449–455. [Google Scholar] [CrossRef]
- Yamada, T.; Matsuda, A.; Koizumi, M.; Shinji, S.; Takahashi, G.; Iwai, T.; Takeda, K.; Ueda, K.; Yokoyama, Y.; Hara, K.; et al. Liquid biopsy for the management of patients with colorectal cancer. Digestion 2018, 99, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Vatandoost, N.; Ghanbari, J.; Mojaver, M.; Avan, A.; Ghayour-Mobarhan, M.; Nedaeinia, R.; Salehi, R. Early detection of colorectal cancer: From conventional methods to novel biomarkers. J. Cancer Res. Clin. Oncol. 2016, 142, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Tanaka, M.; Tanaka, T.; Ishigamori, R. Biomarkers for colorectal cancer. Int. J. Mol. Sci. 2010, 11, 3209–3225. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. Cancer biomarkers: Current issues and future directions. Curr. Opin. Mol. Ther. 2007, 9, 563–571. [Google Scholar]
- Jain, K.K. Recent advances in clinical oncoproteomics. J. BUON 2007, 12 (Suppl. S1), S31–S38. [Google Scholar]
- Lescuyer, P.; Hochstrasser, D.; Rabilloud, T. How shall we use the proteomics toolbox for biomarker discovery? J. Proteome Res. 2007, 6, 3371–3376. [Google Scholar] [CrossRef]
- Brooks, J.D. Translational genomics: The challenge of developing cancer biomarkers. Genome Res. 2012, 22, 183–187. [Google Scholar] [CrossRef]
- EL Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Belov, L.; Matic, K.J.; Hallal, S.; Best, O.G.; Mulligan, S.P.; Christopherson, R.I. Extensive surface protein profiles of extracellular vesicles from cancer cells may provide diagnostic signatures from blood samples. J. Extracell. Vesicles 2016, 5, 25355. [Google Scholar] [CrossRef]
- Dong, L.; Lin, W.; Qi, P.; Xu, M.D.; Wu, X.; Ni, S.; Huang, D.; Weng, W.W.; Tan, C.; Sheng, W.; et al. Circulating long RNAs in serum extracellular vesicles: Their characterization and potential application as biomarkers for diagnosis of colorectal cancer. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1158–1166. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Sanderson, M.P.; Stoeck, A.; Altevogt, P. Exosomes: From biogenesis and secretion to biological function. Immunol. Lett. 2006, 107, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- De Toro, J.; Herschlik, L.; Waldner, C.; Mongini, C. Emerging roles of exosomes in normal and pathological conditions: New insights for diagnosis and therapeutic applications. Front. Immunol. 2015, 6, 203. [Google Scholar] [CrossRef]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteomics 2010, 73, 1907–1920. [Google Scholar] [CrossRef]
- Benito-Martin, A.; Di Giannatale, A.; Ceder, S.; Peinado, H. The new deal: A potential role for secreted vesicles in innate immunity and tumor progression. Front. Immunol. 2015, 6, 66. [Google Scholar] [CrossRef]
- Kesimer, M.; Scull, M.; Brighton, B.; DeMaria, G.; Burns, K.; O’Neal, W.; Pickles, R.J.; Sheehan, J.K. Characterization of exosome-like vesicles released from human tracheobronchial ciliated epithelium: A possible role in innate defense. FASEB J. 2009, 23, 1858–1868. [Google Scholar] [CrossRef]
- Logozzi, M.; De Milito, A.; Lugini, L.; Borghi, M.; Calabrò, L.; Spada, M.; Perdicchio, M.; Marino, M.L.; Federici, C.; Iessi, E.; et al. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS ONE 2009, 4, e5219. [Google Scholar] [CrossRef]
- Kleeff, J.; Ishiwata, T.; Kumbasar, A.; Friess, H.; Büchler, M.W.; Lander, A.D.; Korc, M. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J. Clin. Investig. 1998, 102, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Vardaki, I.; Ceder, S.; Rutishauser, D.; Baltatzis, G.; Foukakis, T.; Panaretakis, T. Periostin is identified as a putative metastatic marker in breast cancer-derived exosomes. Oncotarget 2016, 7, 74966–74978. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Jiang, C.; Fu, Y.; Liu, G.; Shu, B.; Davis, J.; Tofaris, G.K. Multiplexed Profiling of Extracellular Vesicles for Biomarker Development. Nanomicro Lett. 2021, 14, 3. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, M.; Wang, L.; Yan, A.; He, W.; Chen, M.; Lan, J.; Xu, J.; Guan, L.; Chen, J. A visible and colorimetric aptasensor based on DNA-capped single-walled carbon nanotubes for detection of exosomes. Biosens. Bioelectron. 2017, 92, 8–15. [Google Scholar] [CrossRef]
- Chiang, S.F.; Tsai, M.H.; Tang, R.; Hsieh, L.L.; Chiang, J.M.; Yeh, C.Y.; Hsieh, P.S.; Tsai, W.S.; Liu, Y.P.; Liang, Y.; et al. Membrane Proteins as Potential Colon Cancer Biomarkers: Verification of 4 Candidates from a Secretome Dataset. Surg. Sci. 2014, 5, 418–438. [Google Scholar] [CrossRef][Green Version]
- Yoshioka, Y.; Kosaka, N.; Konishi, Y.; Ohta, H.; Okamoto, H.; Sonoda, H.; Nonaka, R.; Yamamoto, H.; Ishii, H.; Mori, M.; et al. Ultra-sensitive liquid biopsy of circulating extracellular vesicles using ExoScreen. Nat. Commun. 2014, 5, 3591. [Google Scholar] [CrossRef]
- Wei, P.; Wu, F.; Kang, B.; Sun, X.; Heskia, F.; Pachot, A.; Liang, J.; Li, D. Plasma extracellular vesicles detected by Single Molecule array technology as a liquid biopsy for colorectal cancer. J. Extracell. Vesicles 2020, 9, 1809765. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yang, Y.; Zeng, Y.; He, M. A microfluidic ExoSearch chip for multiplexed exosome detection towards blood-based ovarian cancer diagnosis. Lab Chip 2016, 16, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.G.; Mohamadi, R.M.; Poudineh, M.; Kermanshah, L.; Ahmed, S.; Safaei, T.S.; Stojcic, J.; Nam, R.K.; Sargent, E.H.; Kelley, S.O. Interrogating Circulating Microsomes and Exosomes Using Metal Nanoparticles. Small 2016, 12, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, Y.; Chen, W.; Yin, L.; Zhu, J.; Zhang, H.; Cai, C.; Li, P.; Huang, L.; Ma, P. Exosomal ephrinA2 derived from serum as a potential biomarker for prostate cancer. J. Cancer 2018, 9, 2659–2665. [Google Scholar] [CrossRef] [PubMed]
- Di, H.; Mi, Z.; Sun, Y.; Liu, X.; Liu, X.; Li, A.; Jiang, Y.; Gao, H.; Rong, P.; Liu, D. Nanozyme-assisted sensitive profiling of exosomal proteins for rapid cancer diagnosis. Theranostics 2020, 10, 9303–9314. [Google Scholar] [CrossRef]
- Li, P.; Wang, J.; Gao, M.; Wang, J.; Ma, Y.; Gu, Y. Membrane Feature-Inspired Profiling of Extracellular Vesicles for Pancreatic Cancer Diagnosis. Anal. Chem. 2021, 93, 9860–9868. [Google Scholar] [CrossRef]
- Tian, F.; Zhang, S.; Liu, C.; Han, Z.; Liu, Y.; Deng, J.; Li, Y.; Wu, X.; Cai, L.; Qin, L.; et al. Protein analysis of extracellular vesicles to monitor and predict therapeutic response in metastatic breast cancer. Nat. Commun. 2021, 12, 2536. [Google Scholar] [CrossRef]
- Mathivanan, S.; Lim, J.W.; Tauro, B.J.; Ji, H.; Moritz, R.L.; Simpson, R.J. Proteomics analysis of A33 immunoaffinity-purified exosomes released from the human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Mol. Cell. Proteomics 2010, 9, 197–208. [Google Scholar] [CrossRef]
- Demory Beckler, M.; Higginbotham, J.N.; Franklin, J.L.; Ham, A.J.; Halvey, P.J.; Imasuen, I.E.; Whitwell, C.; Li, M.; Liebler, D.C.; Coffey, R.J. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol. Cell. Proteomics 2013, 2, 343–355. [Google Scholar] [CrossRef]
- Choi, D.S.; Park, J.O.; Jang, S.C.; Yoon, Y.J.; Jung, J.W.; Choi, D.Y.; Kim, J.W.; Kang, J.S.; Park, J.; Hwang, D.; et al. Proteomic analysis of microvesicles derived from human colorectal cancer ascites. Proteomics 2011, 11, 2745–2751. [Google Scholar] [CrossRef]
- Jaffe, J.D.; Keshishian, H.; Chang, B.; Addona, T.A.; Gillette, M.A.; Carr, S.A. Accurate inclusion mass screening: A bridge from unbiased discovery to targeted assay development for biomarker verification. Mol. Cell. Proteomics 2008, 7, 1952–1962. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Li, R.; Di, X.; Jin, X.; Wang, Y.; Lai, B.; Shi, C.; Ji, M.; Zhu, X.; Wang, K. ITRAQ-based proteomic analysis reveals possible target-related proteins in human adrenocortical adenomas. BMC Genomics 2019, 20, 655. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, A.; Branca, R.M.; Lehtiö, J.; Forshed, J. Quantitative accuracy in mass spectrometry based proteomics of complex samples: The impact of labeling and precursor interference. J. Proteomics 2014, 96, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Wiese, S.; Reidegeld, K.A.; Meyer, H.E.; Warscheid, B. Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research. Proteomics 2007, 7, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Vidova, V.; Spacil, Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Anal. Chim. Acta 2017, 964, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Chen, C.L.; Chen, H.W.; Chung, T.; Wu, C.C.; Chen, C.D.; Hsu, C.W.; Chen, M.C.; Tsui, K.H.; Chang, P.L.; et al. Discovery of novel bladder cancer biomarkers by comparative urine proteomics using iTRAQ technology. J. Proteome Res. 2010, 9, 5803–5815. [Google Scholar] [CrossRef]
- Omasits, U.; Ahrens, C.H.; Müller, S.; Wollscheid, B. Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 2014, 30, 884–886. [Google Scholar] [CrossRef]
- Mani, D.R.; Abbatiello, S.E.; Carr, S.A. Statistical characterization of multiple-reaction monitoring mass spectrometry (MRM-MS) assays for quantitative proteomics. BMC Bioinform. 2012, 13 (Suppl. S16), S9. [Google Scholar] [CrossRef]
- Addona, T.A.; Abbatiello, S.E.; Schilling, B.; Skates, S.J.; Mani, D.R.; Bunk, D.M.; Spiegelman, C.H.; Zimmerman, L.J.; Ham, A.J.; Keshishian, H.; et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 2009, 27, 633–641. [Google Scholar] [CrossRef]
- Ko, S.Y.; Naora, H. Extracellular Vesicle Membrane-Associated Proteins: Emerging Roles in Tumor Angiogenesis and Anti-Angiogenesis Therapy Resistance. Int. J. Mol. Sci. 2020, 21, 5418. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Chen, J.; Hu, D.; Xie, F.; Yang, T.; Li, Z.; Wang, X.; Xiao, Y.; Zhong, J.; Jiang, Y.; et al. Advances in Biological Function and Clinical Application of Small Extracellular Vesicle Membrane Proteins. Front. Oncol. 2021, 11, 675940. [Google Scholar] [CrossRef] [PubMed]
- Addona, T.A.; Shi, X.; Keshishian, H.; Mani, D.R.; Burgess, M.; Gillette, M.A.; Clauser, K.R.; Shen, D.; Lewis, G.D.; Farrell, L.A.; et al. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat. Biotechnol. 2011, 29, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Whiteaker, J.R.; Lin, C.; Kennedy, J.; Hou, L.; Trute, M.; Sokal, I.; Yan, P.; Schoenherr, R.M.; Zhao, L.; Voytovich, U.J.; et al. A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat. Biotechnol. 2011, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Hayward, C.; Fong, P.Y.; Dominguez, M.; Hunsucker, S.W.; Lee, L.W.; McLean, M.; Law, S.; Butler, H.; Schirm, M.; et al. A blood-based proteomic classifier for the molecular characterization of pulmonary nodules. Sci. Transl. Med. 2013, 5, 207ra142. [Google Scholar] [CrossRef]
- Yu, J.S.; Chen, Y.T.; Chiang, W.F.; Hsiao, Y.C.; Chu, L.J.; See, L.C.; Wu, C.S.; Tu, H.T.; Chen, H.W.; Chen, C.C.; et al. Saliva protein biomarkers to detect oral squamous cell carcinoma in a high-risk population in Taiwan. Proc. Natl. Acad. Sci. USA 2016, 113, 11549–11554. [Google Scholar] [CrossRef]
- Kim, Y.; Yeo, I.; Huh, I.; Kim, J.; Han, D.; Jang, J.Y.; Kim, Y. Development and Multiple Validation of the Protein Multi-marker Panel for Diagnosis of Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2236–2245. [Google Scholar] [CrossRef]
- Chang, Y.T.; Chu, L.J.; Liu, Y.C.; Chen, C.J.; Wu, S.F.; Chen, C.H.; Chang, I.Y.F.; Wang, J.S.; Wu, T.Y.; Dash, S.; et al. Verification of saliva matrix metalloproteinase-1 as a strong diagnostic marker of oral cavity cancer. Cancers 2020, 12, 2273. [Google Scholar] [CrossRef]
- Gavert, N.; Sheffer, M.; Raveh, S.; Spaderna, S.; Shtutman, M.; Brabletz, T.; Barany, F.; Paty, P.; Notterman, D.; Domany, E.; et al. Expression of L1-CAM and ADAM10 in human colon cancer cells induces metastasis. Cancer Res. 2007, 67, 7703–7712. [Google Scholar] [CrossRef]
- Kalantari, E.; Razmi, M.; Tajik, F.; Asadi-Lari, M.; Ghods, R.; Madjd, Z. Significant co-expression of putative cancer stem cell markers, EpCAM and CD166, correlates with tumor stage and invasive behavior in colorectal cancer. World J. Surg. Oncol. 2022, 20, 15. [Google Scholar] [CrossRef]
- Watson, N.F.; Durrant, L.G.; Madjd, Z.; Ellis, I.O.; Scholefield, J.H.; Spendlove, I. Expression of the membrane complement regulatory protein CD59 (protectin) is associated with reduced survival in colorectal cancer patients. Cancer Immunol. Immunother. 2006, 55, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Bavi, P.; Wang, J.Y.; Roehrl, M.H. Immuno-proteomic discovery of tumor tissue autoantigens identifies olfactomedin 4, CD11b, and integrin alpha-2 as markers of colorectal cancer with liver metastases. J. Proteomics 2017, 168, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Korniluk, A.; Kamińska, J.; Kiszło, P.; Kemona, H.; Dymicka-Piekarska, V. Lectin adhesion proteins (P-, L- and E-selectins) as biomarkers in colorectal cancer. Biomarkers 2017, 22, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wen, Z.; Jiang, Q.; Zhu, L.; Feng, S.; Zhao, Y.; Wu, J.; Dong, Q.; Mao, J.; Zhu, Y. CD58, a novel surface marker, promotes self-renewal of tumor-initiating cells in colorectal cancer. Oncogene 2015, 34, 1520–1531. [Google Scholar] [CrossRef]
- Kume, H.; Muraoka, S.; Kuga, T.; Adachi, J.; Narumi, R.; Watanabe, S.; Kuwano, M.; Kodera, Y.; Matsushita, K.; Fukuoka, J.; et al. Discovery of colorectal cancer biomarker candidates by membrane proteomic analysis and subsequent verification using selected reaction monitoring (SRM) and tissue microarray (TMA) analysis. Mol. Cell. Proteomics 2014, 13, 1471–1484. [Google Scholar] [CrossRef]
- Guo, X.; Liang, X.; Wang, Y.; Cheng, A.; Qin, C.; Zhang, H.; Wang, Z. Construction and Comprehensive Prognostic Analysis of a lncRNA-miRNA-mRNA Regulatory Network and Tumor Immune Cell Infiltration in Colorectal Cancer. Front. Genet. 2021, 12, 652601. [Google Scholar] [CrossRef]
- Rollins, S.A.; Zhao, J.; Ninomiya, H.; Sims, P.J. Inhibition of homologous complement by CD59 is mediated by a species-selective recognition conferred through binding to C8 within C5b-8 or C9 within C5b-9. J. Immunol. 1991, 146, 2345–2351. [Google Scholar]
- Zhang, R.; Liu, Q.; Liao, Q.; Zhao, Y. CD59: A promising target for tumor immunotherapy. Future Oncol. 2018, 14, 781–791. [Google Scholar] [CrossRef]
- Shang, Y.; Chai, N.; Gu, Y.; Ding, L.; Yang, Y.; Zhou, J.; Ren, G.; Hao, X.; Fan, D.; Wu, K.; et al. Systematic immunohistochemical analysis of the expression of CD46, CD55, and CD59 in colon cancer. Arch. Pathol. Lab. Med. 2014, 138, 910–919. [Google Scholar] [CrossRef]
- Ouyang, Q.; Zhang, L.; Jiang, Y.; Ni, X.; Chen, S.; Ye, F.; Du, Y.; Huang, L.; Ding, P.; Wang, N.; et al. The membrane complement regulatory protein CD59 promotes tumor growth and predicts poor prognosis in breast cancer. Int. J. Oncol. 2016, 48, 2015–2024. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.J.; Wang, Z.; Liao, J.; Liu, M.; Zhong, X.R.; Zheng, H.; Wang, Y.P. CD55 and CD59 expression protects HER2-overexpressing breast cancer cells from trastuzumab-induced complement-dependent cytotoxicity. Oncol. Lett. 2017, 14, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chu, L.; Wang, Q.; Dai, W.; Zhang, X.; Chen, J.; Li, L.; Ding, P.; Zhang, L.; Gu, H.; et al. CD59 is a potential biomarker of esophageal squamous cell carcinoma radioresistance by affecting DNA repair. Cell Death Dis. 2018, 9, 887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, Q.; Peng, J.; Wang, M.; Gao, X.; Liao, Q.; Zhao, Y. Pancreatic cancer-educated macrophages protect cancer cells from complement-dependent cytotoxicity by up-regulation of CD59. Cell Death Dis. 2019, 10, 836. [Google Scholar] [CrossRef] [PubMed]
- Andreu, Z.; Yáñez-Mó, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef] [PubMed]
- Nowee, M.E.; Snijders, A.M.; Rockx, D.A.; de Wit, R.M.; Kosma, V.M.; Hämäläinen, K.; Schouten, J.P.; Verheijen, R.H.; van Diest, P.J.; Albertson, D.G.; et al. DNA profiling of primary serous ovarian and Fallopian tube carcinomas with array comparative genomic hybridization and multiplex ligation-dependent probe amplification. J. Pathol. 2007, 213, 46–55. [Google Scholar] [CrossRef]
- Feng, T.; Sun, L.; Qi, W.; Pan, F.; Lv, J.; Guo, J.; Zhao, S.; Ding, A.; Qiu, W. Prognostic significance of Tspan9 in gastric cancer. Mol. Clin. Oncol. 2016, 5, 231–236. [Google Scholar] [CrossRef]
- Qi, Y.; Qi, W.; Liu, S.; Sun, L.; Ding, A.; Yu, G.; Li, H.; Wang, Y.; Qiu, W.; Lv, J. TSPAN9 suppresses the chemosensitivity of gastric cancer to 5-fluorouracil by promoting autophagy. Cancer Cell Int. 2020, 20, 4. [Google Scholar] [CrossRef]
- Ung, T.H.; Madsen, H.J.; Hellwinkel, J.E.; Lencioni, A.M.; Graner, M.W. Exosome proteomics reveals transcriptional regulator proteins with potential to mediate downstream pathways. Cancer Sci. 2014, 105, 1384–1392. [Google Scholar] [CrossRef]
- Zhang, H.; Cai, Y.H.; Ding, Y.; Zhang, G.; Liu, Y.; Sun, J.; Yang, Y.; Zhan, Z.; Iliuk, A.; Gu, Z.; et al. Proteomics, phosphoproteomics and mirna analysis of circulating extracellular vesicles through automated and high-throughput isolation. Cells 2022, 11, 2070. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Hewapathirana, S.; García-Seisdedos, D.; Kamatchinathan, S.; Kundu, D.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A Hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Protein Name | Surrogate Peptide Sequence | Precursor mz | |
|---|---|---|---|---|
| Heavy | Light | |||
| ADAM10 | Disintegrin and metalloproteinase domain-containing protein 10 | AIDTIYQTTDFSGIR | 855.932 | 850.928 |
| ALCAM | CD166 antigen | VLHPLEGAVVIIFK | 514.988 | 512.316 |
| APMAP | Adipocyte plasma membrane-associated protein | GLFEVNPWK | 549.297 | 545.29 |
| ART4 | Ecto-ADP-ribosyltransferase 4 (CD297) | FGQFLSTSLLK | 624.857 | 620.850 |
| CD58 | Lymphocyte function-associated antigen 3 | VAELENSEFR | 602.297 | 597.293 |
| CD59 | CD59 glycoprotein | AGLQVYNK | 450.755 | 446.748 |
| CD9 | CD9 antigen | EVQEFYK | 475.739 | 471.732 |
| ICAM3 | Intercellular adhesion molecule 3 | IALETSLSK | 485.289 | 481.281 |
| ITGAM | Integrin alpha-M | LFTALFPFEK | 610.843 | 606.836 |
| RHAG | Ammonium transporter Rh type A | FLTPLFTTK | 538.317 | 534.310 |
| SELP | P-selectin | NEIDYLNK | 508.760 | 504.753 |
| TSPAN9 | Tetraspanin-9 | EGLLLYHTENNVGLK | 854.461 | 850.454 |
| TSPAN33 | Tetraspanin-33 | DDLDLQNLIDFGQK | 821.414 | 817.407 |
| TTYH3 | Protein tweety homolog 3 | VLHPLEGAVVIIFK | 768.327 | 763.323 |
| Protein | HC (n = 80) | CRC (n = 73) | CRC vs. HC | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ng/mL a | Detectable b | ng/mL a | Detectable b | Fold Change c | p-Value d | AUC | Sensitivity (%) | Specificity (%) | |
| ADAM10 | 1.83 ± 3.37 | 40/80 | 4.92 ± 2.41 | 68/73 | 2.69 | <0.0001 | 0.83 | 93.15 | 77.50 |
| ALCAM | 0.66 ± 1.38 | 31/80 | 1.76 ± 2.58 | 43/73 | 2.65 | 0.0005 | 0.65 | 56.16 | 76.25 |
| APMAP | 0.91 ± 2.91 | 21/80 | 2.64 ± 3.62 | 40/73 | 2.90 | <0.0001 | 0.67 | 39.73 | 92.50 |
| ART4 | 0.00 ± 0.02 | 2/80 | 0.13 ± 0.33 | 11/73 | 50.49 | 0.0027 | 0.56 | 15.07 | 100.00 |
| CD58 | 0.28 ± 0.72 | 31/80 | 1.54 ± 1.92 | 48/73 | 5.49 | <0.0001 | 0.72 | 60.27 | 90.00 |
| CD59 | 0.83 ± 0.94 | 77/80 | 4.35 ± 2.93 | 73/73 | 5.26 | <0.0001 | 0.95 | 100.00 | 82.50 |
| CD9 | 5.50 ± 11.29 | 36/80 | 7.66 ± 7.65 | 46/73 | 1.39 | 0.0035 | 0.63 | 53.42 | 73.75 |
| ICAM3 | 0.93 ± 1.61 | 52/80 | 1.59 ± 2.86 | 22/73 | 1.72 | 0.0424 | 0.59 | 69.86 | 65.00 |
| ITGAM | 0.08 ± 0.22 | 16/80 | 2.72 ± 11.37 | 23/73 | 32.79 | 0.0142 | 0.59 | 31.51 | 98.75 |
| RHAG | 0.10 ± 0.21 | 18/80 | 0.58 ± 2.14 | 33/73 | 5.70 | 0.0059 | 0.61 | 43.84 | 82.50 |
| SELP | 1.19 ± 2.89 | 13/80 | 0.72 ± 1.45 | 15/73 | 0.61 | 0.8257 | 0.51 | 20.55 | 83.75 |
| TSPAN33 | 0.99 ± 1.45 | 37/80 | 4.50 ± 7.81 | 23/73 | 4.53 | 0.9226 | 0.50 | 28.77 | 100.00 |
| TSPAN9 | 1.11 ± 2.06 | 69/80 | 2.42 ± 0.90 | 73/73 | 2.19 | <0.0001 | 0.87 | 100.00 | 71.25 |
| TTYH3 | 2.52 ± 6.19 | 54/80 | 1.54 ± 2.60 | 31/73 | 0.61 | 0.1323 | 0.57 | 57.53 | 67.50 |
| CEA | 3.03 ± 5.35 | 80/80 | 77.75 ± 469.49 | 73/73 | 25.68 | 0.9906 | 0.50 | 31.51 | 95 |
| Characteristics | Case No. | ADAM10 (ng/mL) | p-Value | CD59 (ng/mL) | p-Value | TSPAN9 (ng/mL) | p-Value | CEA (ng/mL) | p-Value |
|---|---|---|---|---|---|---|---|---|---|
| Gender a | - | - | - | - | - | - | - | - | - |
| Female | 37 | 4.82 ± 2.62 | 0.9781 | 4.38 ± 3.11 | 0.9257 | 2.50 ± 0.91 | 0.3766 | 126.51 ± 657.7 | 0.9715 |
| Male | 36 | 5.02 ± 2.20 | - | 4.32 ± 2.77 | - | 2.33 ± 0.90 | - | 27.63 ± 58.38 | - |
| Age (years) a | - | - | - | - | - | - | - | - | - |
| <58 c | 34 | 4.69 ± 1.81 | 0.5629 | 4.29 ± 2.75 | 0.9494 | 2.38 ± 0.88 | 0.8798 | 10.64 ± 31.55 | 0.2391 |
| ≥58 | 39 | 5.12 ± 2.84 | - | 4.41 ± 3.1 | - | 2.45 ± 0.93 | - | 136.24 ± 639.71 | - |
| Tumor stage b | - | - | - | - | - | - | - | - | - |
| T1 | 14 | 4.39 ± 1.36 | 0.4949 | 3.39 ± 0.79 | 0.4428 | 2.07 ± 0.65 | 0.2655 | 0.88 ± 0.53 | 0.0035 d |
| T2 | 9 | 5.14 ± 3.63 | - | 3.63 ± 2.72 | - | 2.25 ± 0.84 | - | 23.06 ± 60.24 | - |
| T3 | 42 | 5.15 ± 2.29 | - | 4.81 ± 3.28 | - | 2.62 ± 0.90 | - | 112.46 ± 612.04 | - |
| T4 | 8 | 4.40 ± 3.04 | - | 4.45 ± 3.42 | - | 2.15 ± 1.21 | - | 91.53 ± 214.20 | - |
| Lymph node metastasis a | - | - | - | - | - | - | - | - | - |
| N0 | 37 | 5.02 ± 2.22 | 0.0789 | 3.96 ± 1.83 | 0.1842 | 2.24 ± 0.71 | 0.0011 d | 6.16 ± 26.38 | 0.0003 d |
| N1 | 36 | 4.82 ± 2.62 | - | 4.76 ± 3.72 | - | 2.60 ± 1.05 | - | 151.32 ± 664.63 | - |
| Distant metastasis a | - | - | - | - | - | - | - | - | - |
| M0 | 57 | 4.84 ± 2.51 | 0.2103 | 4.13 ± 2.84 | 0.0475 d | 2.30 ± 0.86 | 0.0104 d | 6.97 ± 29.67 | 0.0252 d |
| M1 | 16 | 5.18 ± 2.05 | - | 5.15 ± 3.19 | - | 2.85 ± 0.94 | - | 329.87 ± 983.81 | |
| TNM stage b | - | - | - | - | - | - | - | - | - |
| Stage I | 18 | 4.87 ± 2.42 | 0.3214 | 3.50 ± 0.73 | 0.2273 | 2.15 ± 0.64 | 0.0065 d | 1.40 ± 1.67 | 0.0010 d |
| Stage II | 14 | 4.85 ± 2.20 | - | 3.37 ± 0.86 | - | 2.13 ± 0.63 | - | 2.33 ± 1.91 | - |
| Stage III | 25 | 4.83 ± 2.82 | - | 5.00 ± 4.07 | - | 2.50 ± 1.07 | - | 13.59 ± 44.37 | - |
| Stage IV | 16 | 5.18 ± 2.05 | - | 5.15 ± 3.19 | - | 2.85 ± 0.94 | - | 329.87 ± 983.81 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dash, S.; Wu, C.-C.; Wu, C.-C.; Chiang, S.-F.; Lu, Y.-T.; Yeh, C.-Y.; You, J.-F.; Chu, L.J.; Yeh, T.-S.; Yu, J.-S. Extracellular Vesicle Membrane Protein Profiling and Targeted Mass Spectrometry Unveil CD59 and Tetraspanin 9 as Novel Plasma Biomarkers for Detection of Colorectal Cancer. Cancers 2023, 15, 177. https://doi.org/10.3390/cancers15010177
Dash S, Wu C-C, Wu C-C, Chiang S-F, Lu Y-T, Yeh C-Y, You J-F, Chu LJ, Yeh T-S, Yu J-S. Extracellular Vesicle Membrane Protein Profiling and Targeted Mass Spectrometry Unveil CD59 and Tetraspanin 9 as Novel Plasma Biomarkers for Detection of Colorectal Cancer. Cancers. 2023; 15(1):177. https://doi.org/10.3390/cancers15010177
Chicago/Turabian StyleDash, Srinivas, Chia-Chun Wu, Chih-Ching Wu, Sum-Fu Chiang, Yu-Ting Lu, Chien-Yuh Yeh, Jeng-Fu You, Lichieh Julie Chu, Ta-Sen Yeh, and Jau-Song Yu. 2023. "Extracellular Vesicle Membrane Protein Profiling and Targeted Mass Spectrometry Unveil CD59 and Tetraspanin 9 as Novel Plasma Biomarkers for Detection of Colorectal Cancer" Cancers 15, no. 1: 177. https://doi.org/10.3390/cancers15010177
APA StyleDash, S., Wu, C.-C., Wu, C.-C., Chiang, S.-F., Lu, Y.-T., Yeh, C.-Y., You, J.-F., Chu, L. J., Yeh, T.-S., & Yu, J.-S. (2023). Extracellular Vesicle Membrane Protein Profiling and Targeted Mass Spectrometry Unveil CD59 and Tetraspanin 9 as Novel Plasma Biomarkers for Detection of Colorectal Cancer. Cancers, 15(1), 177. https://doi.org/10.3390/cancers15010177